

Abstract
The accumulation of mildly deleterious missense mutations in individual human genomes has been proposed to be a genetic basis for complex diseases. The plausibility of this hypothesis depends on quantitative estimates of the prevalence of mildly deleterious de novo mutations and polymorphic variants in humans and on the intensity of selective pressure against them. We combined analysis of mutations causing human Mendelian diseases, of human-chimpanzee divergence, and of systematic data on human genetic variation and found that ∼20% of new missense mutations in humans result in a loss of function, whereas ∼27% are effectively neutral. Thus, the remaining 53% of new missense mutations have mildly deleterious effects. These mutations give rise to many low-frequency deleterious allelic variants in the human population, as is evident from a new data set of 37 genes sequenced in >1,500 individual human chromosomes. Surprisingly, up to 70% of low-frequency missense alleles are mildly deleterious and are associated with a heterozygous fitness loss in the range 0.001–0.003. Thus, the low allele frequency of an amino acid variant can, by itself, serve as a predictor of its functional significance. Several recent studies have reported a significant excess of rare missense variants in candidate genes or pathways in individuals with extreme values of quantitative phenotypes. These studies would be unlikely to yield results if most rare variants were neutral or if rare variants were not a significant contributor to the genetic component of phenotypic inheritance. Our results provide a justification for these types of candidate-gene (pathway) association studies and imply that mutation-selection balance may be a feasible evolutionary mechanism underlying some common diseases.
Many common human diseases have a strong heritable component. Although the field of modern human genetics has been incredibly successful in determining the genetic causes of rare Mendelian diseases, complex diseases have proven to be a more challenging problem.1,2 Genetic variation that influences an individual’s susceptibility to most such diseases is still largely unidentified. Of special interest are missense mutations, since many of them are believed to have nonmarginal functional effects.1 The effects of a missense mutation on molecular function, phenotype, and organism fitness can be extremely diverse. A missense mutation can be lethal or can cause severe Mendelian disease; alternatively, it can be mildly deleterious, effectively neutral, or beneficial. Knowledge of relative frequencies of these types of mutations and their contributions to population genetic variation is important for understanding the evolutionary background of common disease and can inform design of human genetic studies.
The effect of a missense mutation on an organism is always multifaceted and can be considered from multiple perspectives—biochemical, medical, and evolutionary. The relationship between the effects of amino acid substitution on protein activity, human health, and an individual’s evolutionary fitness is not trivial. A mutation that damages protein structure does not necessarily lead to a detectable human-disease phenotype, and a mutation that predisposes an individual toward a disease is not necessarily evolutionarily deleterious. Fixation of completely deactivating mutations (pseudogenezation) was apparently a common event during recent human evolution,3 indicating that a mutation that abolishes protein activity is not necessarily subject to purifying selection. Substitutions leading to abnormal hemoglobin function that cause sickle-cell anemia are apparently negative from both biochemical and medical points of view. Nevertheless, they cannot be considered negative from an evolutionary point of view, because balancing selection has brought them to high frequency in many parts of the world as a result of malaria resistance in heterozygotes. To clearly distinguish different aspects of negative mutations, we use the term “damaging” to refer to a mutation that decreases protein activity, the term “detrimental” to refer to a mutation that predisposes an individual toward a disease, and the term “deleterious” to refer to a mutation that has been subject to purifying selection.
A high incidence rate for many complex diseases suggests that a surprisingly high cumulative frequency of medically detrimental variants should be present in the human population. It remains uncertain why such polymorphisms can persist without being eliminated by purifying selection. Currently, two major lines of reasoning exist that explain this apparent paradox. The first considers various complex evolutionary scenarios and treats positive or balancing selection as a major force that can drive medically detrimental mutations to high frequencies. The second line of reasoning postulates a high mutation rate as a major factor that determines the cumulative frequency of detrimental polymorphisms in the population.
The first hypothesis states that the majority of polymorphisms predisposing an individual to a complex disease, although medically detrimental at the present time, were not evolutionarily deleterious. There are several possible phenomena that might help a polymorphic variant that confers susceptibility to a disease phenotype escape purifying selection. One of them is a late disease onset, when detrimental phenotypic consequences of a mutation strike after reproductive age, and, thus, do not affect the individual’s number of offspring. Several earlier studies, however, showed that this phenomenon is unlikely to be common. These studies suggest that most of the mutations that affect phenotypes at an old age also have a small pleiotropic effect earlier in life.4,5 Changing direction of selection is another mechanism that can explain how presently detrimental mutations could have escaped purifying selection.6 Human lifestyle, environment, and nutrition have changed dramatically, and some mutations that were neutral or even beneficial in hunter-gatherer societies tens of thousands of years ago might have become medically detrimental in modern human society. The best-known example of this type of reasoning is the “thrifty genes” hypothesis,7 which postulates that polymorphisms that predispose modern humans to obesity and are presently medically detrimental were able to rise to high frequency in the population because of associated selective advantages at the times of scarce food sources. Yet another mechanism, balancing selection, can also maintain deleterious mutations in a population if heterozygous individuals have a strong evolutionary advantage.8 A classic example of this mechanism is the hemoglobin mutation that is protective against malaria in a heterozygote state and simultaneously leads to sickle-cell anemia in a homozygous state.9 The fourth phenomenon that can lead to a high frequency of medically detrimental mutation is antagonistic pleiotropy, in which the negative effect of mutation on one trait is compensated by its positive effect on another.10 None of these evolutionary scenarios, however, have been shown to be frequent enough to account for a large number of human common complex diseases.
The second theory postulates that the majority of medically detrimental polymorphisms are also mildly evolutionarily deleterious, and the observed frequency of disease-predisposing genetic variation is the result of mutation-selection balance. The key assumption here is that the majority of disease-causing mutations are both medically detrimental and evolutionarily deleterious, but the pressure of purifying selection acting on them is reasonably weak. A high rate of mildly deleterious mutations associated with disease risk counterbalances the action of purifying selection.
Mutation-selection balance was, initially, treated theoretically by Kimura.11 Then, it was both advocated12 and argued against13 as a mechanism capable of maintaining a high level of genetic variance in natural populations. In the last decade, partially because of the popularity of the common disease–common variant hypothesis, mutation-selection balance was frequently overlooked as a mechanism that can explain the existence of many common, harmful, heritable disorders. Recently, however, mutation-selection balance has been put forward again as a plausible mechanism for the maintenance of deleterious genetic variation.14–16
Obviously, mutation-selection balance can become a feasible evolutionary explanation for common disease only if a sufficient fraction of de novo mutations in humans are mildly deleterious. The estimation of the fraction of mildly deleterious missense mutations and the corresponding fraction among low-frequency human polymorphisms is the subject of this work.
The evolutionary origin of present-day detrimental polymorphism has an important implication for the spectrum of disease-predisposing alleles. If the majority of medically detrimental mutations were not evolutionarily deleterious, they might have risen to a high frequency in the population, and thus the common disease–common variant hypothesis is viable. On the contrary, if the majority of medically detrimental polymorphisms are mildly deleterious and their cumulative high frequency in the population is being maintained by mutation-selection balance, then the common disease–rare variant alternative hypothesis is likely to hold true. Given the high incidence rate of many complex diseases, the mutation-selection balance hypothesis can be feasible only if a large fraction of de novo mutations is associated with moderate selection coefficients, so that, despite being deleterious, they can still achieve detectable frequencies in the human population.
Earlier theoretical studies in the framework of a mutation-selection model of common disease focused on the frequency spectrum of susceptibility alleles. Pritchard17 and Pritchard and Cox18 argued that low-frequency alleles are major contributors to common disease, whereas Reich and Lander19 advocated the common disease–common variant hypothesis. Pritchard17 suggested that the rate of susceptibility mutations is high and that they are under pressure of weak purifying selection, leading to an abundance of rare variants. Reich and Lander19 did not focus on the question of how an individual deleterious variant could reach high population frequency. Given the high frequency of the phenotype, they considered allelic identity in the disease class, which depended on the rate of mutations conferring disease susceptibility. Thus, both models depend on the rate of deleterious mutations involved in disease and on the strength of selection against new mutations. Both studies used estimates of mutation rate based on examples from a few loci and mutations that cause fully penetrant Mendelian phenotypes; mutations involved in complex disease, on the other hand, may have smaller effects. The difference in conclusions is mostly explained by the difference in numerical values of the rate of deleterious mutations and the strength of selection acting on them. It should be stressed that the deleterious mutation rate depends not only on the raw per-nucleotide substitution rate but also on the fraction of de novo mutations that are deleterious. Reliable estimation of these parameters requires integration of several types of data.
We combined, in our analysis, data on human Mendelian disease–causing mutations, human-chimpanzee divergence, and genetic variation in the modern human population. The availability of new data on human polymorphism detected in 37 obesity-related genes sequenced in 756 individuals allowed us to investigate the important class of mildly deleterious mutations. Purifying selection acting on this type of mutation is strong enough to effectively prohibit their fixation, but they are present in the population at low frequencies and can be detected if a large number of individuals is sequenced.
We estimated that >50% of de novo missense mutations in an average human gene and 70% of missense SNPs detected only once among 1,500 chromosomes are mildly deleterious. Such mildly deleterious mutations are associated with selection coefficients within a surprisingly narrow range of 0.001–0.003.
A high fraction of mildly deleterious mutations among missense mutations suggests that mutation-selection balance can be a possible explanation for the existence of common disease with complex inheritance. The observation that the majority of human rare nonsynonymous variants are deleterious and thus, of significance for function and phenotype, strongly supports a resequencing strategy for candidate-gene association studies; a disease population is expected to have a higher rate of rare amino acid variants in genes involved in disease than that of healthy controls.
Data and Methods
Mutations Associated with Mendelian Diseases
For information on strongly detrimental mutations, we used the Human Gene Mutation Database (HGMD),20 which contains >50,000 disease-causing mutations of various types, including missense, nonsense, and splice-sites mutations. Disease-associated polymorphisms make up only a very small fraction of HGMD, and the majority of mutations included in the database are fully penetrant and cause simple Mendelian diseases.20 HGMD lacks information on how many times each individual mutation has been identified. However, with the assumption that mutations follow the Poisson statistics, the presence of mutations detected multiple times would not significantly affect our estimates unless the majority of all possible nonsense mutations were detected, which is highly unlikely for most genes.
HGMD may include genes with complete loss-of-function mutations that are embryonically lethal. It contains some gain-of-function mutations and mutations with incomplete penetrance. Although the fraction of these mutations in HGMD is probably small, their presence can cause bias of our estimates. Thus, we repeated our analysis on a smaller but well-characterized set of autosomal dominant or X-linked disease genes originally collected by Kondrashov21 from locus-specific databases for estimating the mutation rate in humans. We extracted information on missense and nonsense mutations in these genes from individual HGMD entries. To ensure that complete loss of function is not lethal, we restricted our analysis to 26 genes that had at least five reported nonsense mutations. All missense mutations in these genes are believed to be loss-of-function mutations.
Human Polymorphism Data: New Large Resequencing Data Set
For our analysis of very-low-frequency nonsynonymous SNPs, we used a new large resequencing data set described by Ahituv et al. (in this issue).22 Complete exonic sequences and their splice sites were sequenced in 58 genes with potential involvement in obesity in 379 obese and 378 lean individuals. Complete sequencing of >1,500 chromosomes provided us with an opportunity to study nonsynonymous SNPs at very low frequencies. All individuals included in the study are of white ancestry.
Since this data set is not based on a random population sample and is phenotypically biased, we limited our analysis to 37 autosomal genes for which no evidence of an effect on obesity was detected. In these genes, there was no statistically significant excess of variation in either the obese or the lean group; 71 and 79 missense variants were found in lean and obese cohorts, respectively. Random resampling of subsets matching the SeattleSNP data set in size produced estimates highly similar to those obtained from smaller systematic data sets, which further supported the absence of bias in the chosen subset of 37 genes.
Human Polymorphism Data: Publicly Available Data
In addition to our new resequencing data set, we used three publicly available data sets that contain sufficient information on rare nonsynonymous alleles: (1) a data set generated by the National Institute of Environmental Health Sciences Environmental Genome Project (NIEHS-EGP) (NIEHS SNPs Program Web site),23 in which >500 genes involved in DNA repair and cell-cycle pathways were sequenced in at least 90 unrelated individuals; (2) a data set generated by the SeattleSNPs project (SeattleSNPs), in which >200 genes involved in the inflammatory responses were sequenced in at least 46 individuals; and (3) a data set generated by the database of Japanese Single Nucleotide Polymorphisms (JSNP),24 in which polymorphic sites in >8,000 genes were discovered using a panel of 12 individuals and were later genotyped in 750 individuals.
Mutations Fixed in the Human Lineage after Divergence from Chimpanzee
To obtain information on mutations fixed in the human lineage after divergence from chimpanzee, we used human-chimpanzee and human-macaque whole-genome pairwise alignments constructed with BLASTZ program. These alignments were obtained from the University of California–Santa Cruz (UCSC) site (UCSC Genome Bioinformatics: Sequence and Annotation Downloads). We compared all nucleotide substitutions detected between human and chimpanzee whole-genome assemblies with the dbSNP database,25 which contains information on most of the known human genetic variation. Substitutions that were detected as present-day polymorphisms were excluded from our analysis of human-chimpanzee divergence. Macaque sequence was used as an outgroup to distinguish mutations fixed in the human lineage from mutations fixed in the chimpanzee lineage. Accordingly, we ignored sites at which the macaque nucleotide was different from both human and chimpanzee nucleotides. Because of this procedure, we potentially excluded a small fraction of very rapidly evolving sites, thus slightly underestimating the fraction of “effectively neutral” amino acid substitutions.
Context-Dependent Mutation Model
Accurate estimation of selective constraints in protein-coding regions requires a context-dependent mutational model.26 We constructed a context-dependent mutation-rate matrix that described the probability of de novo mutation occurrence before any selection took place. With such a mutability model, we were able to compare the observed number of various substitutions with the corresponding number expected under neutrality and, from such comparison, to draw a conclusion about the strength of purifying selection.
We restricted our analysis to single-nucleotide substitutions, excluding other less common types of mutations. The real mutational spectrum is quite complex—CpG dinucleotides have a mutation rate an order of magnitude higher than that of other dinucleotides; transitions are notably more frequent than transversions; and some other, more subtle, context-dependent effects were reported to exist.27 Various mutation models of different degrees of sophistication have been used elsewhere.28 The availability of human, chimpanzee, and (incomplete) baboon genomic sequences allowed us to calculate an empirical “directed” 64×3 mutation matrix for triplets (with probabilities for all XY1Z to XY2Z single-nucleotide substitutions). Such a mutational model that takes into account a nucleotide, its closest neighbors, and the direction of substitution should capture most of the known fine-scale mutation-rate context dependencies. We used a second-order model of dependence on neighboring positions, to capture all context-dependent effects on mutation rate. We also introduced statistical correction for back substitutions in the baboon lineage.
We made alignments of human, chimpanzee, and baboon sequence taken from ENCODE regions,29 using the multiple sequence aligner TBA.30 The frequencies of all nucleotide triplets in the aligned sequences were counted (with any triples containing gaps ignored). Directionality of mutation (whether a substitution has occurred in the human or the chimpanzee lineage) was determined using baboon sequence.
The calculated matrix was in agreement with other recently published context-dependent models of neutral evolution.31–33 For example, the r2 correlation coefficient between our matrix and the matrix from Siepel and Haussler31 is 96%.
Gene Sets
Population and evolutionary dynamics of genes located on sex chromosomes differ from the rest of the genome. To avoid unnecessary complications in our analysis, we used only autosomal genes. To calculate the ratio of nonsynonymous to synonymous mutations characteristic of the entire human genome, we used 14,095 reliably annotated autosomal genes from the Consensus CDS (CCDS) Project.
Confidence of the Estimates
Analysis of human-chimpanzee divergence is based on the complete proteome, and the analysis of the HGMD database incorporates the most comprehensive set of disease genes. However, human SNP data are represented by much smaller gene sets. Therefore, it is necessary to assign confidence to statistical estimates obtained from these sets. Even though these data sets have large numbers of SNPs, the mutations were found in different genes that possess different properties. Thus, sampling errors of the estimates are expected to be mostly determined by sampling of genes rather than of individual mutations.
We computed SEs from SDs of estimates obtained from random nonoverlapping subsets of the data. Although SEs are mostly used for estimates of the mean, we tested, in a series of simulations, that SEs computed using nonoverlapping subsets satisfactorily approximate the error of the estimates presented in this work.
Results
Strongly Detrimental Mutations
As a first step in our analysis of the spectrum of potential effects of amino acid mutations, we estimated the fraction of de novo missense changes that are strongly detrimental. We define a missense mutation as strongly detrimental if it causes complete protein function loss, often seen in Mendelian diseases.
Nonsense mutations that introduce premature stop codons can serve as a proxy of “strong detrimentality.” Totals of 26,305 missense and 6,764 nonsense mutations are listed in HGMD. Although HGMD has an inherent ascertainment bias, following the work of Kondrashov21 and Yampolsky et al.,34 we believe that mutations causing the same phenotype are equally likely to be deposited in the database, regardless of their type—nonsense, missense, or splice site. If all missense mutations were as likely to result in complete loss of function and, subsequently, in strong disease phenotype as were nonsense mutations, then the ratio of missense to nonsense mutations in the HGMD database would be similar to the expected theoretical ratio for de novo mutations. In reality, it is significantly lower. Using our mutation-rate model described above, we estimated that ∼19.7 missense substitutions occur genomewide per nonsense substitution (14,095 human genes from the CCDS Project were used for calculation of genomewide values). At the same time, only 3.9 missense changes were observed per nonsense substitution among disease-causing mutations in the HGMD database (fig. 1A). Such a difference indicates that a large fraction of missense mutations is not deleterious enough to be visible through such a “strong phenotype filter” as the detection and inclusion in HGMD. Using the obtained numbers, we can estimate that 20% (3.9 of 19.7) of missense mutations are strongly detrimental.
Figure 1. .

Spectrum of effects of de novo missense mutations. A, Fraction of strongly detrimental mutations among de novo amino acid substitutions. Disease-causing nonsense mutations in HGMD were used as a standard of “strong detrimentality.” B, Fraction of strongly detrimental mutations among de novo amino acid substitutions. Disease-causing splice-site mutations in HGMD were used as a standard of “strong detrimentality.” C, Fraction of effectively neutral mutations among de novo amino acid substitutions. Synonymous substitutions fixed in the human lineage after divergence from chimpanzee were used as a standard of “effective neutrality.”
Mutations that disrupt dinucleotides of the core splicing-site consensus (GT/AG) often lead to an almost complete loss of gene function. These splice-site substitutions can be used as “reference” for strongly detrimental mutations in the same way that we used nonsense mutations. We calculated that, genomewide, ∼36.5 de novo missense mutations are expected to occur per de novo splice-site mutation. However, in HGMD, only 7.6 missense mutations are listed per mutation in the splice site (fig. 1B). Accordingly, we can estimate that the fraction of strongly deleterious mutations among missense mutations is ∼21%. This number is in remarkable correspondence with the value obtained using nonsense mutations as a reference.
Although HGMD provides a comprehensive set of disease mutations, it is very heterogeneous and includes mutations of incomplete penetrance, gain-of-function mutations, and possibly mutations in genes with embryonically lethal complete loss of function. Presence of mutations in the two latter categories would bias our estimate upward—that is, it would lead to a conservative estimate for the purpose of this work. However, to test whether the upward bias can be significant, we analyzed a much smaller set of very well-characterized genes involved in autosomal dominant or X-linked simple Mendelian diseases. All mutations in this set are believed to lead to loss of function and, for them, complete function loss is not lethal. After individual gene lengths were taken into account, 21% of new missense mutations in the set were estimated to be strongly detrimental.
Effectively Neutral Mutations
As the second step in our analysis of the spectrum of potential effects of amino acid substitutions, we estimated a fraction of de novo missense mutations that are effectively neutral. We define a missense mutation as effectively neutral if its probability to be fixed in the ancestral human population after divergence from chimpanzee would have been similar to that of synonymous substitutions. Although synonymous substitutions were shown not to be completely selectively neutral35–37 (reviewed by Chamary et al.38), the effect on fitness of the vast majority of them in the human population is believed to be relatively small.
We estimated (fig. 1C) that 2.23 de novo missense mutations occur per synonymous mutation. However, among substitutions fixed in the human lineage after divergence from chimpanzee (calculated with the use of macaque genomic sequence as an outgroup) only 0.60 missense mutations were present per single synonymous substitution. The rest of the missense mutations were apparently eliminated by purifying selection. From these values, we can estimate that ∼27% (0.6 of 2.23) of missense mutations in human proteins are similar in effect to synonymous substitutions.
This estimate is sensitive to advantageous mutations driven to fixation by positive selection and to very slightly deleterious mutations fixed by drift. Presence of these mutations would bias our estimate of the fraction of mildly deleterious mutations downward—that is, our estimate is conservative for our purpose. It is also conservative with respect to purifying selection at synonymous sites. Thus, this estimate can be viewed as an estimate of the fraction of new missense mutations that are not associated with fitness loss larger than the reciprocal of effective population size.
Mildly Deleterious Polymorphisms
We analyzed two extremes of the potential effects of amino acid changes on fitness and estimated that, among de novo missense mutations in human proteins, ∼20% are strongly detrimental, whereas another 27% are effectively neutral. Simple arithmetic provides us with the conclusion that the majority (53%) of all de novo missense mutations are, in fact, mildly deleterious. Mildly deleterious mutations can reach low but detectable population frequencies. We next posed a question: if the fraction of mildly deleterious mutations among de novo mutations is 53%, what is the fraction of mildly deleterious alleles among rare and common nonsynonymous SNPs in the human population?
First, we considered very rare missense polymorphisms. We analyzed only polymorphic sites at which at least 1,400 of 1,514 chromosomes have been successfully sequenced. More than 60% of nonsynonymous SNPs discovered in our set of 37 autosomal genes sequenced in 757 individuals were detected as singletons (i.e., were found in a heterozygote state in a single individual). Even though singletons represent very-low-frequency SNPs, the probability that they are de novo mutations is diminishingly small. The number of nonsynonymous SNPs detected at higher frequencies was significantly smaller, and, thus, singletons represented the only group of rare missense alleles that had enough data for a statistically significant analysis.
We estimated the relative fraction of de novo missense mutations represented by singletons. This estimate was based on a comparison of the observed number of nonsynonymous substitutions per synonymous mutation (Na/Ns ratio) with the corresponding theoretical number expected under neutral evolution (N0a/N0s ratio). It should be noted that the assumption that natural selection is absent (neutral evolution) is equivalent to the assumption that natural selection has not yet acted (de novo mutations).
As presented in figure 2 and table 1, the N0a/N0s ratio calculated using our neutral model of evolution for 37 genes of large resequencing data set was equal to 2.204. Experimental Na/Ns ratio for singletons calculated for the same data set was equal to 1.49. Given these values, we determined that only 32% (1-1.49/2.20) of missense mutations are deleterious to the extent that there is a very low probability that they will be found even once in the sample of 1,500 chromosomes. SE of this estimate is 2%.
Figure 2. .

Fraction of de novo missense mutations represented at different levels of allele frequency. The normalized fraction of de novo amino acid substitutions detected in a given data set was calculated from the difference of observed Na/Ns ratio and theoretical N0a/N0s ratio expected under neutrality. Data for rare polymorphisms are shown in orange, data for common polymorphisms are in yellow, and data for substitutions (subst.) fixed in the human lineage after divergence from chimpanzee are in green. SEs are shown by gray error bars.
Table 1. .
Ratios of Missense to Synonymous SNPs and Substitutions
|
Na/Ns |
(Na/Ns)/(N0a/N0s) |
||||||||
| Data Set | Description of Data Set | No. of Genes | Theoretical N0a/N0s |
Singletons | SNPs with MAF > 25% | Substitutions in Human Lineage | Singletonsa | SNPs with MAF > 25%a | Substitutions in Human Lineagea |
| CCDS | Genomewide | 14,095 | 2.232 | … | … | .60 | … | … | .266 ± .002 |
| Obesity-related genes | 757 Individuals sequenced | 37 | 2.204 | 1.49 | … | .44 | .68 ± .02 | … | .20 ± .05 |
| NIEHS-EGP | 90–95 Individuals sequenced | 518 | 2.255 | 1.36 | .56 | .50 | .61 ± .02 | .26 ± .04 | .22 ± .01 |
| SeattleSNPs | 46–47 Individuals sequenced | 236 | 2.203 | 1.43 | .66 | .66 | .65 ± .04 | .32 ± .05 | .30 ± .02 |
| JSNP | 750 Individuals genotyped | 8,786 | 2.244 | 1.37b | .54 | .54 | .61 | .24 | .24 |
Data are mean ± SE.
SNPs with observed frequency <1% instead of singletons have been used in analysis of the JSNP data set.
The experimental Na/Ns ratio for amino acid substitutions fixed in the set of 37 genes in the human lineage after divergence from chimpanzee is equal to 0.44. This means that ∼20% of all missense mutations in these genes are effectively neutral—a value close to the genomewide average.
Human genetic variation detected in the set of 37 genes sequenced in 756 individuals provided an opportunity to get an insight into human genetic variation of very low allele frequency. However, it lacks a significant quantity of data on common genetic variation. To fill this gap, we used three publicly available data sets: (1) a data set generated by the NIEHS-EGP, in which >500 genes involved in DNA repair and cell-cycle pathways were sequenced in at least 90 unrelated individuals; (2) a data set generated by SeattleSNPs, in which >200 genes involved in the inflammatory responses were sequenced in at least 46 individuals; and (3) a data set generated by JSNP, in which polymorphic sites in >8,000 genes were discovered using a panel of 12 individuals and were later genotyped in 750 individuals. These data sets also contained information on rare genetic variation that we used, although a large fraction of rare nonsynonymous SNPs with frequency <1% might have been missed in the NIEHS-EGP and SeattleSNP data sets because <200 chromosomes were sequenced and might have been missed in JSNP because only 12 individuals were used for SNP discovery.
Again, using our neutral model of evolution, we calculated predicted N0a/N0s ratios separately for NIEHS-EGP, SeattleSNPs, and JSNP data sets. Then we calculated experimental Na/Ns ratios for three types of sequence changes. The first type is rare nonsynonymous SNPs that either (a) were observed only once among all chromosomes sequenced (in NIEHS-EGP and SeattleSNPs) or (b) had an observed frequency <1% (JSNP). The second type is very common polymorphisms, with a frequency of the least common allele of >25%. The third type is substitutions fixed in the human lineage after divergence with chimpanzee.
We observed that the Na/Ns ratios for very common polymorphisms were only very slightly higher than Na/Ns ratios for fixed substitutions (fig. 2 and table 1). This fact indicates that the fraction of deleterious amino acid changes among common SNPs is very low. It also supports the notion that the fraction of positively selected amino acid substitutions among all mutations in human proteins was low.39
In sharp contrast with common SNPs, Na/Ns ratio for very rare alleles significantly exceeds Na/Ns ratios for fixed substitutions (that are presumably neutral). This fact indicates that a very large fraction of such rare missense SNPs are deleterious. The fraction of deleterious mutations among observed rare substitutions can be estimated as (Nrarea/Nrares)/(Nfixeda/Nfixeds). Simple calculations reveal that the majority (52%–71%) of amino acid substitutions with the observed frequency of <1% are mildly deleterious in all data sets (table 2). This surprising finding indicates that a low frequency of missense mutation per se can serve as a strong predictor of a deleterious effect of polymorphic variants.
Table 2. .
Fraction of Deleterious Substitutions among Rare Missense SNPs
| Set | No. of Sequenced Individuals | Percentage of Deleterious SNPs among Missense Singletonsa |
| Resequencing data set of obesity-related genes | 757 | 71 ± 8 |
| NIEHS-EGP | 90–95 | 64 ± 1 |
| SeattleSNPs | 46–47 | 52 ± 6 |
Data are mean ± SE.
Characteristic Selection Coefficients of the Mildly Deleterious Mutations Theory
We concluded that more than half of newly arising missense mutations are mildly deleterious. Such mutations are not present among common polymorphisms but are very common among substitutions detected only once in ⩾100 chromosomes. On the basis of these observations, we estimated selection coefficients associated with such mutations.
First, we calculated the theoretical expected number of mutations with selection coefficient s observed only once in a set of m randomly sampled chromosomes. Then, we divided it by the corresponding number of neutral mutations to obtain the ratio R1/m(s):
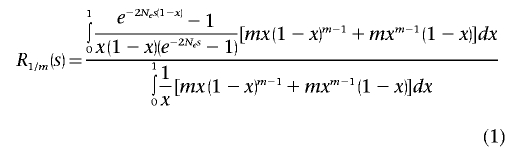 |
In equation (1), Ne represents an effective population size and x is the frequency of an individual allele in the population. The number of detected mutations was obtained by multiplying the theoretical number of all alleles present in a population with frequency x by the probability of a single allele with frequency x being detected as a singleton in a set of m chromosomes and then by integrating over allele frequency x from 0 to 1. It should be noted that an allele is detected as a singleton in a set of m chromosomes if either 1 or m-1 copies are present.
Similarly, we calculated the theoretical ratio RMAF>0.25(s) for mutations that have been detected as polymorphisms in a set of m chromosomes with minor-allele frequency (MAF) >25%.
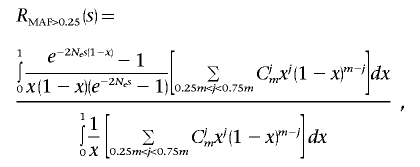 |
where Cnk is the binomial coefficient—the number of combinations of size k from a set with n elements:
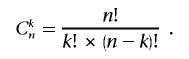 |
These formulas result from an application of diffusion theory to the dynamics of polymorphism in populations.40 Their two major underlying assumptions are constant effective population size and that each novel mutation occurrs at a new, previously monomorphic site (infinite-number-of-sites model). The dominance coefficient was assumed to be equal to 0.5, so, if s is the selection coefficient associated with a heterozygote, then 2s is the selection coefficient associated with a homozygote. The exact values of the dominance coefficient are not very important, as long as they are high enough to allow selection to operate primarily on heterozygotes.
The dependence of RMAF>0.25(s) and R1/m(s) (for various values of m) on selection coefficient s is shown in figure 3A. We determined that roughly equal fractions of missense mutations can be observed as fixed substitutions and as common polymorphisms with MAF >25% (fig. 2). This observation implies that mildly deleterious mutations are virtually absent among frequent SNPs and thus should have selection coefficient values at which RMAF>0.25(s) is very close to zero. On the other hand, the majority of mildly deleterious mutations have high relative chance to be detected as singletons. For each of the three resequencing data sets (obesity-related, NIEHS-EGP, and SeattleSNPs), we estimated the fraction of de novo mildly deleterious missense mutations observed as singletons, relative to neutral expectations:
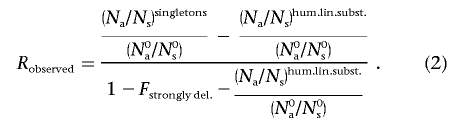 |
Here, the numerator reflects the observed fraction of mutations in the mildly deleterious class detectable as singletons, which is equal to the fraction of all mutations detectable as singletons minus the fraction of effectively neutral de novo mutations (“hum.lin.subst.” indicates human lineage substitutions). The denominator reflects the total fraction of mutations arising in the mildly deleterious class. It is given by subtracting the fraction of strongly deleterious de novo mutations (denoted by Fstrongly del.) and neutral mutations from 1.
Figure 3. .

A, R1/m(s) and RMAF>0.25(s) (see eq. [1]) calculated using equations derived from diffusion theory under the assumption of constant population size and an infinite number of sites. The expected shift of R1/m(s) curves due to recent population expansion is shown by red arrows. Black arrows illustrate estimation of characteristic selection coefficients for a mildly deleterious class of missense mutations (see the “Results” section and eq. [2]). B, R1/m(s) and RMAF>0.25(s) calculated by direct computer simulation of molecular evolution under the assumption of an infinite number of sites and simple population history—a stable-population-size epoch followed by bottleneck and then fast expansion.
Using theoretical dependence of R on the selection coefficient (fig. 3A) for the corresponding number of sequenced chromosomes m, we estimated the characteristic selection coefficients for mildly deleterious de novo missense mutation for genes in each set. All three estimates fall into a relatively narrow range of selection coefficients: 0.002–0.02.
Characteristic Selection Coefficients of Mildly Deleterious Mutations: Computer Simulations
The major drawback of the straightforward theoretical approach described above is the assumption that the effective size of the human population is constant and equals 10,000. This assumption was shown to be appropriate for common SNPs, most of which are ancient in origin. However, an effective population size that is an order of magnitude larger might be more appropriate to describe the dynamics of rare polymorphisms. A larger effective population size leads to more-efficient purifying selection and, as can be seen from equation (1), to lower values of R1/m(s). By underestimating Ne, we are overestimating R1/m(s), and, in reality, the R1/m(s) dependence curve on s is shifted to the area of lower values of selection coefficients (fig. 3A).
To analyze the dependence of R1/m(s) and RMAF>0.25(s) under a more realistic human-population-history scenario, we simulated evolution under the Wright-Fisher model (assuming constant mutation rate and an infinite number of unlinked sites). Our model of human population history was comprised of the four principal epochs: (a) 100,000 generations of stable effective population size 15,000, (b) exponential reduction in 100 generations to the size of 7,000, (c) 500 generations of stable population size 7,000, and (d) exponential growth in 3,000 generations to the effective population size 90,000. Such a scenario, where a long period of stable effective population size is followed by bottleneck and then by rapid expansion, captures key features of human population history. Exact values for the duration of each epoch and the corresponding effective population sizes were chosen in general agreement with recent literature41–43 and then were slightly optimized to better describe allele frequency distributions for noncoding SNPs detected by the NEIHS-EGP. Usage of R1/m(s) and RMAF>0.25(s) dependencies obtained using direct simulation resulted in the estimate that the ∼0.001–0.003 range of selection coefficients corresponds to a mildly deleterious class of de novo missense mutations.
Cumulative Equilibrium Frequency of Mildly Deleterious Polymorphisms in the Human Genome
We determined that mildly deleterious missense mutations that are not present among common polymorphisms are nevertheless numerous among polymorphisms with a detected population frequency <1% and are associated with selection coefficients in the range of 0.001–0.003. Using this result, we estimated the equilibrium frequency of such mildly deleterious alleles in the human population. Under the assumption of strong selection and large population size (Ne×s≫1), the cumulative equilibrium frequency of all mildly deleterious gene alleles in the population is
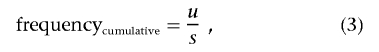 |
where u is mutation rate per gene per generation and s is selection coefficient associated with heterozygotes. We used 2×10-8 as the mutation per nucleotide per generation rate, estimated by Kondrashov.21 Importantly, this estimate for cumulative frequency of deleterious alleles does not depend on population demographic history.
The mutation rate for mildly deleterious missense mutations per gene can be estimated as follows:
 |
or, by substitution of numbers,
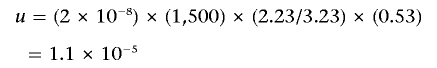 |
Using equation (3), we calculate that, for an average gene, the combined frequency of alleles carrying mildly deleterious missense mutation is ∼1%.
Taking into account that protein-coding sequences compose ∼1.5% of the human genome, we estimated that ∼600 missense mutations with selection coefficient in the range 0.001–0.003 are present in the genome of an average individual. It should be noted that the number of mutations with significantly weaker effect on fitness (with associated selection coefficients <10−4) is likely to be significantly higher.
Discussion
It has been noted that, in some Mendelian diseases, less severe forms of the disease exist that are associated with milder mutations that do not completely abrogate protein activity.1 It was suggested that such mildly deleterious missense mutations may serve as a genetic basis of complex diseases. The plausibility of this hypothesis depends on how frequent such mildly deleterious substitutions are among de novo mutations and how strong the purifying selection that acts on them is. The presence of deleterious missense polymorphisms in the human genome has been reported in many previous studies.44–55 In the present work, we performed a quantitative analysis, calculating the fraction of mildly deleterious mutations among de novo amino acid changes and very rare and very common nonsynonymous SNPs. Until recently, a lack of large resequencing data sets that cover dozens of genes and hundreds of chromosomes precluded such analysis, although several earlier studies analyzed distribution of selection coefficients for new missense mutations in humans and flies by use of the dependence of substitution rate on effective population size,56–58 a comparison of polymorphism and divergence,34,55,58 and a frequency spectrum in smaller samples.54,58,59
We calculated the fraction of mildly deleterious missense mutations among all de novo substitutions as the remaining difference after subtraction of strongly detrimental mutations and effectively neutral mutations. We determined that, among all de novo amino acid substitutions, strongly detrimental mutations comprise ∼20%. The genomewide value that we obtained is similar to the fraction of amino acid replacements that destroy the protein function (25%), obtained by Yampolsky et al.34 Our estimate is also in general agreement with the results of Eyre-Walker et al.,54 who, by fitting a distribution of selection coefficients modeled as a gamma function to the data on human genetic variation, estimated that ∼15% of missense substitutions are strongly deleterious, with selection coefficients >0.1. Earlier, Fay et al.55 proposed that 54% of new missense mutations in humans are strongly deleterious. This estimate referred to the fraction of nonsynonymous SNPs not observed in the sample of 100 chromosomes, so this estimate is not directly comparable to the estimate presented here. Our value for the fraction of effectively neutral amino acid changes (27%) is only slightly higher than a previous estimate51 of 24% and the corresponding KAKS estimate39 for human-chimpanzee comparison (23%). The difference is most likely because of the application of a context-based mutation model. A smaller previous estimate34 of 12% possibly resulted from the use of the too-distant mouse sequence as an outgroup.
On the basis of a very strong difference in the Na/Ns ratios for very rare and very common missense SNPs, we estimated that the majority of rare missense polymorphisms detected in the human population are associated with a surprisingly narrow range of selection coefficients: 0.001–0.003. Because of such relatively mild purifying selection acting on them, they can reach a high cumulative frequency in the human population while still maintaining a highly heterogeneous spectrum of individual alleles.
An increase in the Na/Ns ratio has been noted elsewhere44; however, the extent of such a difference was not always evident, because of the low number of chromosomes sequenced and crude binning by frequency. A strong “threshold-like” increase in the Na/Ns ratio for SNPs with frequency <6% was noted by Wong et al.60 This work,60 however, has not analytically treated the possibility that the observed effect is the consequence of the shape of selection coefficients distribution for de novo mutations.
We estimated that, for an average 500-aa protein, the cumulative equilibrium frequency of alleles carrying deleterious missense SNPs is roughly equal to 1% in the human population. If we consider unusually long proteins or entire pathways as mutational targets, their mutation rate and, thus, the equilibrium frequency of mildly deleterious alleles will be significantly higher than 1%. Some traits, such as susceptibility to complex diseases, can be influenced by dozens, or perhaps even hundreds, of genes. The mutational target for such traits will be exceptionally large. A large mutational target size leads to a large mutation rate per generation and, as a consequence, to a large level of deleterious polymorphism, since, at equilibrium, it is directly proportional to the mutation rate. Even for a simple metabolic pathway that involves several multisubunit enzymes, the percentage of individuals harboring at least one damaging mutation in the pathway can easily exceed 10%. This example shows that a combined frequency of rare mildly deleterious polymorphisms is high enough to serve as the basis for heritable susceptibility even to most of the common complex diseases, such as hypertension or coronary heart disease. This suggests the possibility that mutation-selection balance can be a feasible evolutionary explanation at least for some common diseases.
Candidate-Gene Association Studies
These findings have implications not only for our understanding of the role of rare nonsynonymous SNPs in susceptibility to complex diseases but also for methods of detecting genes that harbor such detrimental genetic variation. Currently, there are two major approaches for identification of genes involved in complex diseases: linkage studies61 and association studies of population samples.61,62 Whereas the former approach is the most effective for identification of Mendelian-like rare genetic variants with high penetrance, the latter approach is better suited for identification of common variants with relatively low penetrance. None of these approaches, however, is able to detect susceptibility loci that harbor numerous, but individually rare, mildly deleterious polymorphisms. However, our analysis of mildly deleterious mutations indicates that this situation is very plausible and might be true for many genes and common diseases.
In the light of our results, a recently proposed alternative approach looks more promising. This approach aims at the detection of enrichment in rare, potentially deleterious missense SNPs in a patient group compared with a control group.63,64 By considering the cumulative frequency of deleterious mutations rather than their individual frequencies, this method is suitable for investigation of common diseases that have a very heterogeneous spectrum of predisposing alleles. Such a missense-accumulation approach to candidate-gene association studies has been successfully used recently in studies of the MC4R gene,65 tyrosine phosphatome in colorectal cancers,66 and genes involved in lipid-metabolism disorders.67
However, two major potential disadvantages of this method have been identified.63 First is the high cost of complete genomic sequencing of candidate genes in a large number of individuals. Second is the analytical challenge of selecting potentially deleterious SNPs from a large quantity of neutral genetic variation.63 The first objection to the method will almost certainly be eliminated in the near future by the continuing dramatic reduction in sequencing costs68 and the development of novel sequencing strategies.69 The second objection is more fundamental, since the inability to distinguish between deleterious and neutral amino acid changes would lead to a very low signal:noise ratio. To study enrichment in deleterious mutations, one should be able to detect them among neutral genetic variation. The number of deleterious alleles in disease phenotypes was previously believed to be low relative to the large number of neutral missense SNPs present in both disease and control groups. Our analysis, however, reveals that the majority of missense substitutions with detected frequency <1% are, in fact, deleterious. Thus, if only substitutions with detected frequency <1% are counted, the enrichment in the mutation number in a disease group should be highly pronounced.
Conclusions
A large fraction of mildly deleterious mutations among missense mutations suggests that mutation-selection balance is a plausible explanation for the existence of common disease with complex inheritance, at least in some cases. Furthermore, it is feasible that some common diseases may be caused by a multitude of rare allelic variants. The observation that the majority of human rare nonsynonymous variants are deleterious, and thus are of significance to function and phenotype, suggests a strategy for candidate-gene association studies. Disease populations are expected to have a higher rate of rare amino acid variants in genes involved in disease than are healthy control populations. This difference can be easily detected in a deep resequencing study. Obviously, this strategy would be highly inefficient if the majority of coding variants at low frequency were neutral. Several recent reports demonstrated an excess of rare missense variants in individuals with phenotypes associated with disease risk. Our analysis provides an explanation for the success of these studies.
Acknowledgments
This work was funded by the National Institutes of Health (NIH) through the NIH Roadmap for Medical Research, grant U54LM008748; by National Institute of General Medical Sciences grant 1R01GM071852; and by Genome Canada foundation. We thank Drs. Jonathan Cohen, Alexey Kondrashov, and John Stamatoyannopoulos for helpful discussions.
Web Resources
The URLs for data presented herein are as follows:
- Consensus CDS (CCDS) Project, http://www.ncbi.nlm.nih.gov/CCDS/
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/
- Human Gene Mutation Database (HGMD), http://www.hgmd.cf.ac.uk/ac/index.php
- JSNP, http://snp.ims.u-tokyo.ac.jp/ (for database of Japanese SNPs)
- NIEHS SNPs Program, http://egp.gs.washington.edu/
- UCSC Genome Bioinformatics: Sequence and Annotation Downloads, http://hgdownload.cse.ucsc.edu/downloads.html
References
- 1.Glazier AM, Nadeau JH, Aitman TJ (2002) Finding genes that underlie complex traits. Science 298:2345–2349 10.1126/science.1076641 [DOI] [PubMed] [Google Scholar]
- 2.Botstein D, Risch N (2003) Discovering genotypes underlying human phenotypes: past successes for Mendelian disease, future approaches for complex disease. Nat Genet 33:228–237 10.1038/ng1090 [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Grus WE, Zhang J (2006) Gene losses during human origins. PLoS Biol 4:e52 10.1371/journal.pbio.0040052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Charlesworth B (2001) Patterns of age-specific means and genetic variances of mortality rates predicted by the mutation-accumulation theory of ageing. J Theor Biol 210:47–65 10.1006/jtbi.2001.2296 [DOI] [PubMed] [Google Scholar]
- 5.Williams PD, Day T, Fletcher Q, Rowe L (2006) The shaping of senescence in the wild. Trends Ecol Evol 21:458–463 10.1016/j.tree.2006.05.008 [DOI] [PubMed] [Google Scholar]
- 6.Di Rienzo A, Hudson RR (2005) An evolutionary framework for common diseases: the ancestral-susceptibility model. Trends Genet 21:596–601 10.1016/j.tig.2005.08.007 [DOI] [PubMed] [Google Scholar]
- 7.Neel JV (1962) Diabetes mellitus: a “thrifty” genotype rendered detrimental by “progress”? Am J Hum Genet 14:353–362 [PMC free article] [PubMed] [Google Scholar]
- 8.Charlesworth D (2006) Balancing selection and its effects on sequences in nearby genome regions. PLoS Genet 2:e64 10.1371/journal.pgen.0020064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Voet D, Voet JG (2004) Biochemistry, 3rd ed. John Wiley & Sons, New York, pp 183–185 [Google Scholar]
- 10.Williams GC (1957) Pleiotropy, natural selection and the evolution of senescence. Evolution 11:398–411 10.2307/2406060 [DOI] [Google Scholar]
- 11.Kimura M (1965) A stochastic model concerning the maintenance of genetic variability in quantitative characters. Proc Natl Acad Sci USA 54:731–736 10.1073/pnas.54.3.731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lande R (1975) The maintenance of genetic variability by mutation in a polygenic character with linked loci. Genet Res 26:221–235 [DOI] [PubMed] [Google Scholar]
- 13.Turelli M (1984) Heritable genetic variation via mutation-selection balance: Lerch’s Zeta meets the abdominal bristle. Theor Popul Biol 25:138–193 10.1016/0040-5809(84)90017-0 [DOI] [PubMed] [Google Scholar]
- 14.Zhang XS, Wang J, Hill WG (2004) Influence of dominance, leptokurtosis and pleiotropy of deleterious mutations on quantitative genetic variation at mutation-selection balance. Genetics 166:597–610 10.1534/genetics.166.1.597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang XS, Hill WG (2005) Genetic variability under mutation selection balance. Trends Ecol Evol 20:468–470 10.1016/j.tree.2005.06.010 [DOI] [PubMed] [Google Scholar]
- 16.Keller MC, Miller G (2006) Resolving the paradox of common, harmful, heritable mental disorders: which evolutionary genetic models work best? Behav Brain Sci 29:385–404 10.1017/S0140525X06009095 [DOI] [PubMed] [Google Scholar]
- 17.Pritchard JK (2001) Are rare variants responsible for susceptibility to complex diseases? Am J Hum Genet 69:124–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritchard JK, Cox NJ (2002) The allelic architecture of human disease genes: common disease-common variant or not? Hum Mol Genet 11:2417–2423 10.1093/hmg/11.20.2417 [DOI] [PubMed] [Google Scholar]
- 19.Reich DE, Lander ES (2001) On the allelic spectrum of human disease. Trends Genet 17:502–510 10.1016/S0168-9525(01)02410-6 [DOI] [PubMed] [Google Scholar]
- 20.Stenson PD, Ball EV, Mort M, Phillips AD, Shiel JA, Thomas NS, Abeysinghe S, Krawczak M, Cooper DN (2003) Human Gene Mutation Database (HGMD): 2003 update. Hum Mutat 21:577–581 10.1002/humu.10212 [DOI] [PubMed] [Google Scholar]
- 21.Kondrashov AS (2003) Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat 21:12–27 10.1002/humu.10147 [DOI] [PubMed] [Google Scholar]
- 22.Ahituv N, Kavaslar N, Schackwitz W, Ustaszewska A, Martin J, Hébert S, Doelle H, Ersoy B, Kryukov G, Schmidt S, et al (2007) Medical sequencing at the extremes of human body mass. Am J Hum Genet 80:XXX–XXX (in this issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Livingston RJ, von Niederhausern A, Jegga AG, Crawford DC, Carlson CS, Rieder MJ, Gowrisankar S, Aronow BJ, Weiss RB, Nickerson DA (2004) Pattern of sequence variation across 213 environmental response genes. Genome Res 14:1821–1831 10.1101/gr.2730004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirakawa M, Tanaka T, Hashimoto Y, Kuroda M, Takagi T, Nakamura Y (2002) JSNP: a database of common gene variations in the Japanese population. Nucleic Acids Res 30:158–162 10.1093/nar/30.1.158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K (2001) dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29:308–311 10.1093/nar/29.1.308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Subramanian S, Kumar S (2006) Higher intensity of purifying selection on >90% of the human genes revealed by the intrinsic replacement mutation rates. Mol Biol Evol 23:2283–2287 10.1093/molbev/msl123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Krawczak M, Ball EV, Cooper DN (1998) Neighboring-nucleotide effects on the rates of germ-line single-base-pair substitution in human genes. Am J Hum Genet 63:474–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li WH (1997) Molecular evolution. Sinauer Associates, Sunderland, MA, pp 80–84 [Google Scholar]
- 29.ENCODE Project Consortium (2004) The ENCODE (ENCyclopedia Of DNA Elements) Project. Science 306:636–640 10.1126/science.1105136 [DOI] [PubMed] [Google Scholar]
- 30.Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al (2004) Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14:708–715 10.1101/gr.1933104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siepel A, Haussler D (2004) Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol Biol Evol 21:468–488 10.1093/molbev/msh039 [DOI] [PubMed] [Google Scholar]
- 32.Hwang DG, Green P (2004) Bayesian Markov chain Monte Carlo sequence analysis reveals varying neutral substitution patterns in mammalian evolution. Proc Natl Acad Sci USA 101:13994–14001 10.1073/pnas.0404142101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arndt PF, Hwa T (2005) Identification and measurement of neighbor-dependent nucleotide substitution processes. Bioinformatics 21:2322–2328 10.1093/bioinformatics/bti376 [DOI] [PubMed] [Google Scholar]
- 34.Yampolsky LY, Kondrashov FA, Kondrashov AS (2005) Distribution of the strength of selection against amino acid replacements in human proteins. Hum Mol Genet 14:3191–3201 10.1093/hmg/ddi350 [DOI] [PubMed] [Google Scholar]
- 35.Hellmann I, Zollner S, Enard W, Ebersberger I, Nickel B, Paabo S (2003) Selection on human genes as revealed by comparisons to chimpanzee cDNA. Genome Res 13:831–837 10.1101/gr.944903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chamary JV, Hurst LD (2005) Evidence for selection on synonymous mutations affecting stability of mRNA secondary structure in mammals. Genome Biol 6:R75 10.1186/gb-2005-6-9-r75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parmley JL, Chamary JV, Hurst LD (2006) Evidence for purifying selection against synonymous mutations in mammalian exonic splicing enhancers. Mol Biol Evol 23:301–309 10.1093/molbev/msj035 [DOI] [PubMed] [Google Scholar]
- 38.Chamary JV, Parmley JL, Hurst LD (2006) Hearing silence: non-neutral evolution at synonymous sites in mammals. Nat Rev Genet 7:98–108 10.1038/nrg1770 [DOI] [PubMed] [Google Scholar]
- 39.Chimpanzee Sequencing and Analysis Consortium (2005) Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87 10.1038/nature04072 [DOI] [PubMed] [Google Scholar]
- 40.Kimura M (1983) Infinite site model. In: The neutral theory of molecular evolution. Cambridge University Press, Cambridge, pp 236–240 [Google Scholar]
- 41.Marth GT, Czabarka E, Murvai J, Sherry ST (2004) The allele frequency spectrum in genome-wide human variation data reveals signals of differential demographic history in three large world populations. Genetics 166:351–372 10.1534/genetics.166.1.351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Williamson SH, Hernandez R, Fledel-Alon A, Zhu L, Nielsen R, Bustamante CD (2005) Simultaneous inference of selection and population growth from patterns of variation in the human genome. Proc Natl Acad Sci USA 102:7882–7887 10.1073/pnas.0502300102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schaffner SF, Foo C, Gabriel S, Reich D, Daly MJ, Altshuler D (2005) Calibrating a coalescent simulation of human genome sequence variation. Genome Res 15:1576–1583 10.1101/gr.3709305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halushka MK, Fan JB, Bentley K, Hsie L, Shen N, Weder A, Cooper R, Lipshutz R, Chakravarti A (1999) Patterns of single-nucleotide polymorphisms in candidate genes for blood-pressure homeostasis. Nat Genet 22:239–247 10.1038/10297 [DOI] [PubMed] [Google Scholar]
- 45.Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, et al (1999) Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet 22:231–238 10.1038/10290 [DOI] [PubMed] [Google Scholar]
- 46.Stephens JC, Schneider JA, Tanguay DA, Choi J, Acharya T, Stanley SE, Jiang R, Messer CJ, Chew A, Han JH, et al (2001) Haplotype variation and linkage disequilibrium in 313 human genes. Science 293:489–493 10.1126/science.1059431 [DOI] [PubMed] [Google Scholar]
- 47.Sunyaev S, Ramensky V, Koch I, Lathe W 3rd, Kondrashov AS, Bork P (2001) Prediction of deleterious human alleles. Hum Mol Genet 10:591–597 10.1093/hmg/10.6.591 [DOI] [PubMed] [Google Scholar]
- 48.Glatt CE, DeYoung JA, Delgado S, Service SK, Giacomini KM, Edwards RH, Risch N, Freimer NB (2001) Screening a large reference sample to identify very low frequency sequence variants: comparisons between two genes. Nat Genet 27:435–438 10.1038/86948 [DOI] [PubMed] [Google Scholar]
- 49.Sunyaev S, Kondrashov FA, Bork P, Ramensky V (2003) Impact of selection, mutation rate and genetic drift on human genetic variation. Hum Mol Genet 12:3325–3330 10.1093/hmg/ddg359 [DOI] [PubMed] [Google Scholar]
- 50.Hughes AL, Packer B, Welch R, Bergen AW, Chanock SJ, Yeager M (2003) Widespread purifying selection at polymorphic sites in human protein-coding loci. Proc Natl Acad Sci USA 100:15754–15757 10.1073/pnas.2536718100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bustamante CD, Fledel-Alon A, Williamson S, Nielsen R, Hubisz MT, Glanowski S, Tanenbaum DM, White TJ, Sninsky JJ, Hernandez RD, et al (2005) Natural selection on protein-coding genes in the human genome. Nature 437:1153–1157 10.1038/nature04240 [DOI] [PubMed] [Google Scholar]
- 52.Crawford DC, Akey DT, Nickerson DA (2005) The patterns of natural variation in human genes. Annu Rev Genomics Hum Genet 6:287–312 10.1146/annurev.genom.6.080604.162309 [DOI] [PubMed] [Google Scholar]
- 53.Gorlov IP, Kimmel M, Amos CI (2006) Strength of the purifying selection against different categories of the point mutations in the coding regions of the human genome. Hum Mol Genet 15:1143–1150 10.1093/hmg/ddl029 [DOI] [PubMed] [Google Scholar]
- 54.Eyre-Walker A, Woolfit M, Phelps T (2006) The distribution of fitness effects of new deleterious amino acid mutations in humans. Genetics 173:891–900 10.1534/genetics.106.057570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fay JC, Wyckoff GJ, Wu CI (2001) Positive and negative selection on the human genome. Genetics 158:1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Eyre-Walker A, Keightley PD, Smith NG, Gaffney D (2002) Quantifying the slightly deleterious mutation model of molecular evolution. Mol Biol Evol 19:2142–2149 [DOI] [PubMed] [Google Scholar]
- 57.Loewe L, Charlesworth B, Bartolome C, Noel V (2006) Estimating selection on nonsynonymous mutations. Genetics 172:1079–1092 10.1534/genetics.105.047217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kryukov GV, Schmidt S, Sunyaev S (2005) Small fitness effect of mutations in highly conserved non-coding regions. Hum Mol Genet 14:2221–2229 10.1093/hmg/ddi226 [DOI] [PubMed] [Google Scholar]
- 59.Bustamante CD, Nielsen R, Hartl DL (2003) Maximum likelihood and Bayesian methods for estimating the distribution of selective effects among classes of mutations using DNA polymorphism data. Theor Popul Biol 63:91–103 10.1016/S0040-5809(02)00050-3 [DOI] [PubMed] [Google Scholar]
- 60.Wong GK, Yang Z, Passey DA, Kibukawa M, Paddock M, Liu CR, Bolund L, Yu J (2003) A population threshold for functional polymorphisms. Genome Res 13:1873–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Freimer N, Sabatti C (2004) The use of pedigree, sib-pair and association studies of common diseases for genetic mapping and epidemiology. Nat Genet 36:1045–1051 10.1038/ng1433 [DOI] [PubMed] [Google Scholar]
- 62.Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 273:1516–1517 10.1126/science.273.5281.1516 [DOI] [PubMed] [Google Scholar]
- 63.Hirschhorn JN, Altshuler D (2002) Once and again-issues surrounding replication in genetic association studies. J Clin Endocrinol Metab 87:4438–4441 10.1210/jc.2002-021329 [DOI] [PubMed] [Google Scholar]
- 64.Hirschhorn JN, Daly MJ (2005) Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6:95–108 10.1038/nrg1521 [DOI] [PubMed] [Google Scholar]
- 65.Vaisse C, Clement K, Durand E, Hercberg S, Guy-Grand B, Froguel P (2000) Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest 106:253–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, Ptak J, Silliman N, Peters BA, van der Heijden MS, et al (2004) Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science 304:1164–1166 10.1126/science.1096096 [DOI] [PubMed] [Google Scholar]
- 67.Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH (2004) Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science 305:869–872 10.1126/science.1099870 [DOI] [PubMed] [Google Scholar]
- 68.Service RF (2006) Gene sequencing; the race for the $1000 genome. Science 311:1544–1546 10.1126/science.311.5767.1544 [DOI] [PubMed] [Google Scholar]
- 69.Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, Rosenbaum AM, Wang MD, Zhang K, Mitra RD, Church GM (2005) Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309:1728–1732 10.1126/science.1117389 [DOI] [PubMed] [Google Scholar]