

Abstract
TREX1 constitutes the major 3′→5′ DNA exonuclease activity measured in mammalian cells. Recently, biallelic mutations in TREX1 have been shown to cause Aicardi-Goutières syndrome at the AGS1 locus. Interestingly, Aicardi-Goutières syndrome shows overlap with systemic lupus erythematosus at both clinical and pathological levels. Here, we report a heterozygous TREX1 mutation causing familial chilblain lupus. Additionally, we describe a de novo heterozygous mutation, affecting a critical catalytic residue in TREX1, that results in typical Aicardi-Goutières syndrome.
TREX1 (GenBank accession numbers AAK07616 and NM_033627) represents the major DNA-specific 3′→5′ exonuclease activity measured in mammalian cells.1 The nonprocessive autonomous mode of action of TREX1 suggested a possible proofreading role during lagging-strand DNA synthesis or a gap-filling role during DNA repair.2 However, Trex1-null mice show no increase in spontaneous mutation frequency or cancer incidence.3 Rather, they develop an inflammatory myocarditis with progressive dilated cardiomyopathy, indicating a previously unrecognized cellular role for the enzyme. Of note, although there was a dramatically increased morbidity of Trex1-null mice postweaning, these mice did not exhibit any obvious neurological defect.
Aicardi-Goutières syndrome (AGS [MIM 225750]) is a genetically determined encephalopathy characterized by calcification of the basal ganglia and white matter, demyelination, and raised levels of lymphocytes in the cerebrospinal fluid.4 Neurological dysfunction becomes clinically apparent in infancy and manifests as progressive microcephaly, spasticity, dystonia, and psychomotor retardation. We identified the AGS1 locus elsewhere,5 and we recently demonstrated that biallelic mutations in TREX1 result in AGS at the AGS1 locus6 and that mutations in genes encoding the three nonallelic components of the RNASEH2 protein complex also cause AGS.7
We and others have drawn attention to features suggesting immune dysfunction in AGS, and a number of reports have specifically highlighted the phenotypic overlap of AGS with the autoimmune syndrome systemic lupus erythematosus (SLE).8–12 Recently, Lee-Kirsch et al.13 described a family segregating a monogenic form of cutaneous lupus in which affected individuals presented with ulcerating lesions of the skin in acral locations, features highly reminiscent of the chilblains seen in some children with AGS.14 They mapped this disease, termed “familial chilblain lupus” (FCL [MIM 610448]), to the AGS1 critical interval and drew attention to the phenotypic overlap with AGS.
Here, we describe a family with FCL that segregates a heterozygous pathogenic mutation in TREX1 as a dominant trait. Additionally, we report a child with typical AGS whose condition results from a de novo heterozygous mutation in TREX1. These novel findings show that certain rare mutations in the TREX1 gene can result in a dominant phenotype with symptoms of FCL and/or AGS.
We ascertained a nonconsanguineous Bangladeshi family in which two brothers and a sister demonstrate features of FCL (fig. 1 and table 1). Their father had been similarly affected throughout his life but was not available for study. There are three siblings without symptoms, one of whom was also studied, and their mother is healthy, with no features of disease in her 8th decade. The onset of the disease was in early childhood. Affected individuals presented with painful bluish-red swelling of the skin affecting mainly the fingers, toes, ears, helices, and, occasionally, the nose. The lesions were induced by cold temperatures and were significantly worse in the winter months. These lesions could ulcerate; in the two affected males, the ulcerations led to a loss of ear cartilage and destruction of the proximal interphalangeal (PIP) joints and distal toes (fig. 2). Ulcerative lesions healed but left areas of atrophic and hypopigmented skin. There was no history of photosensitivity, fever, weight loss, immune deficiency, or malignancy. There was no associated Raynaud phenomenon, and there was no response to nifedipine or to a 3-d infusion of the prostacyclin analog iloprost. Treatment with prednisolone, sulfasalazine, and hydroxychoroquine was also unhelpful. One patient experienced swelling of the knee on two occasions, and joint aspiration demonstrated a sterile effusion consistent with a seronegative inflammatory athropathy. Skin biopsy was never performed. There was no evidence of cryoglobulinemia, cryofibrinogenemia, cold agglutinins, or hepatitis C. Tests for rheumatoid factor, anticardiolipin antibodies, and extractable nuclear antigens were consistently negative. However, antinuclear antibody titres were intermittently raised, and one patient exhibited persistently elevated erythrocyte-sedimentation rate (ESR).
Figure 1. .

Pedigree of the nonconsanguineous family showing vertical transmission of FCL (affected individuals are indicated by blackened symbols). Note that individual II:4 is clinically asymptomatic but carries the familial disease-causing mutation.
Table 1. .
Summary of Clinical Features in the FCL-Affected Family
| Subject and Clinical Description | Laboratory Findings | Comments |
| II:3: | ||
| Presentation as an infant. Lesions affecting hands and feet, especially over PIP joints, sides of fingers, and ends of toes. Active disease associated with extension of swelling and redness along the digit, resulting in functional loss with pain for the duration of the swelling. No involvement of ears or nose. Large joint arthralgias, mainly of the knees, without swelling. In the quiescent periods, she is left with scarred, atrophic, and hypopigmented lesions. | Variable antinuclear antibodies, ranging from normal to 1:1,000 (homogeneous). Persistently raised ESR (20–40; normal <3). Negative extractable nuclear antigens (Sm, RNP, SCl70, Jo1, PM1, Ro, and La), anti–double-stranded DNA (anti-dsDNA), pANCA/cANCA, cardiolipin, rheumatoid factor, hepatitis C, cryoglobulins, and cryofibrinogen. Normal C3 and C4. | Treatment with hydroxychoroquine and sulfasalazine not tolerated. No response to prednisolone or iloprost. |
| II:5: | ||
| Initial presentation at age 4 years, with recurrent cold fingers and toes. Digits swelled in cold weather, becoming red and “peeled.” Received diagnosis of chilblains. Continued episodes associated with pain and tissue infarcts and ulceration involving the digits, tip of the nose, and pinnae. Eventual “autoamputation” of the 4th right toe. Two episodes of frank knee swelling and effusion. He is left with marked tapering of the digits, with tightening of the skin and prominence of the PIP and distal interphalangeal joints. | Antinuclear antibodies 1:100 (homogeneous) on one occasion. Left synovial knee aspirate showed 13,000 white cells/mm3. Negative extractable nuclear antigens, anti-dsDNA, pANCA/cANCA, cardiolipin, rheumatoid factor, cryoglobulins, cryofibrinogen, C3, and C4. | X-ray of hands demonstrated resorption of terminal phalanges of all toes, left little and ring fingers, and right little finger, with subluxation of the PIP joint of the left little finger. No response to nifedipine or epoprostenol. |
| II:6: | ||
| Presentation at age 4 years. Features similar to those of subject II:5, although he has not experienced episodes of large joint swelling. | No data available. |
Figure 2. .

Skin features observed in FCL-affected family. The lesions are quiescent at present, but there are residual areas of atrophic and hypopigmented skin. Previous ulcerations have led to a loss of ear cartilage and destruction of the PIP joints and distal toes. Note the tapering of the fingers, with tight, shiny skin.
Sequencing of the single-exon TREX1 gene revealed that all three affected individuals carried a c.375dupT and a c.50T→C transition that resulted in the replacement of a phenylalanine with a serine at position 17 (p.F17S). Cloning into a pGEM vector system demonstrated that these variants were present on different alleles. The mother is heterozygous for the c.50T→C change only. Sequencing of the RNASEH2A, -B, and -C genes was normal. The TREX1 c.375dupT is predicted to result in a truncated protein missing the last 188 aa. Despite its position within the EXO I domain, one of three sequence motifs containing four acidic residues participating in the coordination of divalent metal ions necessary for catalysis—the phenylalanine at position 17—is not conserved in mouse Trex1 or the related human TREX2.2,15 Moreover, the purified recombinant TREX1 F17S/F17S homodimer exhibited normal 3′→5′ exonuclease activity, indicating that this amino acid change does not adversely affect the catalytic activity of the recombinant TREX1 when measured in vitro (table 2).16 Thus, although the F17S change was not seen in a panel of 50 Asian control individuals, these data—together with the history of chilblains in the father, who must harbor the c.375dupT—led us to conclude that c.50TC is a rare polymorphism. Interestingly, one sister (II:4) was unaffected on clinical examination but carried the same molecular changes seen in her affected siblings. Further testing in this asymptomatic woman revealed a significant lymphopenia (1.16×109/liter; normal 1.5–4.0×109/liter) and raised ESR (23 mm/1st hour; normal 0–7 mm/1st hour), suggesting subclinical penetrance. Results of serum interferon alpha (IFN-α) testing in one symptomatic patient were normal.
Table 2. .
TREX1 Enzyme Activity of Wild-Type and Recombinant F17S/F17S Homodimers
| Dimer | Activitya | Relative Activity |
| Wild-type/wild-type | 2.8 | 1 |
| F17S/F17S | 3.5 | 1.2 |
Measured as fmol nucleotides released per s.
Lymphoblastoid cell lines were established from affected individuals, and TREX1 exonuclease activity in cell extracts was measured, as described elsewhere, with use of a polydeoxynucleotide substrate.3 Extracts of control lymphoblastoid cell lines exhibited readily detected TREX1 activity. In contrast, lymphoblastoid cell lines derived from all three patients with FCL showed exonuclease activity with a consistently marked reduction, to ∼15%–35% that of a normal lymphoblastoid control, but clearly more than seen in an AGS cell line6 with biallelic inactivating TREX1 mutations (fig. 3).
Figure 3. .

TREX1 exonuclease activity. Cell-free protein extracts were assayed for 3′ DNA exonuclease activity with a 3′ labeled poly(dA) substrate after partial purification by column chromatography on single-stranded DNA cellulose. GM0558 is a normal lymphoblastoid control. F39A is a lymphoblastoid cell line from a child with biallelic mutations in TREX1.
We also ascertained a child with a classic history of AGS born to nonconsanguineous Scottish parents. He presented at age 4 mo with developmental delay. Cerebrospinal fluid examination at age 3 years demonstrated 4 white cells/mm3 and a raised titre of IFN-α, 6 IU/liter (normal <2 IU/liter). Magnetic resonance imaging showed demyelination, and calcification of the basal ganglia was seen on CT scan. At age 7 years, he was profoundly delayed, with no meaningful communication, and was fed by gastrostomy tube. He demonstrated severe spasticity with dystonic posturing and was microcephalic. He had never experienced seizures. He had several chilblainlike lesions on his toes and hands and a more generalized patchy mottling of the skin on all four limbs and over his trunk (fig. 4). These lesions first developed at age ∼12 mo and, although present throughout the year, were significantly worse in the winter.
Figure 4. .

Skin features seen in the child with AGS due to a de novo heterozygous D200N TREX1 mutation. Ulcerative lesions are seen at the ends of the toes and fingers, and there is a more generalized patchy mottling of the skin, seen here on the legs.
Sequencing of TREX1 revealed a heterozygous c.598G→A mutation resulting in a D→N substitution at amino acid residue 200 (fig. 5). Both parents had a homozygous wild-type genotype at this position. Differentiation of the maternal and paternal alleles was possible because of a frequently observed C→T SNP at position 531 and subsequent sequencing of single clones, which allowed us to demonstrate that the mutation had arisen on the maternal allele. Genotyping at several loci was consistent with maternity and paternity. This mutation was not present on 210 European control alleles, and sequencing of the RNASEH2A, -B, and -C genes was normal. Using the same method as above, we measured TREX1 activity in a lymphoblastoid cell line from the affected child. Interestingly, and in contrast to AGS-affected patients with disease resulting from biallelic TREX1 mutations, we found that TREX1 exonuclease activity was within 50%–100% of the activity of a normal lymphoblastoid control (fig. 3), comparable to the level seen in cells from unaffected heterozygous parents of subjects with recessive AGS.6
Figure 5. .
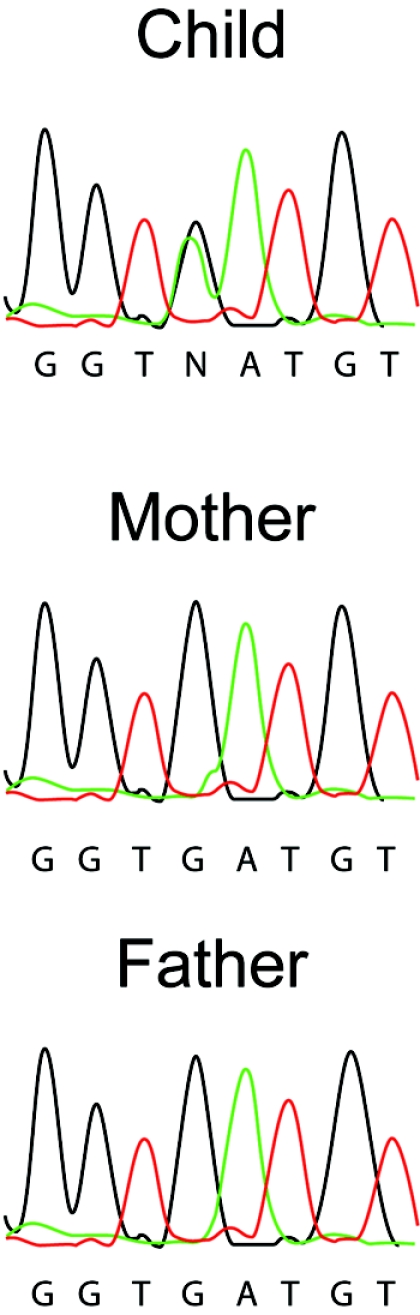
Sequence electropherograms illustrating the de novo c.598G→A TREX1 mutation in the child affected with AGS.
Our previous analyses of AGS-affected patients with biallelic TREX1 mutations indicated that AGS at the AGS1 locus results from an abrogation of TREX1 enzyme activity.6 The TREX1 protein functions as a dimer.17 We documented an ∼75% reduction of TREX1 activity in FCL-affected heterozygotes, which is consistent with the heterozygous inactivating c.375dupT mutation interfering with the function of a dimeric molecule. Importantly, our data show that certain heterozygous mutations lead to strongly diminished levels of TREX1 activity and that FCL can be due to AGS1 mutations. In contrast, enzymatic analysis of the AGS-affected patient we describe here demonstrated close-to-normal activity in a standard exonuclease assay. On the basis of homology with TREX2, the aspartic acid at position 200 in TREX1 represents one of four residues essential for coordinating two magnesium ions involved in DNA binding and catalysis.18 It seems likely that D200N represents a gain-of-function mutation conferring altered substrate specificity, DNA binding, or protein-protein interactions; these would not be detected in the standard TREX1 exonuclease assay.
The observation of heterozygous TREX1 mutations producing an enzymatic and clinical phenotype is novel and relevant. AGS has been considered an autosomal recessive disorder. Our ongoing genetic studies confirm that this is true in most cases, but we show here that AGS can also occur as a result of a de novo mutation in the TREX1 gene, a finding with obvious implications for genetics counseling. No major reduction of enzyme activity was observed in the few TREX1 heterozygotes tested elsewhere,6 and it has been considered that the parents of patients with AGS show no overt phenotype. However, a recent survey of AGS-affected families (Y.J.C., unpublished data) revealed that 4–40 parents experienced intermittent chilblains following exposure to cold temperatures, although none reported the severe and persistent skin lesions described here. The explanation for the severity of the lesions seen in the FCL-affected family we present remains uncertain, but the observation of nonpenetrance in one sibling is consistent with the lack of symptoms experienced by most subjects heterozygous for an AGS mutation. It is of interest that we know one parent of a patient with AGS who has SLE, and we suggest that a systematic study of such parents for biomarkers of SLE is warranted.
In view of the consistent association with elevated levels of IFN-α,4 we predicted elsewhere that elucidation of the molecular basis of AGS would provide insights into the pathogenesis of SLE.8 Our recent finding in AGS-affected patients of mutations in genes encoding the three nonallelic components of the RNASEH2 endonuclease protein complex7 and in the 3′→5′ exonuclease TREX1,6 together with the observation that TREX1 may be involved in caspase-independent apoptosis,19 adds weight to this prediction, since abnormalities of IFN-α metabolism20 and apoptosis21 represent two central themes in the causation of lupus and other autoimmune diseases. We have now identified heterozygous TREX1 mutations in a pedigree segregating a monogenic form of FCL. Of note in the context of AGS, mutations in DNase I are associated with a lupus phenotype,22 and mice deficient in DNase II accumulate undigested DNA in macrophages, with consequent IFN-β up-regulation, and recapitulate the clinical and immunological phenotype of rheumatoid arthritis.23 We have suggested6 that AGS results from a failure of nuclease activities, with consequent accumulation of anomalous nucleic acid species triggering an IFN-α–mediated innate immune response. For these reasons, further evaluation of the specific biochemical defect(s) in TREX1 function underlying dominant AGS and/or FCL should give general insights into the etiology of inherited lupus.
Acknowledgments
We thank the families for their cooperation in this study. This work was supported by the Birth Defects Foundation and the Leeds Teaching Hospitals Charitable Foundation. F.W.P. is in receipt of grant GM069962 from the National Institutes of Health. We thank Professor Pierre Lebon and Dr. Tony Meager, for measurement of IFN-α; Professor David Bonthron, for helpful discussions; and Mr. Bob Smith, for his expert graphics skills.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
- GenBank, http://www.ncbi.nlm.nih.gov/GenBank/ (for TREX1 protein [accession numbers AAK07616 and NM_033627, with the A at 2986 as the first base of the initiating ATG codon])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for AGS and FCL)
References
- 1.Lindahl T, Gally JA, Edelman GM (1969) Properties of deoxyribonuclease 3 from mammalian tissues. J Biol Chem 244:5014–5019 [PubMed] [Google Scholar]
- 2.Hoss M, Robins P, Naven TJ, Pappin DJ, Sgouros J, Lindahl T (1999) A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. EMBO J 18:3868–3875 10.1093/emboj/18.13.3868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morita M, Stamp G, Robins P, Dulic A, Rosewell I, Hrivnak G, Daly G, Lindahl T, Barnes DE (2004) Gene-targeted mice lacking the Trex1 (DNase III) 3′→5′ DNA exonuclease develop inflammatory myocarditis. Mol Cell Biol 24:6719–6727 10.1128/MCB.24.15.6719-6727.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goutieres F, Aicardi J, Barth PG, Lebon P (1998) Aicardi-Goutières syndrome: an update and results of interferon-α studies. Ann Neurol 44:900–907 10.1002/ana.410440608 [DOI] [PubMed] [Google Scholar]
- 5.Crow YJ, Jackson AP, Roberts E, van Beusekom E, Barth P, Corry P, Ferrie CD, Hamel BCJ, Jayatunga R, Karbani G, et al (2000) Aicardi-Goutières syndrome displays genetic heterogeneity with one locus (AGS1) on chromosome 3p21. Am J Hum Genet 67:213–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van BokHoven H, Brunner HG, Hamel BC, et al (2006) Mutations in the gene encoding the 3′–5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 38:917–920 10.1038/ng1845 [DOI] [PubMed] [Google Scholar]
- 7.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, et al (2006) Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat Genet 38:910–916 10.1038/ng1842 [DOI] [PubMed] [Google Scholar]
- 8.Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, Michaud J, Roberts E, Stephenson JB, Woods CG, et al (2003) Cree encephalitis is allelic with Aicardi-Goutières syndrome; implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet 40:183–187 10.1136/jmg.40.3.183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dale RC, Tang SP, Heckmatt JZ, Tatnall FM (2000) Familial systemic lupus erythematosus and congenital infection-like syndrome. Neuropediatrics 31:155–158 10.1055/s-2000-7492 [DOI] [PubMed] [Google Scholar]
- 10.Aicardi J, Goutières F (2000) Systemic lupus erythematosus or Aicardi-Goutières syndrome? Neuropediatrics 31:113 10.1055/s-2000-7533 [DOI] [PubMed] [Google Scholar]
- 11.Rasmussen M, Skullerud K, Bakke SJ, Lebon P, Jahnsen FL (2005) Cerebral thrombotic microangiopathy and antiphospholipid antibodies in Aicardi-Goutières syndrome—reports of two sisters. Neuropediatrics 36:40–44 10.1055/s-2004-830532 [DOI] [PubMed] [Google Scholar]
- 12.De Laet C, Goyens P, Christophe C, Ferster A, Mascart F, Dan B (2005) Phenotypic overlap between infantile systemic lupus erythematosus and Aicardi-Goutières syndrome. Neuropediatrics 36:399–402 10.1055/s-2005-873058 [DOI] [PubMed] [Google Scholar]
- 13.Lee-Kirsch MA, Gong M, Schulz H, Rüschendorf F, Stein A, Pfeiffer C, Ballarini A, Gahr M, Hubner N, Linné M (2006) Familial chilblain lupus, a monogenic form of cutaneous lupus erythematosus, maps to chromosome 3p. Am J Hum Genet 79:731–737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tolmie JL, Shillito P, Hughes-Benzie R, Stephenson JB (1995) The Aicardi-Goutieres syndrome (familial, early onset encephalopathy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis). J Med Genet 32:881–884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mazur DJ, Perrino FW (1999) Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′→5′ exonucleases. J Biol Chem 274:19655–19660 10.1074/jbc.274.28.19655 [DOI] [PubMed] [Google Scholar]
- 16.Perrino FW, Krol A, Harvey S, Zheng SL, Horita DA, Hollis T, Meyers DA, Isaacs WB, Xu J (2004) Sequence variants in the 3′→5′ deoxyribonuclease TREX2: identification in a genetic screen and effects on catalysis by the recombinant proteins. Adv Enzyme Regul 44:37–49 [DOI] [PubMed] [Google Scholar]
- 17.Mazur DJ, Perrino FW (2001) Excision of 3′ termini by the TREX1 and TREX2 3′→5′ exonucleases. J Biol Chem 276:17022–17029 10.1074/jbc.M100623200 [DOI] [PubMed] [Google Scholar]
- 18.Perrino FW, Harvey S, McMillin S, Hollis T (2005) The human TREX2 3′→5′-exonuclease structure suggests a mechanism for efficient nonprocessive DNA catalysis. J Biol Chem 280:15212–15218 10.1074/jbc.M500108200 [DOI] [PubMed] [Google Scholar]
- 19.Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, Perrino FW, Lieberman J (2006) The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol Cell 23:133–142 10.1016/j.molcel.2006.06.005 [DOI] [PubMed] [Google Scholar]
- 20.Banchereau J, Pascual V (2006) Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25:383–392 10.1016/j.immuni.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 21.Cohen PL (2006) Apoptotic cell death and lupus. Springer Semin Immunopathol 2006 28:145–152 10.1007/s00281-006-0038-z [DOI] [PubMed] [Google Scholar]
- 22.Yasutomo K, Horiuchi T, Kagami S, Tsukamoto H, Hashimura C, Urushihara M, Kuroda Y (2001) Mutation of DNASE1 in people with Systemic lupus erythematosus. Nat Genet 28:313–314 10.1038/91070 [DOI] [PubMed] [Google Scholar]
- 23.Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, Yoshikawa H, Nagata S (2006) Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature 443:998–1002 10.1038/nature05245 [DOI] [PubMed] [Google Scholar]