

Abstract
Objective
The existence of Cl- channels in vascular smooth muscle cells (VSMCs) has been increasingly investigated, but the biological functions are not yet clear. Insulin-like growth factor (IGF)-I affects proliferation and migration of VSMCs, and dysregulation of this axis may be involved in atherogenesis and intimal hyperplasia. We examined the effects of Cl- channel blockers on IGF-I-induced proliferation in porcine VSMCs. The siRNA approach was used to support the role of ClC-2, a member of the volume-regulated Cl- channel family, in cell proliferation of VSMCs.
Method and Results
The IGF-I-induced VSMC proliferation was significantly suppressed by the Cl- channel blockers NPPB and IAA94 but not by DIDS. IGF-I-induced cell proliferation parallel a significant increase in the endogenous expression of ClC-2 mRNA and protein. Inhibitors of PI 3-kinase, LY294002 and wortmannin, significantly attenuated the IGF-I-upregulated ClC-2 expression and cell proliferation. We observed ClC-2-like Cl- current, and this current was augmented by IGF-I. SiRNA specifically targeted to ClC-2 abolished IGF-I-induced cell proliferation.
Conclusion
Our data demonstrate that ClC-2 plays a role in IGF-1-induced regulation of VSMC proliferation in cardiovascular diseases.
Keywords: Atherosclerosis, voltage-gated Cl- channels, Cl- channel blocker, Proliferation, Vascular Smooth Muscle Cells
Introduction
Abnormal vascular smooth muscle cell (VSMC) proliferation and directed migration from the media into the intima play major role in the pathogenesis of atherosclerosis [1]. These cellular events are regulated by a number of peptide growth factors including insulin-like growth factor-I (IGF-I), which is a polypeptide growth factor that binds to the type I IGF-I receptor present on many cell types, including VSMC and endothelial cells [2,3]. IGF-I plays a role in cellular growth and survival in cardiovascular tissue, especially in pathological states [4]. It has been recognized that some of the Cl- channel blockers affect the proliferation of a variety of cell types such as endothelial cells, glioma cells, intestinal enterocytes, hepatocytes and peripheral T lymphocytes [5-9]. However, it is not known whether the IGF-induced growth of VSMC can be affected by Cl- channel blockers.
Several Cl- channels have been electrophysiologically demonstrated in VSMCs. Volume-regulated Cl- channels (ClC-2, ClC-3) were expressed in human VSMCs [10]. Proliferating VSMCs have higher rate of mitosis and migration, both of which require change in cell volume and shape. It is reasonable to speculate that volume-regulated Cl- channels might play an important role in the proliferation of VSMCs. IGF-I is a mitogen and strong chemoattractant for cultured VSMCs. IGF-I mediated dephosphorylation pathways control the activity and the pharmacological properties of skeletal muscle chloride channels [11,12]. We hypothesized that volume-regulated chloride channel might contribute to IGF-I induced VSMCs proliferation and survival. To test our hypothesis, we investigated the expression and functional role of volume-regulated chloride channel in IGF-I induced VSMCs proliferation. Our findings demonstrated that Cl- channels blockers, NPPB and IAA94, inhibited both IGF-I and des-IGF-I induced cell proliferation. ClC-2 expression was up-regulated by IGF-I stimulation. We also recorded whole-cell Cl- current that showed an inward rectification which related to ClC-2 and this current was increased by IGF-I. IGF-I induced cell proliferation and Cl- current was inhibited after pre-treated with PI3 kinase inhibitor. Furthermore, knockdown of ClC-2 by siRNA prevented IGF-I induced cell proliferation. These findings have major implications for linking a specific Cl- channel to VSMC proliferation and apoptosis.
Methods
Tissue dissection and cell culture
Pig hearts and external iliac artery were excised from large-White pigs (22-28 Kg, male or female) and placed in ice-cold sterilized PBS solution. Left anterior descending coronary arteries were dissected free of surrounding tissue while maintained in ice-cold PBS solution. Both coronary and iliac arteries were rinsed in PBS supplemented with antibiotics. The endothelial layer of each artery was removed by scraping the luminal surface with a scalpel, and the adventitia was separated from the medial layer. The media was further dissected into small explants ≈ 3×3 mm in diameter, which showed outgrowth of VSMC after a few days in culture. Cells were cultured in 75-mm flasks using medium 199 with 10% FBS and 100 U/ml penicillin, 100 μg/ml streptomycin, and 250 ng/ml amphotericin B (Invitrogen) in a humidified 5% CO2 environment at 37°C. Cell cultures were identified as pure VSMC by uniform immunostaining with an antibody to smooth muscle α-actin and were used for experimentation at passage 2 or 3. Cell viability was determined by trypan blue exclusion.
The research protocol of this study was approved by the Institutional Animal Care and Use Committee of Creighton University. The investigation also conformed with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Electrophysiological recordings
The whole-cell patch-clamp technique was employed to record Cl- currents in porcine vascular smooth muscle cells. The data were analyzed with PCLAMP and whole-cell recordings were performed using an Axopatch 200B patch-clamp amplifier, pClamp 8.2 software, and a Digidata 1322A interface (Axon Instruments, Foster City, CA). The whole-cell recordings frequency response was set 5 kHz with the 8-pole Axopatch amplifier Bessel filter. The series resistance and cell capacitance were compensated for through the use of the internal circuit of the amplifier. Ag-AgCl wires were immersed in the bath as well as pipette solutions, and connected to the patch-clamp amplifier. The bath and the Ag-AgCl reference electrodes were used to minimize changes in liquid junction potential during some experiments. To obtain Cl- voltage-current relations, whole-cell currents were recorded during voltage pulses, 500 ms in length, that were applied from the holding potential of 0 mV. Potential values were corrected for the liquid-junctional potential between the bath and the patch pipette solutions. Electrophysiological experiments were performed at room temperature (20-25°C). The electrical resistances of the pipettes, when filled with pipette solution, ranged from 3.0 to 4.5 MΩ.
For the whole-cell mode of patch-clamp, the external bathing medium was CsCl saline consisted of (in mM): 140 CsCl, 1 CaCl2, 1 MgCl2, and 10 HEPES (pH 7.4). The pipette solution was CsCl saline consisting of (in mM): 140 CsCl, 1 EGTA, 0.05 CaCl2, 1 MgCl2, and 10 HEPES (pH 7.4). All reagents were purchased from Sigma Chemical Company (St Louis, MO, U.S.A). Osmolality was set to 285 ± 5 mOsmol with sucrose. For the whole-cell recording, solution osmolality was monitored using a freezing point osmometer (Microosmette, Precision systems, Natick, MA). The bathing medium was exchanged by continuous perfusion.
Cell Proliferation Assay
VSMCs were plated on 48-well culture plates with complete media. After 24 h, the media was removed and replaced with serum-free media for 24 h to achieve synchronous growth. Cells were then incubated for 48 hours after experimental stimulation, and final cell number in each well was determined.
VSMCs were also plated at a density of 50,000 cells/well in 96-well culture plates with complete media. Forty-eight hours after the experimental stimulation, VSMC bromodeoxyuridine (BrdU) uptake was determined using the cell proliferation ELISA BrdU assay (Roche; Penzberg, Germany). This assay has been previously demonstrated to be a marker for VSMC proliferation.[13] BrdU incorporation into DNA was measured by photometric analysis using a microplate reader (Bio-Rad; Hercules, CA) and was quantified by change in optical density (O.D.). Chloride channel blockers NPPB (5-nitro-2-(3-phenylpropylamino), IAA94 (R(+)-[(6,7-dichloro-2-cyclopentyl-2,3-dihydro-2-methyl-1-oxo-1H-inden-5yl)-oxy]acetic acid) and DIDS(4,4′-diisothiocyanostilbene-2,2′-disulphonic acid) were used in our experiment. Cell counts were also made at different time during cell culture exposure to drugs. In order to exclude the direct cytotoxic effects of the drugs on VSMCs, the measurement of LDH enzymatic activity was done with CytoTox96 Non-Radioactive Cytotoxicity Assay (Promega,WI). Analysis protocol was performed following the manufacturer′s instructions. In brief, the LDH in culture supernatants is measured with a 30-minute coupled enzymatic assay, which results in the conversion of a tetrazolium salt into a red formazan product. The amount of color formed is proportional to the number of lysed cells. Visible wavelength absorbance data are collected using a standard 96-well plate reader. However, there was no change in LDH activity, suggesting no cytotoxic effect of the drugs.
RT-PCR Analysis
Total RNA was extracted from VSMCs using Trizol (Invitrogen, CA) and treatment with DNase (Ambion) using the manufacturer′s protocol. The total RNA was reverse transcribed with an OmniScript RT-PCR System (QIAGEN, CA). PCR were performed with a thermal cycler (PE2400, Applied Biosystems) under the following conditions: 1 cycle at 94°C for 5 min, followed by 30-36 cycles consisting of 1 min denaturation at 94°C, 1 min annealing at 60°C, and 2 min extension at 72°C. The last cycle was extended to 5 min at 72°C. Products were analyzed by 2% (w/v) agarose gel electrophoresis. The sequences of the 5′ sense primers and the 3′ antisense primers used in this study were as follows: CIC-2, sense, 5′-AGCAGCAACTAGATGAGCCTGTCA-3′, and antisense, 5′-TCGATAGCTTTCCGGAGCTCTTT-3′ (product size, 193bp); and ClC-3, sense, 5′-CATGTCAATGGGGAGG-3′, and antisense, 5′-GCAAGAAAGGCAAAACT-3′ (product size, 164 bp).
Real-time PCR Analysis
Reverse transcription was performed using 1 μg of total RNA and oligo(dT) primers in a 20-μl reaction according to the manufacturer’s protocol (PE Applied Biosystems,Foster City, CA). Primers for CIC-2, CIC-3 and GAPDH were designed using Primer Express software (PE Applied Biosystems). The following are sequences: for CIC-2 forward primer, 5′-AGCAGCAACT AGATGAGCCTGTCA-3′; reverse primer, 5′-TGACATAAGCATGGTCCACTCCCA-3′; for ClC-3 forward primer, 5′-ATCGTCCAGCAGGCATTGGAGTAT-3′; and reverse primer, 5′-GCCTGATGGAACCTTGATGCCAAA-3′; and for GAPDH forward primer, 5′-AGGTC GGAGTGAACGGATTTGG-3′; and reverse primer, 5′-TCGCTCCTGGAAGATG GTGATG-3′. Real-time PCR was performed on the ABI Prism 7000 sequence detection system (PE Applied Biosystems) by using SYBR green (Applied Biosystems) as a dsDNA-specific binding dye. The PCR were cycled 40 times after initial denaturation (95°C, 2 min) with the following parameters: denaturation, 95°C, 15s; and annealing and extension, 60°C, 1 min. The threshold cycle (CT) was recorded for each sample to reflect the mRNA expression level. A validation experiment proved the linear dependency of the CT value for both CIC-2 or CIC-3 and GAPDH concentration and consistency of δ CT (CIC-2 or CIC-3 average CT minus GAPDH average CT) in a given sample at different RNA concentration. Therefore,δ CT was used to reflect the relative ClC-2 or ClC-3 expression levels. To determine the effects of different stimuli on CIC-2 or CIC-3 gene expression as compared with unstimulated cells,δδCT was calculated (δδCT = δCT stimulus - δCT non-stimulated cells). CIC2 or CIC3 mRNA was indexed to the GAPDH using the following formula: 1/(2 δCT) × 100%. The value of 2 δCT was calculated to demonstrate the fold changes of CIC-2 or CIC-3 gene expression in stimulated cells as compared with unstimulated cells.
Western Immunoblots
Cells were lysed using eukaryotic membrane protein extraction reagent kit (Pierce, Rockford, IL) supplemented with protease inhibitor mixture. Homogenates were centrifuged for 10 min at 12,000 × g at 4°C. Protein quantification was performed on the supernatant using a DC protein assay kit from Bio-Rad (Hercules, CA). Protein was boiled for 5 min in Laemmli-SDS sample buffer containing 600 mM β- mercaptoethanol. Equal amounts of protein were loaded into each lane of SDSPAGE gel (Bio-Rad) and resolved at 120 V constant. Gels were transferred onto PVDF membrane (Bio-Rad) at 400 mA constant for 1 hr at room temperature, and membranes were blocked in blocking buffer [13]. Blots were incubated in ClC-2 antibody and rinsed three times followed by incubation with HRP-conjugated mouse anti-rabbit secondary antibodies for 90 min. After washing blots were developed with enhanced chemiluminesence on Hyperfilm (Amersham Biosciences, Arlington Heights, IL). ClC-2 and secondary HRP-conjugated antibodies were obtained from Santa Cruz Biotechnology. GAPDH antibody was purchased from Novus Biologicals. Densitometric analysis was performed directly from the blotted membrane using a Bio-Rad Molecular Imager system.
RNA Interference and Cell Transfection
We tried to use small interfering RNA (siRNA), an effective tool to down regulate the expression of target genes in cultured mammalian cells. Two different high-performance purity grade small interfering RNAs (siRNA) to knock down ClC-2 were synthesized by Ambion Inc. One of the siRNA sequence was 5′GGGCCCUUUUGUGCAUAUCtt3′ and its corresponding complementary strand 5′GAUAUGCACAAAAGGGCCCtc3′ siRNA. Another was 5′GCCAUUACUGCUGUGA UA Gtt3′ and its corresponding complementary strand 5′CUAUCAGCAGUAAUGG Ctg3′ siRNA. Nonsilencing oligonucleotide sequence (nonsilencing siRNA) that does not recognize any known homology to mammalian genes was also generated as a negative control. Lipofectamine 2000 reagent (Invitrogen, CA) was used for transient transfection of VSMCs. Briefly, 4 × 105 cells were seeded into each well of a 6-well plate and cultured to 40-50% confluency. siRNAs were diluted in RNase-free water to a final concentration of 20 μM (20 pmol/μl). For each well, siRNA stock was mixed with 5 μl Lipofectamine 2000 in DMEM medium (serum- and antibiotic-free) to a final volume of 800 μl. After 30 min, the siRNA (200nm)/Lipofectamine 2000 complex was added to the well. Forty eight hours after transfection, gene silencing was monitored at the mRNA and protein levels by RT-PCR and Western blotting, respectively. Cell proliferation assay were also repeated in these transfected cells.
Statistical Analysis
Values for all measurements are expressed as the mean ± SEM. One-way ANOVA was used to determine the difference among various experimental groups. Statistical difference between two groups was performed by the Student t test. Values of p < 0.05 were considered as statistically significant.
Results
Cl-Channel Blockers Attenuate IGF-induced VSMC Proliferation
IGF-I stimulated a dose-dependent increase in coronary VSMC proliferation after 48 hours incubation. Cl- channel blockers alone have no effect on cell proliferation but they significantly suppressed IGF-I-induced cell proliferation (P <0.05) (Fig. 1A and 1B).
Figure 1.

Concentration-dependent stimulation of VSMC proliferation induced by IGF-I. A: VSMCs were exposed to serum free media in the presence of absence of chosen concentrations of IGF-I. B: Effect of Cl- channel blockers on VSMC proliferation. Both NPPB and IAA94 inhibited IGF-I induced VSMCs proliferation, whereas DIDS had no effect. #, p < 0.05 compared with corresponding control group. *,p < 0.05 compared with corresponding IGF-I group. Data represent mean ± SEM (n = 5).
Cl- Channel Blockers Inhibit IGF-induced DNA Synthesis
IGF-I (100 ng/ml) stimulated DNA synthesis in coronary artery VSMC s (Fig. 2). This effect of IGF-1 was attenuated by Cl- channels blockers, NPPB and IAA94 (Fig. 2). However, DIDS had no effect. The effect of both NPPB and IASA94 was dosedependent (Fig. 3) (P < 0.05).It is known that IGF-binding proteins significantly affect IGF-1-induced response. Therefore we used an IGF1 analog, Des-IGF-I, that binds to IGF-IR with the same affinity as IGF-1, but does not bind IGF-binding proteins, and examined the effect of various Cl- channels blockers on des-IGF-induced DNA synthesis in coronary artery VSMCs. Both NPPB and IAA94 inhibited des-IGF-I induced VSMC growth but DIDS had no significant effect (data not shown).
Figure 2.

Measurement of porcine VSMC DNA synthesis using bromo-deoxyuridine (BrdU). VSMCs were stimulated with IGF-I (100 ng/ml) in the presence and absence of Cl- channel blockers for 48 h, and BrdU incorporation was analyzed by photometric immunoassay. Data represent mean ± SEM (n = 5). #p < 0.05 vs control, *P < 0.05 vs. IGF-I stimulation.
Figure 3.

Dose response effect of Cl-channel blockers on IGF-I induced DNA synthesis. Both NPPB and IAA94 dose-dependently inhibited coronary VSMC proliferation induced by 100 ng/ml IGF-I. (*P < 0.05 vs. IGF-I stimulation alone). Data represent mean ±SEM of n = 5.
Expression of CIC-2 and CIC-3 mRNA in VSMCs
The mRNA expression of volume-regulated chloride channels (ClC-2 and ClC-3) in VSMCs was examined by RT-PCR. Non-stimulated coronary artery VSMCs expressed both ClC-2 and ClC-3 mRNA (Fig. 4). Real-time PCR revealed that ClC-2 mRNA expression in coronary artery VSMC was significantly up-regulated by IGF-I (100ng/ml) stimulation in a time-dependent manner (Fig. 4A). However, IGF-I stimulation did not have any significant effect on ClC-3 expression in VSMCs (data not shown).
Figure 4.

RT-PCR and Western blot analyses of ClC expression in porcine vascular smooth muscle cells. A: RT-PCR of CIC-2 and ClC-3 mRNA from unstimulated porcine coronary artery SMCs (top). qRT-PCR analysis of CIC-2 mRNA expression after 24h and 48h stimulation by IGF-I reported in relative quantitative expression (bottom panel). Total protein lysates from porcine coronary VSMCs were prepared for immunoblotting. Membranes were probed with anti-ClC-2 antibody and reprobed with anti-GAPDH antibody. Densitometric analysis shows dose- and time-dependent response for IGF-I induced ClC-2 upregulation (B and C). Data represent mean ± SEM (n = 5).
IGF-I Induced Endogenous ClC-2 expression
We examined whether IGF-I could functionally enhance endogenous ClC-2 expression, as examined by immunoblotting using a polyclonal antibody directed against ClC-2. IGF-I stimulated the expression of ClC-2 protein in a time- and dosedependent manner (Fig. 4B and 4C). These results suggest that IGF-I induced VSMC proliferation is closely associated with a corresponding increase in endogenous ClC-2 protein expression.
LY294002 and Wortmannin Inhibit IGF-I-induced ClC2 Expression in VSMCs
To determine whether the IGF-I-stimulated ClC-2 up-regulation is mediated via the PI3-kinase or MAP-kinase signaling pathway, two structurally distinct PI3-kinase inhibitors, wortmannin and LY294002, were used to block PI3-kinase activity, and PD98059 was used to block the activation of MAPK. As shown in Fig. 5, pretreatment of coronary artery VSMC with LY294002 (10 μM) or wortmannin (20 μM) attenuated IGF-I-induced ClC-2 protein expression. In contrast, PD98059 (40 μM) had no significant effect. We also examined the effect of PI3 kinase inhibitors on IGF-I-induced VSMC cell proliferation. Both LY294002 (10 μM) and wortmannin diminished IGF-I-induced cell proliferation (Fig 6).
Figure 5.
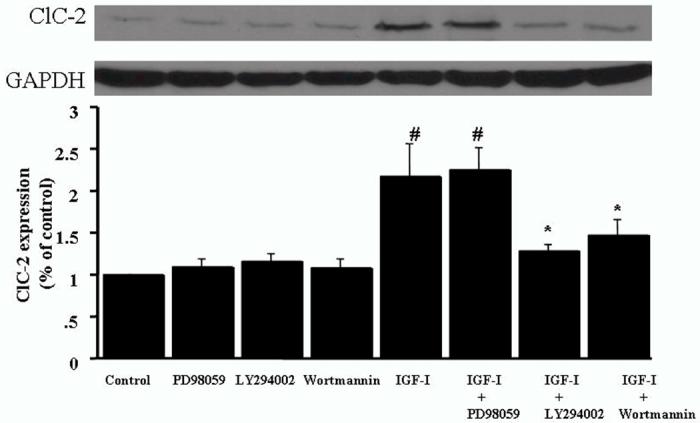
LY294002 and wortmannin inhibit IGF-I-induced ClC-2 protein expression in cultured VSMCs. Serum-starved cells were pretreated with either LY294002 (10 μM), wortmannin (20 μM), or PD98059 (40 μM) for 2 h prior to 24h stimulation by IGF-I (100 ng/ml). Western blotting was performed on the cell lysates utilizing antibody specific for ClC-2. Bar graphs correspond to representative immunoblots. Data represent mean ± SEM (n= 5). #P < 0.05 vs control, *P < 0.05 vs. IGF-I stimulation.
Figure 6.

Effect of PI3 kinase and MAP kinase inhibitors on IGF-I-induced proliferation in cultured VSMCs. Serum-starved cells were pretreated with either LY294002 (10 μM), wortmannin (20 μM), or PD98059 (40 μM) for 2 h prior to 24h stimulation by IGF-I (100 ng/ml). BrdU incorporation was analyzed by photometric immunoassay. Data represent mean ± SEM (n = 5). *P < 0.05 vs. IGF-I stimulation.
Whole-cell Cl- currents in porcine VSMCs
Whole-cell Cl- currents were recorded at different voltage before in control cells (Fig. 7A) without IGF-I and after (Fig. 7B) perfusion of 100 ng IGF-I in pig coronary artery smooth muscle cells. The whole-cell Cl- currents were also recorded from ClC-2 knock out pig coronary artery smooth muscle cells (Fig. 7 C). The whole-cell Cl- currents in normal porcine vascular smooth cells (Fig. 7A) showed an inward rectification. The I-V curve showed relationship between control (Fig. 7A) and after (Fig. 7B) perfusion of 100 ng IGF-I in porcine vascular smooth cells (Fig. 7D). We also studied the effect of wortmannin on IGF-I-induced Cl- current. IGF-I-induced Cl- current was decreased after pre-incubation with wortmannin (Fig 8). The bathing medium and the pipette solution contained CsCl saline. Holding potential was 0 mV. Voltage pulses, 500 ms in duration, were applied at 20 mV increments from -100 to +100 mV.
Figure 7.

Effect of IGF-1 on whole-cell Cl- currents in porcine VSMCs. Whole-cell Cl- currents were recorded at different voltage before in control (A) and after (B) perfusion of IGF-I (100ng/ml) in coronary VSMCs. (C) Whole-cell Cl- currents were also recorded from ClC-2 siRNA transfected coronary SMCs. The I-V curve (D) showed relationship between control (A) and after (B) perfusion of IGF-I (100ng/ml) in pig coronary artery smooth muscle cells. Whole-cell Cl- currents in control cells (A) showed an inward rectification. Representative recording of Cl-currents from individual experimental groups are shown (n = 7). The bathing medium and the pipette solution contained CsCl saline. Holding potential was 0 mV. Voltage pulses, 500 ms in duration, were applied at 20 mV increments from -100 to +100 mV.
Figure 8.
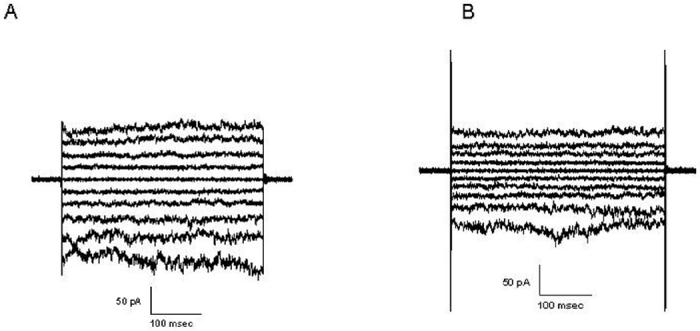
Effect of wortmannin on IGF-I induced whole-cell Cl- current in porcine VSMCs. Whole-cell Cl- currents were recorded after perfusion of IGF-I (100ng/ml) in VSMCs (A) and pre-incubated with 20 μM wortmannin (B). Representative recording of Cl-currents from individual experimental groups are shown (n = 7).
Downregulation of ClC-2 abrogates IGF-I induced Cell Proliferation
The functional significance of ClC-2 in the regulation of VSMC proliferation was assessed in VSMCs in which ClC-2 gene was silenced by siRNA. ClC-2 mRNA expression and protein abundance were markedly reduced in porcine coronary artery VSMCs transfected with siRNA, but not in cells transfected with nonsilencing siRNA. The ClC-3 expression was not affected after ClC-2 siRNA transfection. (Fig. 9A) The VSMC DNA synthesis induced by IGF-I was significantly inhibited in siRNA-transfected cells compared with nonsilencing siRNA-transfected cells (Fig. 9B).
Figure 9.

Effect of ClC-2 siRNA on ClC-2 expression in coronary artery SMCs. (A) mRNA expression and protein abundance were assessed by RT-PCR (top) and Western blotting (bottom), respectively, in VSMC transfected with ClC-2 siRNA or nonsilencing (NS) siRNA. (B) Essential role of ClC-2 in IGF-I stimulated VSMC DNA synthesis was examined. #p < 0.05 vs control, *P < 0.05 vs. IGF-I stimulation
Discussion
We demonstrated that IGF-I induced proliferation of porcine coronary VSMCs was significantly suppressed by NPPB and IAA94, and resistant to DIDS. We further observed that IGF-I-induced cell proliferation was accompanied by an increase in ClC-2 mRNA and protein expression in similar time-dependent manner. PI3-Kinase inhibitor reduced IGF-I-upregulated ClC-2 expression and cell proliferation. ClC-2 like chloride current was also observed and induced by IGF-I stimulation in our study. To our knowledge, this is the first report on the involvement of ClC-2 in IGF-I associated cell proliferation. Taking the data together, we conclude that ClC-2 Cl- channels may be linked to IGF-I induced VSMC proliferation.
In present study, we recorded whole cell Cl- currents and this current was abolished by siRNA specially target to ClC-2. These data indicate that ClC-2 may be a dominating chloride channel in porcine VSMCs. According to our result, we speculate that the activation of IGF-I might be increasing number of ClC-2 channels. The absence of knowledge of the precise function of ClC-2 makes it difficult to evaluate the physiological significance of the modulation of ClC-2 by IGF-I. However, it has been reported that the activation of ClC-2 channels require the activation of PI 3-kinase and PKC in T84 cells. We also found that PI3K inhibitor reduced IGF-I-induced ClC-2 expression as well as IGF-I promoted cell proliferation. At the same time, IGF-I induced Cl- current was inhibited by PI3 kinase inhibitor. It is possible that the binding of IGF-I to its receptors stimulates a series of rapid response, including activation of PI 3-kinase and the activity of the latter activates PI3 kinase, which in turn triggers two effects; it induce cell proliferation, and it activates chloride channel. Zhou et al reported that ClC-3 current was observed in A10 VSMCs [14]. The clonal cell line A10 was derived from the thoracic aorta of DB1X embryonic rat and may possess different properties characteristic of other type of VSMCs. Further study to clarify the different pathways involved in the regulation of ClC channels in VSMCs is warranted.
It is well known that most cells swell during the early phase of cell proliferation probably caused by water influx that accompanies changes in cell metabolism in the cell cycle. An increase in cell volume usually initiates the so-called regulatory volume decrease (RVD) process through activation of ion (K+ and Cl-) channels and transporters, which moves K+ and Cl- out of the cell to balance the water influx and returns the cell volume to its normal size[15,16]. Therefore, activation of volume-regulated chloride channel may play an essential role in cell proliferation. Amongst the most specific chloride channel blockers available which have been tested in smooth muscle are IAA-94 and NPPB. They can depress myogenic constriction and inhibit Cl- conductance. Several studies have demonstrated that selectivity of NPPB to the chloride channel in different cell type such as portal vein and cerebral arteries is concentration dependent[17,18]. It has shown that IAA-94 is effective in substantially inhibiting RVD in cardiomyocytes [19]. An IC50 ranging of NPPB and IAA94 are between 5 and 100 μM. There are several NPPB and IAA94-sensitive Cl- channels including ClC-2, ClC-3, volume-regulated anion channel (VRAC), Ca2+-activated Cl- channel (CaCC) and maxi-Cl- channel. However, the contribution of ClC-3, CaCC and maxi-chloride channel in porcine vascular smooth cell proliferation appears less because these channels are sensitive to DIDS and in our study we did not observe any significant effect of DIDS. There are conflicting reports about the responsiveness of VRAC to DIDS, probably because this channel may not be a single entity, but instead may represent several different channels that are expressed to a variable degree in different tissues [20, 21]. Another candidate of Cl- channels involved in VSMC proliferation is ClC-2, which is sensitive to NPPB and IAA94 and resistant to DIDS. ClC-2 is the only one structurally identified NPPB-sensitive and DIDS-insensitive Cl- channel. We hypothesize that ClC-2 chloride channels might be the major chloride channel that involved in cell proliferation.
IGF-I exerts its effects by interacting with IGF-IR, whose expression varies with the cellular growth status. Different expression of IGF-binding proteins may also regulate the effect of IGF-1. Des-IGF-I is an IGF-I analog that binds to IGF-IR with affinity similar to IGF-1 but does not bind to IGF-binding proteins [22]. Both Des-IGF-I and IGF-I induced cell proliferation was attenuated by chloride channel blockers. Our data indicated that the function of IGF-I is linked with ClC-2 protein through IGF-IR. IGF-IR is a transmembrane tyrosine kinase that activates two primary intracellular pathways, PI3K and extracellular signal-regulated MAPK (ERK1/2). Previous reports indicate that the activation of ClC-2 requires the activation of PI3K and protein kinase C [23]. We have also shown that PI3K inhibitor can suppress IGF-I induced ClC-2 up-regulation, suggesting the possibility that ClC-2 channels are targets for IGF-IR signaling in VSMCs and PI3K pathway may play an important role in ClC-2 protein regulation.
The critical role of ClC-2 in cell proliferation is still unclear. During cell cycle progression, cells undergo a significant increase in size especially at the G1/S phase, which perturbs cell volume homeostasis and is counterbalanced by decrease in regulatory volume [24]. Cl- channel together with the voltage-dependent K+ channel is a major player of regulatory volume decrease. Cl- permeability reportedly varies with the cell cycle phase, being low in G0 and S phases and increasing in G1/S [25]. CLH-3, an ortholog of ClC-2, is required to induce oocyte maturation [26]. The activation of CLH-3 channels occurs by serine-threonine dephosphorylation via a type 1 protein phosphatase, a feature that was also been demonstrated for rat ClC-2. These examples demonstrate intriguing functional interaction of Cl- channels with the cell cycle machinery. On the other hand, increased Cl- channel activity has been shown to coincide with entry into the cell cycle in human cervical cancer cells, and Cl- channel blockers have been shown to modulate Schwann cell proliferation [27-29]. Similarly the proliferation of C6 glioma cells and mouse liver cells is inhibited after ClC knockdown. These studies suggest that the inability to regulate cell volume may be the underlying mechanism that leads to impaired cell proliferation. However, transgenic knock-out mice for ClC-2 has not been able to detect any defects in cell volume regulation [30]. This apparent discrepancy may be attributable to a compensatory mechanism and must be reconciled by additional studies. It has also been reported that the growth of leukemic cells was suppressed most efficiently by NPPB, but the knocking down of ClC-2 protein by antisense oligonucleotide did not affect the growth of leukemic cells [24]. However, in our study normal VSMCs were used for proliferation assay. Thus the potential reasons for discrepancy could be related to the difference in the cell type and leukemic transformed cells versus primary human cells.
Wang and colleagues examined the effects of Cl- channel blockers on aortic smooth muscle cell proliferation and found that endothelin-1 induced aortic smooth muscle cell proliferation was inhibited by DIDS [31]. However in this study we did not observe any significant effect of DIDS in porcine VSMCs. This discrepancy in the responsiveness of VSMCs to DIDS could be due to difference in species and/or the mitogen used. Wang and colleagues used rat aortic VSMC and examined the effects of Cl- channel blockers on the proliferative response to endothelin-1. On the other hand, we used porcine coronary artery VSMCs and examined the effects of Cl- channel blockers on IGF-I induced cell proliferation. It is of interest that Cl- channels involved in VSMC proliferation may be stimulus- and species-specific. Several growth factors such as PDGF, FGF and VEGF have been reported to be involved in vascular smooth muscle cell proliferation and remodeling in atherosclerosis [32]. PDGF was shown to increase ClC-3 expression in canine cultured pulmonary artery smooth muscle cells [33]. Proteomic analysis of vascular endothelial growth factor-induced endothelial cell differentiation reveals a role for chloride intracellular channel 4 in tubular morphogenesis [34]. FGF-responsive telencephalic precursor cells also express functional GABA(A) receptor/Cl-channels[35]. We have not investigated the effect of these growth factors on cell proliferation and chloride channel activity. These studies may help to unveil the relationship between the growth factors and chlorides and the involvement of growth factors in the pathogenesis of atherosclerosis.
In summary, Cl- channel blockers inhibited IGF-I induced cell proliferation. ClC-2 expression was up-regulated by IGF-I stimulation. Although it is still not clear how the ClC-2 channels are involved in VSMC proliferation, our findings suggest that ClC-2 channels are important modulator of cell cycle in VSMCs and may play a role in pathological processes including intimal hyperplasia, restenosis and atherosclerosis where IGF-I induced VSMC proliferation is important in the development of vascular modeling. Clearly, further studies are necessary to elucidate the role of Cl- channels in cell proliferation and apoptosis in general.
Acknowledgement
This work was supported by the National Institutes of Health Grants R01HL070885 (to D.K.A.) and R01HL073349 (to D.K.A.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross R. Atherosclerosis: an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Libby P. Coronary artery injury and the biology of atherosclerosis: inflammation, thrombosis, and stabilization. Am J Cardiol. 2000;86:3–8. doi: 10.1016/s0002-9149(00)01339-4. [DOI] [PubMed] [Google Scholar]
- 3.Delafontaine P. Insulin-like growth factor I and its binding proteins in the cardiovascular system. Cardiovasc Res. 1995;30:825–34. [PubMed] [Google Scholar]
- 4.Bayes-Genis A, Conover CA, Schwartz RS. The insulin-like growth factor axis: a review of atherosclerosis and restenosis. Circ Res. 2000;86:125–30. doi: 10.1161/01.res.86.2.125. [DOI] [PubMed] [Google Scholar]
- 5.Voets T, Szucs G, Droogmans G, Nilius B. Blockers of volume-activated Cl- currents inhibit endothelial cell proliferation. Pflugers Arch. 1995;431:132–134. doi: 10.1007/BF00374387. [DOI] [PubMed] [Google Scholar]
- 6.Rouzaire-Dubois B, Milandri JB, Bostel S, Dubois JM. Control of cell proliferation by cell volume alterations in rat C6 glioma cells. Pflugers Arch. 2000;440:881–88. doi: 10.1007/s004240000371. [DOI] [PubMed] [Google Scholar]
- 7.O′Loughlin E-V, Pang GP, Noltorp R, Koina C, Batey R, Clancy R. Interleukin 2 modulates ion secretion and cell proliferation in cultured human small intestinal enterocytes. Gut. 2001;49:636–43. doi: 10.1136/gut.49.5.636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wondergem R, Gong W, Monen SH, Dooley SN, Gonce JL, Conner TD, et al. Blocking swelling-activated chloride current inhibits mouse liver cell proliferation. J Physiol. 2001;532:661–72. doi: 10.1111/j.1469-7793.2001.0661e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schumacher P-A, Sakellaropoulos G, Phipps DJ, Schlichter LC. Smallconductance chloride channels in human peripheral T lymphocytes. J Membr Biol. 1995;145:217–32. doi: 10.1007/BF00232714. [DOI] [PubMed] [Google Scholar]
- 10.Lamb F-S, Clayton GH, Liu BX, Smith RL, Barna TJ, Schutte BC. Expression of CLCN voltage-gated chloride channel genes in human blood vessels. J Mol Cell Cardiol. 1999;31:657–66. doi: 10.1006/jmcc.1998.0901. [DOI] [PubMed] [Google Scholar]
- 11.De luca A, Pierno S, Liantonio A, Camerino C, Conte Camerino D. Phosphorylation and IGF-1-mediated dephosphorylation pathways control the activity and the pharmacological properties of skeletal muscle chloride channels. Br J Pharmacol. 1998;125(3):477–82. doi: 10.1038/sj.bjp.0702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Law R-E, Meehan WP, Xi SP, Graf K, Wuthrich DA, Coats W, et al. Troglitazone inhibits vascular smooth muscle cell growth and intimal hyperplasia. J Clin Invest. 1996;98:1897–1905. doi: 10.1172/JCI118991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Allen R-T, Krueger KD, Dhume A, Agrawal DK. Sustained Akt/PKB activation and transient attenuation of c-jun N-terminal kinase in the inhibition of apoptosis by IGF-I in vascular smooth muscle cells. Apoptosis. 2005;10:525–35. doi: 10.1007/s10495-005-1882-3. [DOI] [PubMed] [Google Scholar]
- 14.Zhou J-G, Ren JL, Qin QY, He H, Guan YY. Regulation of intracellular Cl-concentration through volume-regulated ClC-3 chloride channels in A10 vascular smooth muscle cells. J Biol Chem. 2005;280:7301–8. doi: 10.1074/jbc.M412813200. [DOI] [PubMed] [Google Scholar]
- 15.Sardini A, Amey JS, Weylandt KH, Nobles M, Valverde MA, Higgins CF. Cell volume regulation and swelling-activated chloride channels. Biochim Biophys Acta. 2003;30:1618–153. doi: 10.1016/j.bbamem.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 16.Nilius B, Droogmans G. Amazing chloride channels: an overview. Acta Physiol Scand. 2003;177:119–47. doi: 10.1046/j.1365-201X.2003.01060.x. [DOI] [PubMed] [Google Scholar]
- 17.Jentsch T-J, Stein V, Weinreich F, Zdebik AA. Molecular structure and physiological function of chloride channels. Physiol Rev. 2002;82:503–568. doi: 10.1152/physrev.00029.2001. [DOI] [PubMed] [Google Scholar]
- 18.Large W, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl- conductance in smooth muscle. Am J Physiol Cell Physiol. 1996;271:C435–54. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- 19.Nelson M-T, Conway MA, Knot HJ, Brayden JE. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. J Physiol (Lond) 1997;502:259–64. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shen M-R, Wu SN, Chou CY. Volume-sensitive chloride channels in the primary culture cells of human cervical carcinoma. Biochim Biophys Acta. 1996;1315:138–44. doi: 10.1016/0925-4439(95)00115-8. [DOI] [PubMed] [Google Scholar]
- 21.Wang Z, Mitsuiye T, Rees SA, Noma A. Regulatory volume decrease of cardiac myocytes induced by β-adrenergic activation of the Cl- channel in guinea pig. J Gen Physiol. 1997;110:73–82. doi: 10.1085/jgp.110.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Francis G-L, Ross M, Ballard FJ, Milner SJ, Senn C, McNeil KA, et al. Novel recombinant fusion protein analogues of insulin-like growth factor (IGF)-I indicate the relative importance of IGF-binding protein and receptor binding for enhanced biological potency. J Mol Endocrinol. 1992;8:213–23. doi: 10.1677/jme.0.0080213. [DOI] [PubMed] [Google Scholar]
- 23.Bali M, Lipecka J, Edelman A, Fritsch J. Regulation of ClC-2 chloride channels in T84 cells by TGF-alpha. Am J Physiol Cell Physiol. 2001;280(6):C1588–98. doi: 10.1152/ajpcell.2001.280.6.C1588. [DOI] [PubMed] [Google Scholar]
- 24.Jiang B-H, Hattori N, Liu B, Nakayama Y, Kitagawa K, Inagaki C. Suppression of cell proliferation with induction of p21 by chloride channel blockers in human leukemic cells. Eur J Pharmacol. 2004;488:27–34. doi: 10.1016/j.ejphar.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 25.Bubien J-K, Kirk KL, Rado TA, Frizzell RA. Cell cycle dependence of chloride permeability in normal and cystic fibrosis lymphocytes. Science. 1990;248:1416–19. doi: 10.1126/science.2162561. [DOI] [PubMed] [Google Scholar]
- 26.Rutledge E, Denton J, Strange K. Cell cycle- and swelling-induced activation of a Caenorhabditis elegans ClC channel is mediated by CeGLC-7alpha/beta phosphatases. J Cell Biol. 2002;158:435–44. doi: 10.1083/jcb.200204142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shen M-R, Droogmans G, Eggermont J, Voets T, Ellory JC, Nilius B. Differential expression of volume-regulated anion channels during cell cycle progression of human cervical cancer cells. J Physiol (Lond) 2000;529:385–94. doi: 10.1111/j.1469-7793.2000.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pappas C-A, Ritchie JM. Effect of specific ion channel blockers on cultured Schwann cell proliferation. Glia. 1998;22:113–20. [PubMed] [Google Scholar]
- 29.Wilson GF, Chiu S-Y. Mitogenic factors regulate ion channels in Schwann cells cultured from newborn rat sciatic nerve. J Physiol (Lond) 1993;470:501–20. doi: 10.1113/jphysiol.1993.sp019872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nehrke K, Arreola J, Nguyen HV, Pilato J, Richardson L, Okunade G, et al. Loss of hyperpolarization-activated Cl(-) current in salivary acinar cells from Clcn2 knockout mice. J Biol Chem. 2002;277:23604–11. doi: 10.1074/jbc.M202900200. [DOI] [PubMed] [Google Scholar]
- 31.Wang G-L, Wang XR, Lin MJ, He H, Lan XJ, Guan YY. Deficiency in ClC-3 chloride channels prevents rat aortic smooth muscle cell proliferation. J Neurosci. 2003;23(13):5572–82. doi: 10.1161/01.res.0000042062.69653.e4. [DOI] [PubMed] [Google Scholar]
- 32.Eyries M, Collins T, Khachigian LM. Modulation of growth factor gene expression in vascular cells by oxidative stress. Endothelium. 2004;11(2):133–9. doi: 10.1080/10623320490482691. [DOI] [PubMed] [Google Scholar]
- 33.Dai YP, Bongalon S, Hatton WJ, Hume JR, Yamboliev IA. ClC-3 chloride channel is upregulated by hypertrophy and inflammation in rat and canine pulmonary artery. Br J Pharmacol. 2005;145(1):5–14. doi: 10.1038/sj.bjp.0706135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bohman S, Matsumoto T, Suh K, Dimberg A, Jakobsson L, Yuspa S, et al. Proteomic analysis of vascular endothelial growth factor-induced endothelial cell differentiation reveals a role for chloride intracellular channel 4 (CLIC4) in tubular morphogenesis. J Biol Chem. 2005;280(51):42397–404. doi: 10.1074/jbc.M506724200. [DOI] [PubMed] [Google Scholar]
- 35.Ma W, Liu QY, Maric D, Sathanoori R, Chang YH, Barker JL. Basic FGF-responsive telencephalic precursor cells express functional GABA(A) receptor/Cl-channels in vitro. J Neurobiol. 1998;35(3):277–86. doi: 10.1002/(sici)1097-4695(19980605)35:3<277::aid-neu5>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]