

Abstract
Population stratification remains an important issue in case-control studies of disease-marker association, even within populations considered to be genetically homogeneous. Campbell et al. (Nature Genetics 2005;37:868–872) illustrated this by showing that stratification induced a spurious association between the lactase gene (LCT) and tall/short status in a European American sample. Furthermore, existing approaches for controlling stratification by use of substructure-informative loci (e.g., genomic control, structured association, and principal components) could not resolve this confounding. To address this problem, we propose a simple two-step procedure. In the first step, we model the odds of disease, given data on substructure-informative loci (excluding the test locus). For each participant, we use this model to calculate a stratification score, which is that participant’s estimated odds of disease calculated using his or her substructure-informative–loci data in the disease-odds model. In the second step, we assign subjects to strata defined by stratification score and then test for association between the disease and the test locus within these strata. The resulting association test is valid even in the presence of population stratification. Our approach is computationally simple and less model dependent than are existing approaches for controlling stratification. To illustrate these properties, we apply our approach to the data from Campbell et al. and find no association between the LCT locus and tall/short status. Using simulated data, we show that our approach yields a more appropriate correction for stratification than does principal components or genomic control.
Case-control studies of disease-marker association are susceptible to the confounding effects of population stratification, which originate from the coupling of allele-frequency heterogeneity to disease-risk heterogeneity within a population. To avoid stratification, studies often use data from individuals from a single race or ethnicity group (or, at the very least, they analyze data stratified on the basis of participants’ race or ethnicity) in the hope of achieving a genetically homogeneous population. Recent results1 disputed this perception by demonstrating the existence of stratification in a case-control sample of Americans of European origin who were selected for extreme values of height; in these data, both tall/short status and allele frequencies at a SNP located within the lactase gene (LCT [MIM 603202]) (involved in lactase persistence) varied considerably from northwestern to southeastern Europe. A naive association analysis between this LCT SNP and height resulted in a strongly significant finding (P=3.6×10-7). In efforts to determine whether this result was spurious, the association analyses were repeated by conditioning on grandparental ancestry, and a much weaker signal was observed (P=.0074).1 Furthermore, additional association analyses in a case-control study from Poland (P=.92) and a case-parent trio study from Scandinavia (P=.93) failed to confirm the initial significant association. These results led to the conclusion that the initial association result between the LCT SNP and height within the European American sample was largely or completely due to population stratification.1
Although the demonstration of stratification in subjects of European American ancestry is of concern, conventional wisdom suggests that such stratification can be corrected by applying appropriate statistical methods that use panels of genetic markers that provide information on population structure. However, neither genomic control2,3 nor structured association4–6 could properly correct for the confounding effects of stratification with the use of a collection of 111 missense and noncoding SNPs and 67 ancestry-informative SNPs.1 More recently, an approach based on principal components7–10 also failed to resolve this stratification.10 These results suggest that improved statistical methods for correcting population stratification in genetic association studies of complex disease are needed.
We describe here a novel statistical approach for controlling population stratification in case-control studies of disease. Our approach consists of two steps. In the first step, we model the odds of disease, given data on substructure-informative loci (excluding the test locus). For each participant, we use this model to calculate a stratification score, which is that participant’s estimated odds of disease calculated using his or her substructure-informative–loci data in the disease-odds model. In the second step, we assign subjects to strata defined by stratification score and then test for association between the disease and the test locus within these strata. The resulting association test is valid even in the presence of population stratification. Our stratification-score approach circumvents many of the modeling assumptions and analytical limitations inherent in existing procedures, such as genomic control, structured association, and principal components. Using the height data described above, as well as simulated data, we show that subclassification based on the stratification score provides an appropriate and powerful correction for confounding due to population stratification in situations where other approaches fail.
Material and Methods
Subclassification Based on the Stratification Score
Assume a retrospective study design that collects marker data from unrelated case and control subjects. For a given subject, let D denote a disease indicator (1=case; 0=control). Let G denote the genotype at a SNP of interest. Let Z denote a vector of genotype data for a set of substructure-informative loci. Finally, let
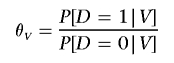 |
denote the odds of disease for a given set of variables V.
We assume that we can account for population stratification by an unmeasured (possibly vector-valued) variable U. We assume that U is not an effect modifier, so, if U were observed, we would have θG,U=exp[α+β(G)+γ(U)], where β(·) and γ(·) are known functions (up to parameters to be estimated). As a result, stratification on values of γ(U) yields the true association between D and G. Because U is unmeasured, we instead use the substructure-informative loci Z as a surrogate for this stratification variable (note that Z can also be generalized to include additional environmental covariates that provide information on U). We assume that Z provides enough information on substructure that G provides no additional information on U in the presence of Z within controls—that is, P[U|G,Z,D=0]=P[U|Z,D=0]. In this situation, we write11 the odds of disease given G and Z as
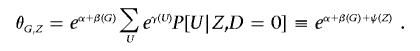 |
As a result, stratification on the unknown function ψ(Z) yields the true association between D and G.12
The null hypothesis of no association between G and D implies that β(G)=0, and hence ψ(Z)=ln{θZ}-α. Thus, under the null hypothesis, stratification on values of ln{θZ} (or θZ) is equivalent to stratifying on ψ(Z). This result implies that, when the null hypothesis is true, stratification on θZ appropriately estimates the true (null) association between D and G. We conclude that a test of β(G)=0 in strata with constant values of the score ln{θZ} is valid in the presence of population stratification. A more detailed demonstration of the above result can be found in appendix A.
These results motivate the application of our two-step procedure for controlling population stratification in case-control studies. In the first step, we compute θZ by applying a user-defined model that can range from the simple (e.g., logistic regression) to the complex (e.g., machine-learning algorithms). For all calculations in this article, we compute θZ by first using generalized partial least squares13 (PLS) to identify new variables that are linear combinations of marker genotypes and then using these new variables in a logistic-regression model for disease. Like principal components, PLS finds orthogonal linear combinations of the marker genotypes that explain variability in the data. However, unlike principal components, PLS attempts to simultaneously explain variability in both the marker data and the trait data; hence, the linear combinations found by PLS are always correlated with the trait. Generalized PLS extends the PLS model, which was originally formulated for quantitative data, to categorical outcomes. We chose the number of PLS variables by selecting the model that minimized the Bayesian information criterion (BIC).14
In the second step of our two-step approach, we use the quartiles of the stratification scores based on θZ to assign each subject to one of five strata (of approximately equal size), and then we test for association between G and D in the stratified data (e.g., using stratified logistic regression). Use of five strata is motivated by studies that show that this choice accounts for at least 90% of bias when a continuous variable is categorized, for a variety of distributions.15–17
Application to Height Data from Campbell et al.
Using data from Campbell et al.,1 we compared our stratification-score approach to genomic control, structured association, principal components, and a naive approach that ignores stratification. We used data from 192 tall and 176 short participants who were genotyped at a SNP of interest (rs4988235) in the LCT gene, as well as at a panel of substructure-informative loci consisting of 111 missense or noncoding SNPs and 67 ancestry-informative markers (AIMs).
We first conducted a naive Armitage trend test between the LCT SNP and height. Using the substructure-informative loci, we then attempted to resolve the stratification in the sample, using genomic control and principal components. For genomic control, we estimated the inflation factor 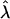 by dividing the median of the Armitage trend tests for the substructure-informative loci by the median of the χ21 distribution2 and then by taking18
by dividing the median of the Armitage trend tests for the substructure-informative loci by the median of the χ21 distribution2 and then by taking18 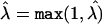 . We used this estimate to scale down the naive Armitage trend test of the LCT SNP. For principal components, we used the eigenvectors of the variance-covariance matrix of the substructure-informative loci as covariates in a linear-regression model that examines the relationship between height and the LCT SNP. As recently recommended,10 we included 10 covariates corresponding to the first 10 principal components of the variance-covariance matrix in the model. We used the likelihood-ratio statistic to test the coefficient of genotype at the test locus (coded as an additive model); significance was assessed by comparing the test statistic to the appropriate quantile of the χ2 distribution with 1 df. Results for these data calculated by use of STRUCTURE have been reported elsewhere.1
. We used this estimate to scale down the naive Armitage trend test of the LCT SNP. For principal components, we used the eigenvectors of the variance-covariance matrix of the substructure-informative loci as covariates in a linear-regression model that examines the relationship between height and the LCT SNP. As recently recommended,10 we included 10 covariates corresponding to the first 10 principal components of the variance-covariance matrix in the model. We used the likelihood-ratio statistic to test the coefficient of genotype at the test locus (coded as an additive model); significance was assessed by comparing the test statistic to the appropriate quantile of the χ2 distribution with 1 df. Results for these data calculated by use of STRUCTURE have been reported elsewhere.1
Finally, we calculated the stratification score for each participant, using generalized PLS variables in logistic regression, as described above. We then divided the data into five strata that have equal numbers of observations in each stratum, on the basis of the quartiles of the stratification scores. Using these strata, we tested for association between height and the LCT SNP, using stratified logistic regression.
Simulation Design
We conducted additional simulations to compare our proposed approach for correcting stratification to genomic control and principal components. We simulated data sets with 500 cases and 500 controls that were sampled retrospectively from a population consisting of three equally frequent latent subpopulations. Within the population, we simulated a test SNP, assuming different values for the inbreeding coefficient FST (0.03 or 0.15, with the latter value corresponding to the estimated inbreeding coefficient in the height data1) and the minor-allele frequency (MAF). For a test SNP with FST=0.03, we considered the models 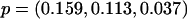 ,
, 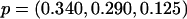 , and
, and 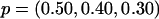 , where
, where 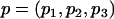 and pj denote the MAF of the locus in latent subpopulation j. These values correspond to pooled population MAFs of ∼0.10, 0.25, and 0.40, respectively. For a test SNP with FST=0.15, we considered the models
and pj denote the MAF of the locus in latent subpopulation j. These values correspond to pooled population MAFs of ∼0.10, 0.25, and 0.40, respectively. For a test SNP with FST=0.15, we considered the models 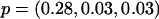 ,
, 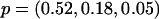 , and
, and 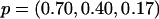 , which again correspond to pooled population MAFs of ∼0.10, 0.25, and 0.40, respectively.
, which again correspond to pooled population MAFs of ∼0.10, 0.25, and 0.40, respectively.
We assumed that control participants have the same allele-frequency distribution as the overall population (a rare-disease approximation). Case participants were sampled in different proportions from the three subpopulations. To induce severe stratification, we sampled cases in the proportions 0.45, 0.33, and 0.22 from subpopulations 1, 2, and 3, respectively. To induce more moderate stratification, we sampled cases in the proportions 0.40, 0.33, and 0.27. In addition, we also considered a situation of no confounding by sampling cases in the same proportions (0.33, 0.33, and 0.33) as the controls. We implemented this last sampling scheme to assess the performance of our stratification-score approach in situations where it is not actually required for valid analysis, since there is no difference in baseline disease risk (a requirement for confounding to occur) when cases and controls are sampled in the same proportion. Further, the substructure-informative loci are unrelated to disease risk, resulting in a stratification based entirely on noise.
All simulations assumed Hardy-Weinberg equilibrium (HWE) within each subpopulation and thus among controls in each subpopulation. We assumed a multiplicative model of allele effect for the tested locus, such that the case samples in each subpopulation were also in HWE with risk-allele frequency in subpopulation j given by eβpj/(eβpj+1-pj), where β is the log-odds of disease per copy of the risk allele. We considered simulations under both a null model (β=0) and an alternative model (β=ln(1.4)). We assumed that the value of β was constant across strata.
We generated panels of 100 substructure-informative markers under two different scenarios. The first scenario assumed the marker data consisted of AIMs with large FST values in the population, whereas the second scenario assumed that the marker data consisted of random SNPs, all with FST=0.03. Under both scenarios, we generated appropriate SNP data, using a large list19 of candidate-gene SNPs with variable allele-frequency differences among three subpopulations consisting of East Asians, African Americans, and European Americans. For sampling AIMs, we chose the 100 most informative SNPs (i.e., those with the highest FST values) from this list that were polymorphic in each subpopulation. The FST values of these candidate-gene SNPs ranged from 0.55 to 0.84. For simulation of random SNPs, we chose 100 markers from the list with an FST value of 0.03.
Results
Analysis of Height Data
Ignoring stratification, we found a significant association between the LCT SNP and height, using a naive Armitage trend test (P=.0038). This P value differs from that reported elsewhere1 (P=3.6×10-7), because the latter result is from the analysis of a much larger sample (1,057 short and 1,132 tall subjects, also including participants who were not genotyped at the AIMs) that further assumed HWE in both case and control participants.20
We found that neither genomic control2,3 nor principal components7–10 resolved the confounding in the sample. For genomic control, the scaled-down Armitage trend test was still significant (e.g., P=.0038), regardless of whether we used the 111 missense and noncoding SNPs alone, the 67 ancestry-informative SNPs alone, or all 178 loci together, because, in each case, the median trend test for marker SNPs was less than the median of the χ21 distribution. For principal components, we duplicated results published elsewhere10—that the first 10 principal components of the variance-covariance matrix for the substructure-informative loci failed to resolve the confounding between height and the LCT SNP (P=.003). Campbell et al.1 reported that the structured-association package STRUCTURE6 found only one population in the height data by use of the entire panel of 178 substructure-informative loci. Hence, the association test based on structured association is the naive (unstratified) test, which is significant (P=.0038).
Unlike genomic control, structured association, and principal components, our stratification score approach resolved the confounding in the height data from Campbell et al.1 We calculated the stratification score for each subject, using the first six PLS components (based on minimization of the BIC). We then ranked the stratification scores of all subjects and used the ranking to divide the subjects into five strata of approximately equal size. Using stratified logistic regression, we found no association between the LCT SNP and tall/short status (P=.44). Table 1 shows the genotype counts of tall or short subjects within each stratum formed using the stratification score, as well as the accompanying trend test result. Results show little association between genotype and disease within each stratum.
Table 1. .
LCT SNP Genotype Distribution among Strata
| No. of Subjects with LCT Genotype |
|||||
| Stratum and Height Status |
CC | CT | TT | Armitage χ21 | P |
| Stratum 1: | .99 | .32 | |||
| Tall | 0 | 2 | 2 | ||
| Short | 14 | 31 | 23 | ||
| Stratum 2: | .06 | .80 | |||
| Tall | 3 | 5 | 3 | ||
| Short | 17 | 25 | 21 | ||
| Stratum 3: | .07 | .79 | |||
| Tall | 5 | 23 | 13 | ||
| Short | 8 | 12 | 13 | ||
| Stratum 4: | 2.37 | .12 | |||
| Tall | 5 | 30 | 30 | ||
| Short | 3 | 2 | 3 | ||
| Stratum 5: | .61 | .43 | |||
| Tall | 4 | 35 | 32 | ||
| Short | 0 | 1 | 2 | ||
| Strata ignored: | 8.43 | .0037 | |||
| Tall | 17 | 95 | 80 | ||
| Short | 42 | 71 | 62 | ||
To ensure that our null finding was not because of insufficient power resulting from the pattern of tall/short subjects within each stratum, we conducted additional simulations of stratified data with the same row marginal totals as in table 1. Short participants were assumed to be in HWE and to have T allele frequency p=39/70, the observed frequency of the T allele among short participants. Tall participants were assumed to be in HWE and have T allele frequency eβp/(eβp+1-p); in this expression, β is the log relative risk of being tall per copy of the T allele. We found that this pattern allows an 85% power to detect a two-fold increase in risk per allele in a multiplicative model, which suggests that our null finding is not because of low power.
Simulations Results: Type I Error
Table 2 provides type I error results for simulated data sets that assume a test locus with a moderate FST of 0.03 under substantial stratification (see the “Simulation Design” section). We show empirical type I error rates for five statistics that test for association between the genotype at a SNP of interest and disease: a naive χ21 association test that ignores stratification, a χ21 association test stratified by the true yet unknown subpopulation status (the gold standard when stratification exists), a χ21 association test based on our proposed stratification-score approach, a χ21 association test based on principal components, and a χ21 association test based on genomic control.
Table 2. .
Type I Error Rates under Substantial Stratification[Note]
| Marker Type and Test Locus MAF |
No Adjustment | Known Strata | Stratification Score | Principal Components | Genomic Control |
| AIM: | |||||
| .10 | .121 | .055 | .043 | .054 | .017 |
| .25 | .195 | .057 | .048 | .062 | .026 |
| .40 | .132 | .058 | .051 | .064 | .022 |
| Random: | |||||
| .10 | .126 | .049 | .049 | .049 | .023 |
| .25 | .169 | .039 | .050 | .041 | .031 |
| .40 | .139 | .048 | .049 | .054 | .028 |
Note.— Empirical type I error results at nominal α=0.05 for 500 cases and 500 controls under the assumption of a test-locus FST of 0.03. The simulation design is described in the “Material and Methods” section. Stratification score, principal components, and genomic control tests use 100 substructure-informative loci to correct for population stratification.
Table 2 shows that, as anticipated, naive association tests that ignore stratification have inflated type I error (∼0.12–0.20 when the nominal significance is α=0.05, depending on the MAF of the test locus), whereas association tests stratified by known subpopulation have appropriate type I error. We found that both our proposed stratification-score procedure and principal components yielded appropriate type I error regardless of the control MAF and the nature of the substructure-informative loci used (AIMs with large FST values or random markers with the same FST=0.03 as the locus of interest). On the other hand, we observed that genomic control can overcorrect for stratification, particularly when AIMs are used. This result is anticipated, because genomic control implicitly assumes that the FST value (or λ) of the substructure-informative loci is the same as the FST value (or λ) of the tested locus. The use of AIMs would lead to an estimate of λ that is much larger than the inherent λ of the tested SNP (unless the SNP is an AIM itself), thereby leading to an overcorrection in the test of genomic control. We observed similar trends for more moderate levels of stratification as well (table 3).
Table 3. .
Type I Error Rates under Moderate Stratification[Note]
| Marker Type and Test Locus MAF |
No Adjustment | Known Strata | Stratification Score | Principal Components | Genomic Control |
| AIM: | |||||
| .10 | .084 | .054 | .045 | .056 | .038 |
| .25 | .094 | .055 | .054 | .062 | .038 |
| .40 | .082 | .056 | .057 | .068 | .038 |
| Random: | |||||
| .10 | .085 | .051 | .049 | .056 | .046 |
| .25 | .089 | .041 | .042 | .042 | .046 |
| .40 | .081 | .047 | .050 | .054 | .046 |
Note.— Empirical type I error results at nominal α=0.05 for 500 cases and 500 controls under the assumption of a test-locus FST of 0.03. The simulation design is described in the “Material and Methods” section. Stratification score, principal components, and genomic control tests use 100 substructure-informative loci to correct for population stratification.
Table 4 shows simulation results under substantial stratification when the test locus is under stronger selective pressure (FST=0.15) and, hence, shows larger variation across subpopulations than do the substructure-informative loci (FST=0.03). We investigated this simulation design in part because it mimics the height data from Campbell et al.1 In this situation, both principal components and genomic control failed to preserve the nominal size, with principal components yielding empirical type I error rates between 0.076 and 0.091 at α=0.05 (depending on MAF) and genomic control yielding empirical type I error rates up to seven times the nominal rate. In contrast, our stratification-score approach had appropriate type I error in these situations. We also observed similar trends under more modest levels of stratification (table 5). These results suggest that our two-step approach provides a more appropriate correction for population stratification when the test locus demonstrates more variation across subpopulations than do the substructure-informative loci. Such a scenario can easily arise in the study of candidate genes or other regions that are under strong selective pressure.
Table 4. .
Type I Error Rates under Substantial Stratification[Note]
| Marker Type and Test Locus MAF |
No Adjustment | Known Strata | Stratification Score | Principal Components | Genomic Control |
| Random: | |||||
| .10 | .380 | .052 | .052 | .076 | .121 |
| .25 | .661 | .057 | .050 | .091 | .332 |
| .40 | .641 | .058 | .054 | .090 | .350 |
Note.— Empirical type I error results at nominal α=0.05 for 500 cases and 500 controls under the assumption of a test-locus FST of 0.15. The simulation design is described in the “Material and Methods” section. Stratification score, principal components, and genomic control tests use 100 substructure-informative loci to correct for population stratification.
Table 5. .
Type I Error Rates under Moderate Stratification[Note]
| Marker Type and Test Locus MAF |
No Adjustment | Known Strata | Stratification Score | Principal Components | Genomic Control |
| Random: | |||||
| .10 | .160 | .053 | .043 | .070 | .110 |
| .25 | .240 | .050 | .047 | .059 | .167 |
| .40 | .253 | .047 | .046 | .070 | .175 |
Note.— Empirical type I error results at nominal α=0.05 for 500 cases and 500 controls under the assumption of a test-locus FST of 0.15. The simulation design is described in the “Material and Methods” section. Stratification score, principal components, and genomic control tests use 100 substructure-informative loci to correct for population stratification.
Finally, table 6 shows simulations results when no confounding actually exists in the sample. Across all models considered, we found that our stratification-score approach and principal components both had appropriate type I error rates that were similar to that of the naive (yet valid) association test. Genomic control, on the other hand, appeared to yield conservative inference across these simulations, with empirical type I error rates ranging between 0.022 and 0.036 at nominal α=0.05.
Table 6. .
Type I Error Rates under No Confounding[Note]
| Marker Type and Test Locus MAF |
No Adjustment | Known Strata | Stratification Score | Principal Components | Genomic Control |
| AIM: | |||||
| .10 | .054 | .044 | .048 | .055 | .036 |
| .25 | .047 | .045 | .043 | .053 | .034 |
| .40 | .045 | .043 | .041 | .046 | .027 |
| Random: | |||||
| .10 | .048 | .051 | .044 | .047 | .025 |
| .25 | .050 | .047 | .045 | .042 | .022 |
| .40 | .053 | .053 | .044 | .047 | .029 |
Note.— Empirical type I error results at nominal α=0.05 for 500 cases and 500 controls under the assumption of a test-locus FST of 0.03. The simulation design is described in the “Material and Methods” section. Stratification score, principal components, and genomic control tests use 100 substructure-informative loci to correct for population stratification.
Simulations Results: Power
Table 7 shows power results at nominal significance α=0.05 for simulated data sets under an alternative model of true disease-marker association, under the assumption of a test locus with a moderate FST of 0.03 under substantial stratification. We show empirical power for four association statistics: a χ21 association test stratified by the true yet unknown subpopulation status that serves as a gold standard, a χ21 association test based on our proposed stratification-score approach, a χ21 association test based on principal components, and a χ21 association test based on genomic control. For AIMs, table 7 demonstrates that our proposed stratification-score procedure and principal components had comparable power and both procedures consistently had improved power relative to genomic control for detecting the disease-marker association. For random markers, table 7 shows that all three methods have comparable power. We also observed similar trends for more moderate levels of stratification (table 8).
Table 7. .
Power under Substantial Stratification[Note]
| Marker Type and Test Locus MAF |
Known Strata | Stratification Score | Principal Components | Genomic Control |
| AIM: | ||||
| .10 | .679 | .667 | .687 | .422 |
| .25 | .903 | .905 | .920 | .668 |
| .40 | .958 | .951 | .960 | .695 |
| Random: | ||||
| .10 | .690 | .732 | .751 | .702 |
| .25 | .918 | .923 | .940 | .934 |
| .40 | .957 | .962 | .970 | .951 |
Note.— Empirical power results at nominal α=0.05 for 500 cases and 500 controls under the assumption of a test-locus FST of 0.03. The simulation design is described in the “Material and Methods” section. Stratification score, principal components, and genomic control tests use 100 substructure-informative loci to correct for population stratification.
Table 8. .
Power under Moderate Stratification[Note]
| Marker Type and Test Locus MAF |
Known Strata | Stratification Score | Principal Components | Genomic Control |
| AIM: | ||||
| .10 | .701 | .680 | .708 | .538 |
| .25 | .890 | .863 | .900 | .744 |
| .40 | .963 | .952 | .967 | .813 |
| Random: | ||||
| .10 | .670 | .690 | .707 | .703 |
| .25 | .897 | .870 | .912 | .933 |
| .40 | .956 | .940 | .961 | .962 |
Note.— Empirical power results at nominal α=0.05 for 500 cases and 500 controls under the assumption of a test-locus FST of 0.03. The simulation design is described in the “Material and Methods” section. Stratification score, principal components, and genomic control tests use 100 substructure-informative loci to correct for population stratification.
We also conducted power calculations under substantial stratification when the test locus showed more variation across subpopulations (FST=0.15) than did the substructure-informative loci (FST=0.03). We found that our proposed approach maintained good power in these situations, with results quite similar to those shown in table 7 for random markers. We did not make power comparisons with principal components and genomic control because of their inappropriate size in this situation.
Finally, table 9 shows power results under no confounding in the sample. In this situation, we find that both our stratification-score approach and principal components have power similar to that of the (valid) naive test and the known-subpopulation test, regardless of the MAF of the test locus and the nature of the substructure-informative loci. Genomic control had power similar to that of these approaches when the substructure-informative loci were random markers but had less power when the substructure-informative loci were AIMs. These simulations demonstrate that the use of our stratification-score approach appears to have negligible effect on power when there is no confounding within the sample.
Table 9. .
Power under No Confounding[Note]
| Marker Type and Test Locus MAF |
No Adjustment | Known Strata | Stratification Score | Principal Components | Genomic Control |
| AIM: | |||||
| .10 | .597 | .635 | .593 | .648 | .472 |
| .25 | .862 | .900 | .871 | .903 | .742 |
| .40 | .951 | .961 | .936 | .948 | .856 |
| Random: | |||||
| .10 | .622 | .634 | .617 | .657 | .578 |
| .25 | .866 | .890 | .851 | .906 | .846 |
| .40 | .949 | .936 | .946 | .966 | .931 |
Note.— Empirical power results at nominal α=0.05 for 500 cases and 500 controls under the assumption of a test-locus FST of 0.03. The simulation design is described in the “Material and Methods” section. Stratification score, principal components, and genomic control tests use 100 substructure-informative loci to correct for population stratification.
Discussion
We have proposed a powerful new approach for controlling population stratification in case-control studies of disease: subclassification based on the stratification score. We showed that our proposed approach corrected for population stratification in a case-control data set of extreme height,1 using a panel of 101 AIMs and 67 missense or noncoding SNPs. This is in contrast to the methods of genomic control, structured association, and principal components, all of which failed to control for stratification in these data. This example, together with our simulation results, shows that our procedure provides an improved correction for population stratification, compared with existing approaches. Our approach can be easily implemented using existing software, such as SAS or R. We have provided samples of such code for implementing our approach on our Web site (Epstein software).
Our approach is based on a flexible modeling framework that requires fewer assumptions than do existing methods used for valid inference in the presence of stratification. Unlike principal components, our approach properly controls for stratification when the test locus exhibits more variation among subpopulations than do the substructure-informative loci used to correct the confounding, as for the LCT locus in the height data. Unlike genomic control2,3 and similar methods,21 our approach is applicable to situations in which the tested locus and the substructure-informative loci have different FST values. Furthermore, our approach can accommodate multiallelic test and substructure-informative loci and can be further extended to adjust for population stratification in multilocus genotype or haplotype association analysis. Unlike structured-association methods,5,6,22,23 our approach does not require that we assume a population composed of discrete subpopulations. This is important because the concept of discrete subpopulations in a population-based study is probably an oversimplification, since the population itself likely consists of a continuous mixture of ancestral subgroups. Finally, unlike structured-association approaches that are typically computationally intensive, our approach is computationally simple to implement.
In this article, we have advocated subclassification of data into five strata based on the stratification score. We stress that this choice does not correspond to a belief that there are five subpopulations, but instead is based on studies that show that this choice removes 90% of bias when a continuous variable is categorized.15 This strategy is also analogous to that used in observational studies that subclassify data into five propensity-score–based strata.16,17 If this seems arbitrary, one could treat the stratification score as a continuous covariate in a logistic-regression model when testing for association between D and G. This choice avoids the arbitrary selection of five strata but requires that the stratification score be correctly estimated; subclassification requires only that the ordering of stratification scores be correct. However, use of the stratification score as a quantitative variable is especially appealing for small studies, where subclassification into five strata may result in many empty cells.
For some of our simulations, we assumed a test locus with FST=0.15. Both allele frequencies and disease risk must covary before population stratification can produce a spurious association. Because a small FST implies homogeneous allele frequencies at that locus even if the population is structured, associations involving loci with large FST are more likely be spurious. A value of FST=0.15 may seem unlikely, considering that within-continent average FST values are <0.01 for most populations.24 However, although average FST values are small, locus-specific FST values vary widely. Empirical studies19,25 have identified many marker loci with estimated FST>0.15, suggesting that substantial variation across subpopulations can regularly occur in large-scale or genomewide association studies. In fact, FST calculated among short subjects from the height study of Campbell et al.1 is ∼0.15.
For proper inference, both genomic control and structured association require “null” substructure-informative loci that are not associated with disease. For genomic control, the inclusion of a null marker that is truly associated with disease within the method will overestimate the inflation factor and will lead to an overcorrection of the test statistic. For structured association, inclusion of null markers truly associated with disease will distort the HWE among case and control populations. Since structured-association methods allocate subpopulation status on the basis of minimizing the deviation of HWE, this inclusion can result in inappropriate subpopulation assignment. On the other hand, our proposed approach, as well as principal components, can handle substructure-informative loci that are truly associated with disease (with the assumption that they do not interact with the test locus of interest). This is appealing since, with an increasing number of substructure-informative loci used for correcting of stratification, there is an increase in the probability of a substructure-informative locus being truly associated with disease.
Bayesian or stepwise logistic regression has been proposed to assess association between disease and a test locus, with adjustment for the confounding effects of population stratification by use of substructure-informative loci.18 We feel that our proposed approach is preferred over these logistic-regression procedures. Unlike our approach, stepwise logistic-regression procedures often fail to preserve a nominal type I error rate for testing association. Of course, stepwise logistic regression could be recalibrated to give the proper size, but this would require extensive permutation analysis to select an appropriate cutoff value to use when significance is assessed. Bayesian logistic regression is computationally intensive, and, furthermore, it failed to properly correct for population stratification under extreme sampling of cases from a particular subpopulation.18 We found that our proposed approach properly corrected for stratification in such a situation (data not shown). Thus, given the nontrivial computational effort required for these logistic-regression procedures, our approach will be far more efficient computationally than either the stepwise or Bayesian logistic-regression proposals of Setakis et al.18
Our stratification-score approach for controlling stratification has a parallel in the propensity-score approach for controlling confounding in prospective studies.16,17 Stratification on the propensity score, which is defined as the probability of exposure given potential confounders, removes confounding from the relationship between disease and a binary exposure. It is noteworthy that stratification on the estimated propensity score does not affect the size of the second-step test statistic.26–28 We observed a similar phenomenon in our approach. This is important, as it allows great flexibility in the choice of the first-step model for the disease odds conditional on the substructure-informative loci. We can choose first-step models that range from the traditional (e.g., logistic regression) to the complex (e.g., high-dimensional procedures, such as generalized PLS or support-vector machines29). In particular, we can apply first-step models, like PLS, that do not provide standard inference (e.g., they fail to produce P values without extensive permutation testing) and yet can still use the second-step model to calculate an appropriate P value for testing association between the test locus and disease. This is appealing because we can then apply our procedure to data sets consisting of large numbers of correlated substructure-informative loci (with varying allele number, allele frequency, and FST values), such as those available in whole-genome association studies.
Our approach is also related to the confounder score: the odds of disease given covariates among persons with the same exposure level. Poststratification on the confounder score removes the effects of confounding within case-control studies.12 In the genetic context, implementation of the confounder score consists of stratifying on the disease odds given the substructure-informative loci among subjects with the same genotype at the tested locus of interest. The confounder-score approach leads to an unbiased estimator of the true association between disease and genotype but can lead to inflated type I error30 due to colinearity between the test locus and the substructure-informative loci in the presence of population stratification. Our proposed approach avoids this colinearity issue by stratifying on the disease odds among all subjects, regardless of the test-locus genotype. Using simulated data, we showed that our proposed approach has appropriate type I error in the presence of population stratification.
Our stratification-score approach can be extended to more general settings in genetic association studies. For example, within the first step of our procedure, we model and calculate the odds of disease conditional on substructure-informative loci. However, we can also incorporate additional (environmental) covariates that provide information on population substructure within this model, assuming that such covariates do not interact with the test-locus genotype. Also, in the second step of our two-step procedure, we can accommodate multilocus genotype or haplotype data. We will explore these extensions, as well as methods for detecting gene-gene and gene-environment interaction effects, in a subsequent paper.
Acknowledgments
We thank Drs. Catarina Campbell and Joel Hirschhorn for providing us with the marker and height data from their study. We thank Drs. Eleanor Feingold and Kathryn Garber for their helpful comments on previous versions of the manuscript. This work was supported by National Institutes of Health grants HG003618 (to M.P.E.) and HL077663 (to A.S.A.).
Appendix A: Removing the Effects of Confounding by Stratifying on θZ
We define strata in such a way that we assume θZ is constant (i.e., θZ=κ) for each subject in a given stratum. As a result, for a given stratum S, we can write
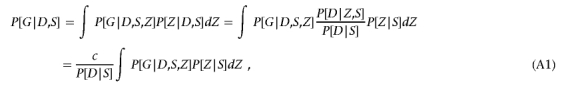 |
where c denotes the constant P[D|Z,S], which is a function of θZ.
If we consider the odds ratio Ψ(S)G,G′ that compares the odds of G to some reference genotype G′ within stratum S, we can use equation (A1) to write
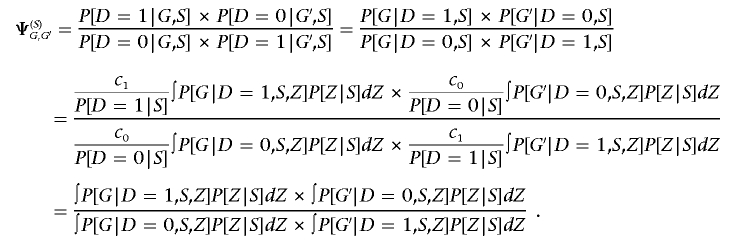 |
Note that, if β(G)=0, then we have P[G|D=1,S,Z]=P[G|D=0,S,Z]=P[G|S,Z] and Ψ(S)G,G′=1 immediately. To show the converse, if there is no association between G and D in each stratum, then P[G|D=1,S,Z]=P[G|D=0,S,Z]; if this is the case, then β(G)=0 in a model in which we stratify on S. Therefore, we conclude that stratifying on a confounder score defined by θZ leads to a valid association test of D and G, even when population stratification exists.
Footnotes
Any opinions expressed in this article are those of the authors and do not necessarily represent the views of the Centers for Disease Control and Prevention.
Web Resources
The URLs for data presented herein are as follows:
- Epstein software, http://www.genetics.emory.edu/labs/epstein/software
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for LCT) [PubMed]
References
- 1.Campbell CD, Ogburn EL, Lunetta KL, Lyon HN, Freedman ML, Groop LC, Altshuler D, Ardlie KG, Hirschhorn JN (2005) Demonstrating stratification in a European American population. Nat Genet 37:868–872 10.1038/ng1607 [DOI] [PubMed] [Google Scholar]
- 2.Devlin B, Roeder K (1999) Genomic control for association studies. Biometrics 55:997–1004 10.1111/j.0006-341X.1999.00997.x [DOI] [PubMed] [Google Scholar]
- 3.Devlin B, Roeder K, Wasserman L (2001) Genomic control, a new approach to genetic-based association studies. Theor Popul Biol 60:155–166 10.1006/tpbi.2001.1542 [DOI] [PubMed] [Google Scholar]
- 4.Pritchard JK, Rosenberg NA (1999) Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet 65:220–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu X, Zhang S, Zhao H, Cooper RS (2002) Association mapping using a mixture model for complex traits. Genet Epidemiol 23:181–196 10.1002/gepi.210 [DOI] [PubMed] [Google Scholar]
- 8.Zhang S, Zhu X, Zhao H (2003) On a semi-parametric test to detect associations between quantitative traits and candidate genes using unrelated individuals. Genet Epidemiol 24:44–56 10.1002/gepi.10196 [DOI] [PubMed] [Google Scholar]
- 9.Chen H-S, Zhu X, Zhao H, Zhang S (2003) Qualitative semi-parametric test to detect genetic association in case-control design under structured population. Ann Hum Genet 67:250–264 10.1046/j.1469-1809.2003.00036.x [DOI] [PubMed] [Google Scholar]
- 10.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D (2006) Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38:904–909 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- 11.Satten GA, Kupper LL (1993) Inferences about exposure-disease associations using probability-of-exposure information. J Am Stat Assoc 88:200–208 10.2307/2290714 [DOI] [Google Scholar]
- 12.Miettinen O (1976) Stratification by a multivariate confounder score. Am J Epidemiol 104:609–620 [DOI] [PubMed] [Google Scholar]
- 13.Marx BD (1996) Iteratively reweighted partial least squares estimation for generalized linear regression. Technometrics 38:374–381 10.2307/1271308 [DOI] [Google Scholar]
- 14.Schwarz G (1978) Estimating the dimension of a model. Ann Stat 6:461–464 [Google Scholar]
- 15.Cochran WG (1968) The effectiveness of subclassification in removing bias in observational studies. Biometrics 24:295–313 10.2307/2528036 [DOI] [PubMed] [Google Scholar]
- 16.Rosenbaum PR, Rubin DB (1983) The central role of the propensity score in observational studies for causal effects. Biometrika 70:41–55 10.1093/biomet/70.1.41 [DOI] [Google Scholar]
- 17.Rosenbaum PR, Rubin DB (1984) Reducing bias in observational studies using subclassification on the propensity score. J Am Stat Assoc 79:516–524 10.2307/2288398 [DOI] [Google Scholar]
- 18.Setakis E, Stirnadel H, Balding DJ (2006) Logistic regression protects against population stratification in genetic association studies. Genome Res 16:290–296 10.1101/gr.4346306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Akey JM, Zhang G, Zhang K, Jin L, Shriver MD (2002) Interrogating a high-density SNP map for signatures of natural selection. Genome Res 12:1805–1814 10.1101/gr.631202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Saseini P (1997) From genotype to genes: doubling the sample size. Biometrics 53:1253–1261 10.2307/2533494 [DOI] [PubMed] [Google Scholar]
- 21.Reich DE, Goldstein DB (2001) Detecting association in a case-control study while correcting for population stratification. Genet Epidemiol 20:4–16 [DOI] [PubMed] [Google Scholar]
- 22.Hoggart CJ, Parra EJ, Shriver MD, Bonilla C, Kittles RA, Clayton DG, McKeigue PM (2003) Control of confounding of genetic associations in stratified populations. Am J Hum Genet 72:1492–1504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Satten GA, Flanders WD, Yang Q (2001) Accounting for unmeasured population structure in case-control studies of genetic association using a novel latent-class model. Am J Hum Genet 68:466–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.National Research Council (1996) The evaluation of forensic DNA evidence. National Academy Press, Washington, DC [Google Scholar]
- 25.Zhivotovsky LA, Ahmed S, Wang W, Bittles AH (2001) The forensic DNA implications of genetic differentiation between endogamous communities. Forensic Sci Int 119:269–272 10.1016/S0379-0738(00)00442-4 [DOI] [PubMed] [Google Scholar]
- 26.Agodini R, Dynarski M (2001) Are experiments the only option? A look at dropout prevention programs. Technical report. Mathematica Policy Research, Princeton, NJ [Google Scholar]
- 27.Benjamin DJ (2003) Does 401(k) eligibility increase saving? Evidence from propensity score subclassification. J Public Econ 87:1259–1290 10.1016/S0047-2727(01)00167-0 [DOI] [Google Scholar]
- 28.Lunceford JK, Davidian M (2004) Stratification and weighting via the propensity score in estimation of causal treatment effects: a comparative study. Stat Med 23:2937–2960 10.1002/sim.1903 [DOI] [PubMed] [Google Scholar]
- 29.Hastie T, Tibshirani R, Friedman J (2001) The elements of statistical learning. Springer, New York [Google Scholar]
- 30.Pike MC, Anderson J, Day NE (1979) Some insight into Miettinen’s multivariate confounder score approach to case-control studies. J Epidemiol Community Health 33:104–106 [DOI] [PMC free article] [PubMed] [Google Scholar]