

Abstract
We conducted a genomewide linkage screen of a simple heavy-smoking quantitative trait, the maximum number of cigarettes smoked in a 24-h period, using two independent samples: 289 Australian and 155 Finnish nuclear multiplex families, all of which were of European ancestry and were targeted for DNA analysis by use of probands with a heavy-smoking phenotype. We analyzed the trait, using a regression of identity-by-descent allele sharing on the sum and difference of the trait values for relative pairs. Suggestive linkage was detected on chromosome 22 at 27–29 cM in each sample, with a LOD score of 5.98 at 26.96 cM in the combined sample. After additional markers were used to localize the signal, the LOD score was 5.21 at 25.46 cM. To assess the statistical significance of the LOD score in the combined sample, 1,000 simulated genomewide screens were conducted, resulting in an empirical P value of .006 for the LOD score of 5.21. This linkage signal is driven mainly by the microsatellite marker D22S315 (22.59 cM), which had a single-point LOD score of 5.41 in the combined sample and an empirical P value <.001 from 1,000 simulated genomewide screens. This marker is located within an intron of the gene ADRBK2, encoding the beta-adrenergic receptor kinase 2. Fine mapping of this linkage region may reveal variants contributing to heaviness of smoking, which will lead to a better understanding of the genetic mechanisms underlying nicotine dependence.
Long-term cigarette smoking (susceptibility to tobacco addiction [MIM 188890]) remains the leading behavioral risk factor for early morbidity and mortality from disorders such as lung cancer, heart disease, emphysema, and bronchitis in both men and women,1–3 in both industrialized and developing countries,4 with risk for many of these illnesses reported to increase sharply with the number of cigarettes smoked.3,5,6 Whereas some have suggested that research on the social causes of cigarette smoking should be the priority,7 this argument is based largely on data concerned with the initiation of smoking rather than on outcomes in those who have become smokers. Although the prevention of cigarette smoking among young people is of undeniable importance, even complete success in global elimination of all new onsets of cigarette smoking would leave 4 million existing smokers per year to die from smoking-related diseases.8
Very consistent evidence of genetic effects on long-term persistence in smoking is found across sex, birth cohort, and society in twin samples from Sweden, Finland, Australia, and the United States. With controlling for factors associated with risk for becoming a cigarette smoker, estimates of heritability for smoking persistence versus quitting successfully have been found to be as high as 52%–71%.9–14 Difficulty quitting cigarette smoking is believed to be due to dependence on nicotine and perhaps other components of tobacco. Substantial heritability15 has been found for both DSM-based16 categorical and quantitative measures of nicotine dependence, such as Fagerström-based measures.17 Two important findings concerning the heritability of nicotine dependence have been derived through the multivariate genetic factor analysis of DSM- and Fagerström-based symptom data obtained in a large sample survey of adult Australian twins15: (1) a strong genetic factor is, in large part, responsible for the clustering of symptoms associated with nicotine dependence, and (2) measures of the number of cigarettes smoked (including a two-level variable derived from MaxCigs24 [the maximum number of cigarettes smoked in a 24-h period], used as an indicator of DSM-IV nicotine tolerance, and daily smoking rate, used for Fagerström-based scales) are among the items with the highest factor loadings in the genetic analyses of these data (and, in women, are the highest). This suggests that the degree of nicotine tolerance achieved may be an important marker for genes contributing to risk of nicotine dependence (most smokers achieve a level of daily cigarette consumption that would have been toxic when they first started smoking).
To our knowledge, there is only a small number of published linkage studies that have targeted smoking persistence or nicotine dependence, with findings mostly suggestive and lacking replication. Using nicotine dependence as a binary phenotype derived from the Fagerström Tolerance Questionnaire (FTQ)17 with the cutoff FTQ score ⩾7, Straub and his colleagues18 found suggestive linkage on chromosomes 2 and 10. More recently, Swan et al.19 found suggestive linkage on chromosome 6 for the Fagerström Test for Nicotine Dependence (FTND)17 (LOD=2.7) and a measure of nicotine withdrawal severity (LOD=2.7), on chromosome 7 for a quantitative measure of nicotine dependence (LOD=3.0), on chromosome 8 for a binary measure of DSM-IV nicotine dependence (LOD=2.7), and on chromosome 16 for a pattern of short-term quitting of cigarettes (LOD=4.0). A recent study of African Americans found evidence of linkage for several smoking phenotypes, including the FTND, to chromosomes 9q31, 11p11, and 13q13.20 Another study found linkage for the FTND in an African American sample on chromosome 5 and in a European American sample on chromosome 7.21 Suggestive linkage on nearly all the chromosomes has been reported for measures of quantity of cigarettes smoked, including measures of the average number of cigarettes smoked per day19 and the largest value reported for this same measure in a longitudinal study across baseline and series of follow-up assessments.22–27 Despite the importance of nicotine tolerance to dependence on nicotine, social restraints on times and places that cigarettes may be smoked may have reduced the informativeness of these daily consumption measures.28,29 A more complicated phenotype has also been used, where two dimensions of the smoking habit—numbers of cigarettes smoked and number of years smoked—are combined in the same measure.30–34 Overall, among the strongest signals from these findings is the multipoint LOD of 4.17 for typcial quantity smoked on chromosome 10 at 92 cM.20 Additionally, there is overlap on chromosome 8 between the studies of Ehlers et al.34 (LOD=2.00) and Saccone et al.22 (LOD=2.31) at ∼115 cM, on chromosome 9 between the studies of Li et al.24 (P=.004) at 101.64 cM and Gelernter et al.33 (P=.001) at 112.85 cM, and on chromosome 11 between the studies of Li et al.24 (P=1.8×10-5) and Morley et al.27 (P=.004) at ∼65 cM. Additional linkage on chromosome 11 has been reported at 3 cM (P=.0003).33
In the present study, we conducted a genetic linkage analysis of a simple quantitative trait, MaxCigs24. This trait represents a measure of tolerance to the effects of nicotine, a key component of nicotine dependence.16 Although the underlying genetic structure of MaxCigs24 is unlikely to be identical to that of nicotine dependence, it is expected to share common genetic risk factors15 and has the advantage of grading both dependent and nondependent individuals. Whereas recent restrictions on the times and locations where smoking is allowed have had a negative impact on the informativeness of reports of typical daily cigarette consumption, it is reasonable to expect that MaxCigs24 would be more robust than typical measures of cigarette use to such kinds of limitations.
As part of an international consortium known as “the Nicotine Addiction Genetics (NAG) project,” investigators from Australia, Finland, and the United States are using population-based samples to identify genes harboring risk loci for heavy cigarette smoking and nicotine dependence and to understand the relative importance of these genes in determining risk of becoming a long-term, dependent cigarette smoker. Toward this goal, we conducted genome scans using DNA from >450 Australian and Finnish families. Presented here are findings for a measure of nicotine tolerance that, as prior research suggests,15 may be a strong behavioral marker for genes associated with nicotine dependence and is a phenotype for which we find replicated evidence of linkage.
Subjects and Methods
Family Ascertainment
The study participants for the NAG project were enrolled at two different sites: the Queensland Institute of Medical Research (QIMR) in Australia and the University of Helsinki (UH) in Finland (see table 1). Families were identified through heavy-smoking index cases by use of previously administered interview and questionnaire surveys of the community-based Australian and population-based Finnish registers of twins. The Finnish arm of the NAG project recruited families from the Finnish Twin Cohort, which consists of all Finnish twin pairs born between 1938 and 1957. Families chosen for the Australian arm of the NAG study were identified from two cohorts of the Australian Twin Panel, which included spouses of the older of these two cohorts, for a total of ∼12,500 families with information about smoking. The ancestry of the Australian samples is predominantly Anglo-Celtic or northern European (>90%).
Table 1. .
Summary Statistics for the Three Samples[Note]
| Summary Statistics by Sample |
|||
| Characteristic | Australian NAGa | Finnish NAGa | Australian BigSibb |
| No. of families: | 289 | 154 | 1,254 |
| Offspring, mean±SD | 3.2±1.4 | 3.2±1.4 | 4.1±1.8 |
| No. (%) of men | 590 (45.0) | 260 (50.8) | 2,389 (44.3) |
| No. (%) of women | 721 (55.0) | 252 (49.2) | 3,003 (55.7) |
| Total no. of subjects | 1,311 | 512 | 5,392 |
| Age range (years) | 21–86 | 29–91 | 18–91 |
| Age, mean±SD (years) | 48.8±14.1 | 57.9±7.3 | 46.1±10.7 |
| No. (%) of subjects considered heavy smokers: | |||
| Men | 449 (76.1) | 122 (47.5) | 862 (38.4) |
| Women | 389 (57.8) | 110 (46.6) | 720 (27.3) |
| No. (%) of subjects who meet DSM-IV criteria for nicotine dependencec: | |||
| Men | 402 (75.1) | 117 (47.5) | 824 (56.3) |
| Women | 398 (71.4) | 95 (47.3) | 903 (56.8) |
| No. of subjects who meet FTND12 criteria for nicotine dependencec (cigarette ranged): | |||
| Men: | 535 (0–10) | 246 (0–10) | 1,463 (0–10) |
| Mean±SD no. of cigarettes | 4.8±2.5 | 3.8±2.2 | 3.4±2.7 |
| Women: | 557 (0–10) | 201 (0–10) | 1,589 (0–10) |
| Mean±SD no. of cigarettes | 4.0±2.6 | 3.2±2.5 | 2.8±2.6 |
| No. of subjects who meet MaxCigs24 criteria (cigarette ranged): | |||
| Men: | 537 (3–98) | 247 (2–98) | 1,462 (1–98) |
| Mean±SD no. of cigarettes | 44.3±19.7 | 34.8±16.7 | 35.0±19.5 |
| Women: | 561 (2–98) | 202 (2–60) | 1,597 (2–98) |
| Mean±SD no. of cigarettes | 33.1±16.1 | 22.4±10.8 | 26.7±14.9 |
Note.— All families are nuclear.
The figures reported are for genotyped individuals.
A community-based Australian sample that was used to determine population estimates of parameters related to our quantitative trait; we report only on those subjects who were interviewed.
Regular smokers only (at least 100 cigarettes, lifetime).
Cigarette range indicates no. of cigarettes, lifetime. Values of 99 or greater were rounded to 98.
We also used data obtained from a third Australian community-based family study, the Australian Big Sibship Study (BigSib), for the purpose of obtaining population estimates of our quantitative trait. The BigSib sample comprises families ascertained through the Australian Twin Panel selected for five or more offspring sharing both biological parents (see table 1). This is a reasonable approach, since we have found sibship size to be uncorrelated with cigarette smoking outcomes in ever-smokers (those who have at least tried cigarettes, even a puff). The BigSib sample includes 1,254 families that were unselected with respect to alcohol use, tobacco use, or any other form of psychopathology. Families for the BigSib sample were recruited from the same Australian Twin Panel sources as were the NAG Australian families, and phenotypic information was obtained using the same assessment protocol as for the NAG.
All data-collection procedures were approved by institutional review boards at Washington University (WU), the QIMR, and the UH, including the use of appropriate and approved informed-consent procedures. Participants gave informed consent for an interview, for providing a blood sample for DNA extraction and cell lines, and for the sharing of their anonymous clinical and genotypic records and DNA with other scientists outside of this project’s research team of investigators. If the subject was an index case, permission was requested to contact other family members.
Clinical Assessment
The data-collection protocol included two phases—a telephone screening and a diagnostic interview phase—after which a sample of blood for DNA extraction was requested. Eligible families from both sites were required to have at least one adult sib pair (not including MZ twin pairs) concordant for a history of cigarette smoking, determined by earlier questionnaire or interview surveys. At the Australian site, families were targeted for screening with use of the criterion that they have at least one sib pair concordant for a lifetime history of heavy smoking (defined as either a history of smoking 20 cigarettes per day during the period of heaviest smoking or smoking at least 40 cigarettes in any 24-h period). Priority was given to families with available biological parents. Unless both parents were available, an attempt was made to obtain clinical data and a blood specimen from at least one unaffected full biological sibling. At the Finnish site, because of the absence of information on cigarette-smoking history in other nontwin siblings, families with DZ cotwins concordant for a lifetime history of regular smoking were screened and enrolled in the study. Priority was given to families of DZ twins with histories of heavier cigarette consumption. At both sites, during the screening interview, sufficient information on a lifetime history of cigarette smoking was obtained from the index case, to confirm family eligibility, and permission was requested to contact available parents and full biological siblings for participation in the study.
Clinical data were collected using a computer-assisted telephone diagnostic interview (CATI), an adaptation of the Semi-Structured Assessment for the Genetics of Alcoholism (SSAGA)35,36 for telephone administration. The tobacco section of the CATI was derived from the Composite International Diagnostic Interview (CIDI)37 and incorporated standard FTND, DSM-IIIR, and DSM-IV assessments of nicotine dependence. It also included a detailed history of the first use of cigarettes and other tobacco products; the quantity and frequency of use for current, most recent, and heaviest period of use; and supplemental items concerning attempts at cessation. Information on other comorbid psychiatric disorders, such as major depression and anxiety disorders, and conduct and antisocial personality disorders (with use of DSM-IIIR and DSM–IV criteria) was elicited by the CATI. The mailed questionnaire survey included personality scales (NEO38) and obtained additional information on cigarette use and nicotine dependence (the Nicotine Dependence Syndrome Scale39). Interviewers were selected from trained panels of interviewers at QIMR and the UH, which include nurses or graduates in Psychology or a related field.
At the Australian site, a diagnostic telephone interview was completed by 3,425 Australians (1,800 women), and 2,912 blood samples were obtained (including 1,557 from women and including 807 from parents) from these individuals. DNA from 953 Australian individuals in 289 families and from 623 Finnish individuals in 155 families who were identified as informative for the heaviness-of-smoking phenotype was sent for a genome scan. DNA was available from an additional 133 who refused to give blood but provided a buccal sample. At the Finnish site, a diagnostic telephone interview was completed by 2,265 Finnish subjects (1,058 women), and 2,224 samples of blood were collected (1,057 from female subjects and 171 from subject parents).
If it was determined during the interview that the subject was a regular smoker (smoked ⩾100 cigarettes, lifetime; 10 additional Australian genotyped individuals and 1 Finnish genotyped individual were also considered to be regular smokers, since they smoked 26–99 cigarettes, lifetime, and smoked weekly for ⩾2 mo), the subject was then asked “What is the largest number of cigarettes you have ever smoked in a 24-h period?” (MaxCigs24). If a subject reported that he or she had smoked >98 cigarettes in a 24-h period, then MaxCigs24 was coded 98. MaxCigs24 is a quantitative phenotype that has acceptable long-term test-retest reliability and high heritability in both men and women. Using a volunteer cohort of adult Australian twins,15 some of whom were included in the linkage analyses, we computed the extent to which individual differences in the liability to MaxCigs24 in regular smokers was due to heritable and environmental influences, and we estimated the heritability to be 55% (95% CI 48%–60%). The 5-year test-retest reliability observed in Australian twins is 0.72. The correlation of MaxCigs24 with measures of nicotine dependence is substantial. In the BigSib sample, among regular smokers, both men and women, the correlation of MaxCigs24 with FTND is 0.67, and the correlation with the DSM-IV symptom count is 0.64.
Table 1 shows the distributions of smoking outcomes for the Australian NAG, Finnish NAG, and BigSib samples. For the offspring generation (not shown), the approximate mean age (±SD) at onset of weekly smoking (smoked at least once per wk for at least 2 mo) is 16.1±3.5 in the Australian NAG sample, 18.2±4.7 in the Finnish NAG sample (an older sample), and 17.4±4.1 in the BigSib sample. The number of years smoked is ∼19.0±8.8 in the Australian NAG sample, 28.8±12.7 in the Finnish NAG sample, and 18.0±10.8 in the BigSib sample.
Genotyping and Cleaning
The genome screen for the Australian NAG sample was based on a set of 381 autosomal microsatellite markers spaced at ∼10 cM throughout the autosomal genome. At the time of analysis, genotype data for the Finnish NAG sample were available for a subset of 362 of these markers. The excluded markers were distributed randomly throughout the genome, and these exclusions did not affect the overall mean spacing in the Finnish marker set. The genetic map used for our analysis is based on the deCODE genetic map.40 A few of the markers used in our study were not part of the deCODE set and were assigned genetic map positions on the basis of an interpolation of the physical map obtained from build 36.2 of the human reference genome (National Center for Biotechnology Information [NCBI]). The Australian NAG sample was genotyped at the Australian Genome Research Facility (AGRF), and the Finnish NAG sample was genotyped at the Finnish Genome Center (FGC). The AGRF used an ABI (Applied Biosystems) genotyping platform, and the FGC used both ABI and MegaBACE (Amersham Biosciences) platforms. To refine the linkage signal found on chromosome 22, two additional microsatellite markers flanking D22S315—namely, D22S1174 and D22S1144—were genotyped for the Finnish NAG sample at the FGC and for the Australian NAG sample at WU, with use of an ABI 3100 platform.
Low-level processing and database management for the genotype data were performed using the statistical/database package SAS and the Perl programming language. The genotype data were then screened for Mendelian errors and familial misspecifications, such as cases of nonpaternity, with the computer programs PedCheck (v. 1.1),41 RelCheck (v. 0.67),42,43 and PREST (v. 3.0).44 After measures were taken to correct for familial misspecifications, any genotypes producing Mendelian errors were deleted, and the error rate was computed as the percentage of nonmissing alleles that were removed. We then tested the markers for Hardy-Weinberg equilibrium (HWE), using a χ2 statistic as implemented in the program PedStats (v. 0.4.6).45 One marker, D21S1256, had a significance level of P<.0001 in the HWE test and an error rate of 38% (the median rate was 0.3%) and was dropped from the analysis.
Determination of the Trait in the Australian Sample
To minimize the effects of ascertainment in the genotyped Australian NAG sample, we used the community-based BigSib sample to determine how to correct for skewness, adjust for outliers, and correct for covariates in the genetic analysis of MaxCigs24. The distribution of MaxCigs24 in both the BigSib and Australian NAG samples is shown in figure 1A.
Figure 1. .

Distribution of the MaxCigs24 in the Australian BigSib (top) (N=3,059) and NAG (middle) (N=1,098) samples and in the Finnish NAG sample (bottom) (N=449). After 60 cigarettes, the bars represents bins of 5 cigarettes (e.g., the bar labeled “100” is for MaxCigs >95).
We now describe how the trait was determined in the BigSib sample. To reduce skewness in the distribution of MaxCigs24, we considered the transformations log(x), log(x+1), and 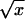 . The square-root transformation was clearly the most effective. It reduced the skewness from 0.97 to 0.03 and the kurtosis from 1.60 to 0.08. We then corrected the square root of MaxCigs24 for covariates with a linear regression by use of SAS. To determine the best model, the F statistic was used to measure the overall fit by testing the null hypothesis that all coefficients in the linear regression except the intercept are 0, and a t test was used to test the significance of individual terms in the linear regression by testing the null hypothesis that the coefficient of the term is 0. Each of these tests was implemented in SAS with use of the REG procedure. The covariates considered were sex, age, sex×age, and age2. Although sex×age and age2 were both significant covariates, the sex and age model gave the best fit. After the regression was performed, the distribution of the residual was examined to identify outliers. The original values of MaxCigs24 corresponding to residuals >3 SDs from the mean were rounded down to 80/d for the upper end of the distribution and rounded up to 5/d for the lower end of the distribution. The linear regression was repeated to achieve a better fit, and the final trait, which we denote as “CMaxCigs24,” was defined as the residual between the measurement and its predicted value.
. The square-root transformation was clearly the most effective. It reduced the skewness from 0.97 to 0.03 and the kurtosis from 1.60 to 0.08. We then corrected the square root of MaxCigs24 for covariates with a linear regression by use of SAS. To determine the best model, the F statistic was used to measure the overall fit by testing the null hypothesis that all coefficients in the linear regression except the intercept are 0, and a t test was used to test the significance of individual terms in the linear regression by testing the null hypothesis that the coefficient of the term is 0. Each of these tests was implemented in SAS with use of the REG procedure. The covariates considered were sex, age, sex×age, and age2. Although sex×age and age2 were both significant covariates, the sex and age model gave the best fit. After the regression was performed, the distribution of the residual was examined to identify outliers. The original values of MaxCigs24 corresponding to residuals >3 SDs from the mean were rounded down to 80/d for the upper end of the distribution and rounded up to 5/d for the lower end of the distribution. The linear regression was repeated to achieve a better fit, and the final trait, which we denote as “CMaxCigs24,” was defined as the residual between the measurement and its predicted value.
To determine CMaxCigs24 in the Australian NAG sample, we applied the same rule to adjust MaxCigs24 for outliers, took the square root, and used the coefficients from the linear regression determined by the BigSib sample to compute the residual in the NAG sample. The distribution of CMaxCigs24 in the BigSib and NAG samples is plotted in figure 2.
Figure 2. .

Distribution of the CMaxCigs24 trait in the Australian NAG and BigSib samples. CMaxCigs24 is the quantitative trait used for genetic linkage analysis. Shown is the distribution in the community-based Australian BigSib sample and the ascertained (and genotyped) Australian NAG sample. Also shown is the normal curve, with mean and variance estimated from the BigSib sample. As should be expected, the distribution in the ascertained sample is shifted to the right.
Determination of the Trait in the Finnish Sample
Since we did not have access to a community-based Finnish sample and since population estimates of mean and variance of MaxCigs24 in the Finnish population (see fig. 1B for the distribution) were not available, the determination of the quantitative trait in the Finnish NAG sample was based solely on the sample itself (i.e., 2,265 Finnish diagnostic telephone interviews). Otherwise, the methodology was analogous to that of the Australian sample.
Determination of the Trait in the Combined Australian and Finnish Sample
We defined the quantitative trait in the combined Australian and Finnish NAG sample by converting CMaxCigs to Z scores in each sample individually and by analyzing the combined sample using a mean of 0 and a variance of 1. The estimates of mean and variance in the Australian NAG sample were taken from the BigSib sample.
Linkage Analysis
We tested CMaxCigs24 for genetic linkage with a method introduced by Sham et al.,46 which is a derivative of the classic regression-based technique of Haseman and Elston.47 We performed a linear regression of the number of alleles identical by descent (IBD) for a relative pair on the square of the sum and the square of the difference of the trait values. This method is implemented by the computer program Merlin-Regress,46 a component of the Merlin (v. 0.10.2) software suite,48 and computes both single-point and multipoint LOD scores. Estimates of IBD were computed internally by the program Merlin-Regress. To assess the amount of genetic information provided by the data, we computed an entropy-based measure as implemented by the program Merlin, which provides this measure on a multipoint basis so that it can be plotted simultaneously with the LOD scores.
This regression method allows for the specification of three parameters of the quantitative trait: mean, variance, and heritability. In light of our own estimates of heritability (see the “Clinical Assessment” section), we used the default heritability of 50% in Merlin-Regress. For the analysis of the Australian NAG sample, we used the estimates of mean and variance from the Australian BigSib sample; for the Finnish NAG sample, we used the estimates from the sample itself.
In the combined Australian and Finnish NAG sample, allele assignments and frequencies were calculated separately for each of the genotyped samples and therefore sum to 2 rather than 1. Normally, Merlin-Regress forces allele frequencies to sum to 1. We therefore made a slight modification to the program’s source code to circumvent this issue (G. Abecasis, personal communication). Because of the large number of alleles observed, it was also necessary to compile the program with the special 64-bit integer option USE_LONG_INT.
Simulations and Empirical P Values
Deviations from normality, such as skewness and outliers, are a concern when the regression method described in the “Linkage Analysis” section is used, and it is prudent to perform simulations to determine empirical P values.46 Merlin,48 was used to generate simulated genotype data that preserve the original family structure, phenotypes, and allele frequencies. To run simulations on the combined sample, it was necessary to first generate simulated data sets for the Australian and Finnish samples individually. Once the simulated data were generated for each of the two samples, the two simulated data sets were combined using the method described in the “Linkage Analysis” section. We generated 1,000 simulated samples of combined Australian and Finnish genotype data. The samples were each tested for linkage with use of the same regression method implemented by Merlin-Regress, with use of the same trait and trait parameters as for the analysis of the real genotype data. The empirical P value 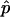 for a LOD score L was calculated by counting the number of times, r, the LOD score exceeded L in the simulated data and by applying the formula
for a LOD score L was calculated by counting the number of times, r, the LOD score exceeded L in the simulated data and by applying the formula 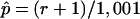 .49,50
.49,50
Results
The maximum multipoint LOD score in the Australian NAG sample was 3.05 on chromosome 22 at 26.96 cM (table 2 and fig. 3). No other chromosome showed a peak LOD score >2 for this phenotype in the Australian sample. In the Finnish NAG sample, the maximum multipoint LOD score for MaxCigs24 was 4.22 on chromosome 20 at 72.90 cM. There was also a LOD score of 3.23 on chromosome 22 at 28.96 cM and a LOD score of 1.78 on chromosome 12 at 46.00 cM. In the combined sample, the maximum LOD score was 5.98 on chromosome 22 at 26.96 cM. There was another peak on chromosome 22 with a LOD score of 2.05 at 44.96 cM, and the LOD score for the peak on chromosome 20 at 72.90 cM from the Finnish sample dropped to 2.82 in the combined sample. The linkage peak on chromosome 22 at 26.96 cM appears to be driven mainly by marker D22S315 (22.59 cM), which had a single-point LOD score of 5.41 in the combined sample.
Table 2. .
Multipoint Linkage Results from the Analysis of CMaxCigs24[Note]
| LOD Score by Sample |
||||
| Chromosome, and Positiona (cM) |
Flanking Marker(s) | Australian | Finnish | Combined |
| Original genomewide screen: | ||||
| 1: | ||||
| 48.00 | D1S234 and D1S255 | .99 | .16 | 1.08 |
| 68.00 | D1S255 and D1S2797 | 1.49 | .40 | 1.84 |
| 3: | ||||
| 102.89 | D3S1566 and D3S3681 | −.01 | 1.87 | .37 |
| 5: | ||||
| 125.19 | D5S471 and D5S2115 | 1.25 | −.03 | .62 |
| 7: | ||||
| 115.51 | D7S515 and D7S486 | 1.27 | .07 | 1.08 |
| 145.51 | D7S640 and D7S684 | 1.53 | .70 | 2.23 |
| 12: | ||||
| 32.00 | D12S364 and D12S310 | 1.05 | .46 | 1.51 |
| 46.00 | D12S1617 and D12S345 | .00 | 1.79 | .63 |
| 74.00 | D12S5368 and D12S83 | 1.23 | .20 | 1.32 |
| 13: | ||||
| 123.85 | D13S285 | 1.16 | −.36 | .32 |
| 17: | ||||
| 30.63 | D17S1852 and D17S799 | .01 | 1.39 | .62 |
| 20: | ||||
| 44.90 | D20S112 and D20S195 | .81 | 1.53 | 2.08 |
| 72.90 | D20S119 and D20S178 | .49 | 4.22 | 2.82 |
| 22: | ||||
| 26.96 | D22S315 and D22S280 | 3.05 | 3.16 | 5.98 |
| 44.96 | D22S283 and D22S423 | 1.79 | .36 | 2.05 |
| After localization of linkage peak on chromosome 22: | ||||
| 22: | ||||
| 25.46 | D22S315 and D22S1144 | 2.48 | 3.03 | 5.21b |
Note.— LOD scores >3 are shown in bold.
Chromosomal locations where the LOD score of a multipoint linkage peak was >1 in at least one of the three samples: Australian, Finnish, and combined. The chromosomal positions are determined by the largest of the three LOD scores and are based on the deCODE genetic map.32 For example, the table shows the result on chromosome 22 at 26.96 cM, because this is the location of the peak in the combined sample, whereas the Finnish sample actually peaks at 28.96 cM, with a LOD of 3.23.
Empirical P value of .006 from 1,000 genomewide multipoint linkage simulations.
Figure 3. .

Maximum multipoint LOD scores for each chromosome from the genomewide linkage screen of CMaxCigs24.
Additional Genotyping
Because the linkage peak on chromosome 22 lies between markers D22S315 and D22S280, which are separated by a gap of 17 cM, two additional markers flanking D22S315—namely, D22S1174 and D22S1144—were genotyped in an effort to localize the linkage signal. The resulting linkage peak in the combined sample shifted toward D22S315 (22.59 cM), from a LOD score of 5.98 at 26.96 cM to a LOD score of 5.21 at 25.46 cM (fig. 4). The multipoint LOD score in the Australian sample peaked at 23.96 cM, with a LOD score of 2.54, and, for the Finnish sample, the peak was at 26.46 cM, with a LOD score of 3.09.
Figure 4. .

The linkage peak on chromosome 22. Shown are the multipoint LOD scores for CMaxCigs24 after two markers (shown in boldface) flanking D22S315 were added to the original genomewide screening set.
Simulations
We analyzed 1,000 simulated combined Australian and Finnish samples for MaxCigs24, using genotype data that included both the original screening set of 381 markers and the two additional markers on chromosome 22. There were five multipoint LOD scores from the simulated data >5.21, which ranged from 5.28 to 6.53. Hence, the empirical P value for the multipoint LOD score of 5.21 found on chromosome 22 in the real data is .006. There were no single-point LOD scores from simulated data >5.41, which implies that the empirical P value for the single-point LOD score of 5.41 at marker D22S315 in the real data is <.001.
Discussion
We tested MaxCigs24 for genetic linkage, using a genomewide approach in two independent samples. The most compelling evidence of linkage was on chromosome 22 at 27–29 cM, where the multipoint LOD scores in the Australian and Finnish samples were 3.05 at 26.96 cM and 3.23 at 28.96 cM, respectively (table 2 and fig. 3). This overlap is particularly noteworthy, given that, whereas coordinated across sites, the data collection, genotyping, and cleaning occurred at different sites for each sample. The combined Australian and Finnish sample produced a multipoint LOD score of 5.98 at 26.96 cM that appears to be driven mainly by marker D22S315 (22.59 cM), which had a single-point LOD score of 5.41. Because the multipoint linkage peak occurred between two microsatellite markers that are separated by a wide gap of 17 cM, the location of the peak is uncertain. To address this issue, we genotyped two additional microsatellite markers in both the Australian and Finnish samples, one on each side of D22S315. The result of this additional information was that the location of the linkage peak on chromosome 22 in the combined sample shifted to a LOD score of 5.21 at 25.46 cM, which is closer to D22S315 (fig. 4). The empirical P value of this LOD score from 1,000 genomewide multipoint simulations score is .006, and the empirical P value for the single-point LOD score of 5.41 is <.001. Hence, it is unlikely that these signals are due to random effects. We also see elevated single-point LOD scores for typical daily cigarette consumption (Finland only: LOD 2.59; data not shown) and for the FTND measure of nicotine dependence (combined sample: LOD 2.20; data not shown) at this location.
Two previous studies that examined other measures of tobacco use22,34 and two studies of alcohol dependence (MIM 103780)51,52 (a commonly associated phenotype) found small signals on chromosome 22, with one more proximal to our finding.51 It may be the case that refinement of the tobacco-use phenotype in addition to targeting heavy-smoking samples, as exemplified here, helped to hone this area of potential genetic risk for nicotine tolerance.
Previous analyses of Australian twin data suggest that a strong genetic factor underlies the clustering of symptoms associated with nicotine dependence and that the number of cigarettes consumed, or measures of nicotine tolerance, may be an important marker for genes contributing to risk of nicotine dependence in at least some smokers.15 There is no critical value in the distribution of MaxCigs24 that distinguishes those with nicotine dependence. However, the correlation of MaxCigs24 with other nicotine-dependence phenotypes was found to be substantial in a large community sample of Australian families (the BigSib sample). Among regular smokers (those with a history of smoking ⩾100 cigarettes) with a lifetime history of smoking <20 cigarettes in any 24-h period, <3% of male and 6% of female smokers reported a lifetime history of DSM-IV nicotine dependence. Among those with a history of smoking >40 cigarettes in any 24-h period, >86% of male and 92% of female cigarette smokers reported a lifetime history of dependence. Whereas dichotomous phenotypes are clinically important, MaxCigs24 has the advantage of quantitatively grading the severity of cigarette use and making use of the full range of individual differences in cigarette use and nicotine tolerance among regular smokers. In addition, the number of cigarettes smoked in any 24-h period may be less affected by the increasing number of restraints on the times and places where cigarette smoking is permitted, such as smoking restrictions in restaurants and workplaces, which may reduce the informativeness of reports of typical daily cigarette consumption, a measure that has been used in other linkage studies of smoking behavior.
At the time of analysis, the Finnish NAG sample was genotyped for a subset of 362 markers from the full 10-cM screening set used for the Australian NAG sample. The omitted markers were distributed randomly throughout the genome, which resulted in an occasional gap in coverage. Large gaps between markers are a concern, because they can artificially inflate LOD scores and lead to false-positive results. The only LOD score >1 in the Finnish sample that was near a gap in the Finnish marker coverage was on chromosome 20 at 72.90 cM, where the LOD score was 4.22 (table 2). The closest markers to this peak that were genotyped for the Finnish sample were D20S119 (69.40 cM) and D20S100 (88.96 cM). However, the peak is very close to marker D20S119, so inflation of this LOD score is not as much of a concern as it would be if the peak were located near the midpoint of the flanking markers. Additional markers should be genotyped to localize this linkage signal. In the simulated linkage analyses, inflation of the LOD scores would make the empirical P values larger and, if anything, cause us to underestimate the significance of our results.
The genotyped Australian and Finnish NAG samples were ascertained through an affected sibling pair concordant for smoking history. This ascertainment scheme may influence the results of our quantitative linkage analysis. We controlled for the effects of ascertainment in the Australian sample with the BigSib sample, a large collection of Australian families chosen simply for large sibship size, which was used to make estimates of the mean and variance of the distribution of MaxCigs24. Since no community-based Finnish sample was available, the corresponding estimates of these parameters for the Finnish analysis were sample based. It was shown by Sham et al.46 that the misspecification of mean and variance will only weaken the power to detect linkage and will not inflate the type I error rate. The only case of model misspecification that Sham et al.46 found that inflated the type I error rate was when the heritability was severely underestimated and a diallelic marker with equal allele frequencies was used; even in this case, the inflation was very slight. Hence, our finding of linkage to chromosome 22 in the Finnish sample, which supports the evidence in the Australian sample at the same locus, is particularly impressive, given that the phenotype may suffer from power loss due to model misspecification.
By its very nature, linkage analysis is a tool that deals with a low genomic resolution, and positive results may implicate many genes. If we consider the refined peak on chromosome 22 after additional markers were genotyped (fig. 4), the 1-LOD support is the 9.50-cM interval from 20.96 cM to 29.46 cM. This corresponds to the physical region extending from 23.843 Mb to 26.020 Mb, according to build 36.2 of the human reference genome (NCBI), a 2.2-Mb region containing ∼25 genes. However, the linkage signal is driven mainly by marker D22S315 (22.59 cM) (UniSTS), which lies in an intron of the gene ADRBK2 (MIM 109636), encoding the beta-adrenergic receptor kinase 2. The ADRBK2 gene product is involved in the desensitization of multiple G-protein receptor systems, such as dopamine and corticotrophin releasing factor,53,54 and is an interesting candidate protein for moderating loci for nicotine dependence via regulating the reinforcing effects of catecholamines. In addition, an ADRBK2-knockout mouse has been shown to alter opiate tolerance55 (although there has been no examination of effects on tolerance to nicotine, to our knowledge).
We also found significant evidence of linkage to chromosome 20 in the Finnish sample, where the peak multipoint LOD score was 4.22 at 72.90 cM (table 2) (2-LOD support 64–86 cM). It is noteworthy that the α4 nicotinic receptor gene, CHRNA4 (MIM 118504), is positioned at ∼98 cM on chromosome 20 (EntrezGene), although the evidence of linkage to this gene from our analysis is weak.
The 2004 Surgeon General’s report highlights the fact that cigarette smoking continues to be the single most preventable cause of death in the United States, its impact pervading all strata of sex, age, culture, and demography.56 The moderate-to-high heritability of both nicotine dependence and heavy smoking indicates that disentangling the sources of genetic risk associated with smoking may provide substantial biological insight into this disorder. To our knowledge, this is the first article to report consistent evidence of genetic linkage with use of the same quantitative smoking phenotype assessed identically in two independent samples. Whereas the linkage region we identified encompasses many genes, the evidence appears to be strongest at ADRBK2, a plausible candidate gene for the study of nicotine dependence. Fine mapping of this region may reveal genetic variants contributing to cigarette smoking and ultimately aid our comprehension of the complex etiologic pathways involved in nicotine dependence.
Acknowledgments
The NAG project is an international collaborative study (P.A.F.M., WU, Principal Investigator) that includes three sites (with respective Principal Investigators): QIMR, Queensland (N.G.M.); UH, Helsinki (J.K.); and WU, St. Louis (P.A.F.M.). Data collection is conducted at two of these sites (QIMR and UH); WU is the coordinating site and lead institution. Genotyping and data analyses are conducted at all three sites. This study is supported by National Institutes of Health grants DA12854 (to P.A.F.M.), AA07728 (to A.C.H.), AA07580 (to A.C.H.), AA13321 (to A.C.H.), AA13320 (to R.D.T.), and DA019951 (to M.L.P.); American Cancer Society grant IRG-58-010-50 (to S.F.S.); grants from the Australian National Health and Medical Research Council; an Academy of Finland postdoctoral fellowship (to A.L.); Doctoral Programs of Public Health, UH, Finland (support to U.B.); and European Union Contract QLG2-CT-2002-01254 (to J.K. and L.P.). J.K. and L.P. are supported by the Academy of Finland Center of Excellence for Complex Disease Genetics. We thank the Australian and Finnish families, for their cooperation, and the staff from all three sites, for their many contributions.
Web Resources
The URLs for data presented herein are as follows:
- EntrezGene, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=gene
- Merlin, http://www.sph.umich.edu/csg/abecasis/Merlin/ (for Merlin and Merlin-Regress)
- NCBI, ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/Mapping_data/ and ftp://ftp.ncbi.nih.gov/genomes/H_sapiens/mapview/ (for additional data to interpolate between genetic and physical maps and to interpolate deCODE genetic map positions and physical map positions [current build, at the time of writing, of the human reference genome was 36.2])
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/ (for susceptibility to tobacco addiction, alcohol dependence, ADRBK2, and CHRNA4)
- PedCheck, http://watson.hgen.pitt.edu/register/docs/pedcheck.html
- PedStats, http://www.sph.umich.edu/csg/abecasis/PedStats/
- PREST, http://fisher.utstat.toronto.edu/sun/Software/Prest/
- RelCheck, http://biostat.jhsph.edu/~kbroman/software/#relcheck
- UniSTS, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db=unists
References
- 1.Doll R, Gray R, Hafner B, Peto R (1980) Mortality in relation to smoking: 22 years’ observations on female British doctors. Br Med J 280:967–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Doll R, Peto R (1976) Mortality in relation to smoking: 20 years’ observations on male British doctors. Br Med J 2:1525–1536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Risch HA, Howe GR, Jain M, Burch JD, Holowaty EJ, Miller AB (1993) Are female smokers at higher risk for lung cancer than male smokers? A case-control analysis by histologic type. Am J Epidemiol 138:281–293 [DOI] [PubMed] [Google Scholar]
- 4.Shafey O, Dolwick S, Guindon GE (eds) (2003) Tobacco control country profiles 2003. American Cancer Society, Atlanta [Google Scholar]
- 5.Kawachi I, Colditz GA, Stampfer MJ, Willett WC, Manson JE, Rosner B, Speizer FE, Hennekens CH (1993) Smoking cessation and decreased risk of stroke in women. JAMA 269:232–236 10.1001/jama.269.2.232 [DOI] [PubMed] [Google Scholar]
- 6.Rosenberg L, Kaufman DW, Helmrich SP, Miller DR, Stolley PD, Shapiro S (1985) Myocardial infarction and cigarette smoking in women younger than 50 years of age. JAMA 253:2965–2969 10.1001/jama.253.20.2965 [DOI] [PubMed] [Google Scholar]
- 7.Merikangas KR, Risch N (2003) Genomic priorities and public health. Science 302:599–601 10.1126/science.1091468 [DOI] [PubMed] [Google Scholar]
- 8.Vineis P, Alavanja M, Buffler P, Fontham E, Franceschi S, Gao YT, Gupta PC, Hackshaw A, Matos E, Samet J, et al (2004) Tobacco and cancer: recent epidemiological evidence. J Natl Cancer Inst 96:99–106 [DOI] [PubMed] [Google Scholar]
- 9.Medlund P, Cederlof R, Floderus-Myrhed R, Friberg LM, Sorenson S (1976) A new Swedish twin registry. Acta Med Scand Suppl 600:1–111 [PubMed] [Google Scholar]
- 10.Kaprio J, Koskenvuo M (1988) A prospective study of psychological and socioeconomic characteristics, health behavior and morbidity in cigarette smokers prior to quitting compared to persistent smokers and non-smokers. J Clin Epidemiol 41:139–150 10.1016/0895-4356(88)90088-1 [DOI] [PubMed] [Google Scholar]
- 11.Heath AC, Martin NG (1993) Genetic models for the natural history of smoking: evidence for a genetic influence on smoking persistence. Addict Behav 18:19–34 10.1016/0306-4603(93)90005-T [DOI] [PubMed] [Google Scholar]
- 12.Madden PA, Heath AC, Pedersen NL, Kaprio J, Koskenvuo MJ, Martin NG (1999) The genetics of smoking persistence in men and women: a multicultural study. Behav Genet 29:423–431 10.1023/A:1021674804714 [DOI] [PubMed] [Google Scholar]
- 13.Heath AC, Madden PAF (1995) Genetic influences on smoking behavior. In: Turner JR, Cardon LR, Hewitt JK (eds) Behavior genetic applications in behavioral medicine research. Plenum, New York, pp 45–66 [Google Scholar]
- 14.True WR, Heath AC, Scherrer JF, Waterman B, Goldberg J, Lin N, Eisen SA, Lyons MJ, Tsuang MT (1997) Genetic and environmental contributions to smoking. Addiction 92:1277–1287 10.1111/j.1360-0443.1997.tb02847.x [DOI] [PubMed] [Google Scholar]
- 15.Lessov CN, Martin NG, Statham DJ, Todorov AA, Slutske WS, Bucholz KK, Heath AC, Madden PA (2004) Defining nicotine dependence for genetic research: evidence from Australian twins. Psychol Med 34:865–879 10.1017/S0033291703001582 [DOI] [PubMed] [Google Scholar]
- 16.American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders, 4th ed (DSM-IV). American Psychiatric Press, Washington, DC [Google Scholar]
- 17.Heatherton HF, Kozlowski LT, Frecker RC, Fagerström KO (1991) The Fagerström Test for nicotine dependence: a revision of the Fagerström Tolerance Questionnaire. Br J Addict 9:1119–1127 10.1111/j.1360-0443.1991.tb01879.x [DOI] [PubMed] [Google Scholar]
- 18.Straub RE, Sullivan PF, Ma Y, Myakishev MV, Harris-Kerr C, Wormley B, Kadambi B, Sadek H, Silverman MA, Webb BT, et al (1999) Susceptibility genes for nicotine dependence: a genome scan and followup in an independent sample suggest that regions on chromosomes 2, 4, 10, 16, 17 and 18 merit further study. Mol Psychiatry 4:129–144 10.1038/sj.mp.4000518 [DOI] [PubMed] [Google Scholar]
- 19.Swan GE, Hops H, Wilhelmsen KC, Lessov-Schlaggar CN, Cheng LS, Hudmon KS, Amos CI, Feiler HS, Ring HZ, Andrews JA, et al (2006) A genome-wide screen for nicotine dependence susceptibility loci. Am J Med Genet B Neuropsychiatr Genet 141:354–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li MD, Payne TJ, Ma JZ, Lou X-Y, Zhang D, Dupont RT, Crews KM, Somes G, Williams NJ, Elston RC (2006) A genomewide search finds major susceptibility loci for nicotine dependence on chromosome 10 in African Americans. Am J Hum Genet 79:745–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gelernter J, Panhuysen C, Weiss R, Brady K, Poling J, Krauthammer M, Farrer L, Kranzler HR (2007) Genomewide linkage scan for nicotine dependence: identification of a chromosome 5 risk locus. Biol Psychiatry 61:119–126 10.1016/j.biopsych.2006.08.023 [DOI] [PubMed] [Google Scholar]
- 22.Saccone NL, Neuman RJ, Saccone SF, Rice JP (2003) Genetic analysis of maximum cigarette-use phenotypes. BMC Genet Suppl 4:S105 10.1186/1471-2156-4-S1-S105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goode EL, Badzioch MD, Kim H, Gagnon F, Rozek LS, Edwards KL, Jarvik GP, Framingham Heart Study (2003) Multiple genome-wide analyses of smoking behavior in the Framingham Heart Study. BMC Genet Suppl 4:S102 10.1186/1471-2156-4-S1-S102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li MD, Ma JZ, Cheng R, Dupont RT, Williams NJ, Crews KM, Payne TJ, Elston RC, Framingham Heart Study (2003) A genome-wide scan to identify loci for smoking rate in the Framingham Heart Study population. BMC Genet Suppl 4:S103 10.1186/1471-2156-4-S1-S103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vink JM, Beem AL, Posthuma D, Neale MC, Willemsen G, Kendler KS, Slagboom PE, Boomsma DI (2004) Linkage analysis of smoking initiation and quantity in Dutch sibling pairs. Pharmacogenomics J 4:274–282 (erratum 4:345–346) 10.1038/sj.tpj.6500255 [DOI] [PubMed] [Google Scholar]
- 26.Wang D, Ma JZ, Li MD (2005) Mapping and verification of susceptibility loci for smoking quantity using permutation linkage analysis. Pharmacogenomics J 5:166–172 10.1038/sj.tpj.6500304 [DOI] [PubMed] [Google Scholar]
- 27.Morley KI, Medland SE, Ferreira MA, Lynskey MT, Montgomery GW, Heath AC, Madden PA, Martin NG (2006) A possible smoking susceptibility locus on chromosome 11p12: evidence from sex-limitation linkage analyses in a sample of Australian twin families. Behav Genet 36:87–99 10.1007/s10519-005-9004-0 [DOI] [PubMed] [Google Scholar]
- 28.Farrelly MC, Evans WN, Sfekas AE (1999) The impact of workplace smoking bans: results from a national survey. Tob Control 8:272–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilpin EA, Pierce JP (2002) The California Tobacco Control Program and potential harm reduction through reduced cigarette consumption in continuing smokers. Nicotine Tob Res Suppl 2 4:S157–S166 10.1080/1462220021000032708 [DOI] [PubMed] [Google Scholar]
- 30.Bergen AW, Korczak JF, Weissbecker KA, Goldstein AM (1999) A genome-wide search for loci contributing to smoking and alcoholism. Genet Epidemiol Suppl 17:S55–S60 [DOI] [PubMed] [Google Scholar]
- 31.Duggirala R, Almasy L, Blangero J (1999) Smoking behavior is under the influence of a major quantitative trait locus on human chromosome 5q. Genet Epidemiol Suppl 17:S139–S144 [DOI] [PubMed] [Google Scholar]
- 32.Bierut LJ, Rice JP, Goate A, Hinrichs AL, Saccone NL, Foroud T, Edenberg HJ, Cloninger CR, Begleiter H, Conneally PM, et al (2004) A genomic scan for habitual smoking in families of alcoholics: common and specific genetic factors in substance dependence. Am J Med Genet A 124:19–27 10.1002/ajmg.a.20329 [DOI] [PubMed] [Google Scholar]
- 33.Gelernter J, Liu X, Hesselbrock V, Page GP, Goddard A, Zhang H (2004) Results of a genomewide linkage scan: support for chromosomes 9 and 11 loci increasing risk for cigarette smoking. Am J Med Genet B Neuropsychiatr Genet 128:94–101 10.1002/ajmg.b.30019 [DOI] [PubMed] [Google Scholar]
- 34.Ehlers CL, Wilhelmsen KC (2006) Genomic screen for loci associated with tobacco usage in Mission Indians. BMC Med Genet 7:9 10.1186/1471-2350-7-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bucholz KK, Cadoret R, Cloninger CR, Dinwiddie SH, Hesselbrock VM, Nurnberger JI Jr, Reich T, Schmidt I, Schuckit MA (1994) A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. J Stud Alcohol 55:149–158 [DOI] [PubMed] [Google Scholar]
- 36.Hesselbrock M, Easton C, Bucholz KK, Schuckit M, Hesselbrock V (1999) A validity study of the SSAGA—a comparison with the SCAN. Addiction 94:1361–1370 10.1046/j.1360-0443.1999.94913618.x [DOI] [PubMed] [Google Scholar]
- 37.Cottler LB, Robins LN, Grant BF, Blaine J, Towle LH, Witthen HU, Sartorius N (1991) The CIDI-core substance abuse and dependence questions: cross-cultural and nosological issues: the WHO/ADAMHA field trial. Br J Psychiatry 159:653–658 [DOI] [PubMed] [Google Scholar]
- 38.Costa PT Jr, McCrae RR (1992) Revised NEO Personality Inventory (NEO_PI_R0 and NEO Five Factor Inventory (NEO FFI)) professional manual. Psychological Assessment Resources, Odessa, FL [Google Scholar]
- 39.Shiffman S, Waters A, Hickcox M (2004) The nicotine dependence syndrome scale: a multidimensional measure of nicotine dependence. Nicotine Tob Res 6:327–348 10.1080/1462220042000202481 [DOI] [PubMed] [Google Scholar]
- 40.Kong A, Gudbjartsson DF, Sainz J, Jonsdottir GM, Gudjonsson SA, Richardsson B, Sigurdardottir S, Barnard J, Hallbeck B, Masson G, et al (2002) A high-resolution recombination map of the human genome. Nat Genet 31:241–247 [DOI] [PubMed] [Google Scholar]
- 41.O’Connell JR, Weeks DE (1998) PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63:259–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boehnke M, Cox NJ (1997) Accurate inference of relationships in sib-pair linkage studies. Am J Hum Genet 61:423–429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Broman KW, Weber JL (1998) Estimation of pairwise relationships in the presence of genotyping errors. Am J Hum Genet 63:1563–1564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McPeek MS, Sun L (2000) Statistical tests for detection of misspecified relationships by use of genome-screen data. Am J Hum Genet 66:1076–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wigginton JE, Abecasis GR (2005) PEDSTATS: descriptive statistics, graphics and quality assessment for gene mapping data. Bioinformatics 21:3445–3447 10.1093/bioinformatics/bti529 [DOI] [PubMed] [Google Scholar]
- 46.Sham PC, Purcell S, Cherny SS, Abecasis GR (2002) Powerful regression-based quantitative-trait linkage analysis of general pedigrees. Am J Hum Genet 71:238–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haseman JK, Elston RC (1972) The investigation of linkage between a quantitative trait and a marker locus. Behav Genet 2:3–19 10.1007/BF01066731 [DOI] [PubMed] [Google Scholar]
- 48.Abecasis GR, Cherny SS, Cookson WO, Cardon LR (2002) Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 30:97–101 10.1038/ng786 [DOI] [PubMed] [Google Scholar]
- 49.North BV, Curtis D, Sham PC (2002) A note on the calculation of empirical P values from Monte Carlo procedures. Am J Hum Genet 71:439–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.North BV, Curtis D, Sham PC (2003) A note on calculation of empirical P values from Monte Carlo procedure. Am J Hum Genet 72:498–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bergen AW, Yang XR, Bai Y, Beerman MB, Goldstein AM, Goldin LR, Framingham Heart Study (2003) Genomic regions linked to alcohol consumption in the Framingham Heart Study. BMC Genet Suppl 4:S101 10.1186/1471-2156-4-S1-S101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foroud T, Edenberg HJ, Goate A, Rice J, Flury L, Koller DL, Bierut LJ, Conneally PM, Nurnberger JI, Bucholz KK, et al (2000) Alcoholism susceptibility loci: confirmation studies in a replicate sample and further mapping. Alcohol Clin Exp Res 24:933–945 10.1111/j.1530-0277.2000.tb04634.x [DOI] [PubMed] [Google Scholar]
- 53.Carmen CV, Benovic JL (1998) G-protein-coupled receptors: turn-ons and turn-offs. Curr Opin Neurobiol 8:335–344 10.1016/S0959-4388(98)80058-5 [DOI] [PubMed] [Google Scholar]
- 54.Teli T, Markovic D, Levine MA, Hillhouse EW, Grammatopoulos DK (2005) Regulation of corticotrophin-releasing hormone receptor type 1α signaling: structural determinants for G protein-coupled receptor kinase-mediated phosphorylation and agonist-mediated desensitization. Mol Endocrinol 19:474–490 10.1210/me.2004-0275 [DOI] [PubMed] [Google Scholar]
- 55.Terman GW, Jin W, Chong Y-P, Lowe J, Caron MG, Lefkowitz RJ, Chavkin C (2004) G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br J Pharmacol 141:55–64 10.1038/sj.bjp.0705595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Centers for Disease Control (2004) 2004 Surgeon General's report: the health consequences of smoking (http://www.cdc.gov/tobacco/data_statistics/sgr/sgr_2004/00_pdfs/chapter1.pdf) (accessed March 23, 2007) [PubMed]