

Abstract
It has been proposed that cell growth and autophagy are coordinated in response to cellular nutrient status, but the relationship between them is not fully understood. Here, we have characterized the fly mutants of Autophagy-specific gene 1 (ATG1), an autophagy-regulating kinase, and found that ATG1 is a negative regulator of the target of rapamycin (TOR)/S6 kinase (S6K) pathway. Our Drosophila studies have shown that ATG1 inhibits TOR/S6K-dependent cell growth and development by interfering with S6K activation. Consistently, overexpression of ATG1 in mammalian cells also markedly inhibits S6K in a kinase activity-dependent manner, and short interfering RNA-mediated knockdown of ATG1 induces ectopic activation of S6K and S6 phosphorylation. Moreover, we demonstrated that ATG1 specifically inhibits S6K activity by blocking phosphorylation of S6K at Thr 389. Taken together, our genetic and biochemical results strongly indicate crosstalk between autophagy and cell growth regulation.
Keywords: autophagy, cell growth, S6K, TOR
Introduction
The nutritional environment is a crucial determinant of important cellular decisions, such as growth, proliferation and development. Recently, a series of outstanding studies have demonstrated that nutrient availability tightly regulates cell growth through an evolutionarily highly conserved signalling pathway, the target of rapamycin (TOR)/p70 S6 kinase (S6K) pathway (reviewed in Hay & Sonenberg, 2004). S6K was originally discovered as a kinase that phosphorylates 40S ribosomal protein S6 at many sites and its activity has been considered as a characteristic of cell growth (Kozma & Thomas, 1994). Studies on the action mechanism of immunosuppressant rapamycin led to a surprising discovery that TOR is the upstream activator of S6K in vivo (Chung et al, 1992; Brown et al, 1994; Sabatini et al, 1994). Recently, studies using mammalian cell lines (Fingar et al, 2002) and knockout mice (Shima et al, 1998) clearly showed that the TOR/S6K signalling pathway controls cell growth in vertebrates. Consistently, Drosophila S6K (dS6K) and Drosophila TOR (dTOR) mutants also showed reduced cell and body size compared with the wild-type (w1118) fly (Montagne et al, 1999; Oldham et al, 2000; Zhang et al, 2000).
TOR is also involved in the regulation of autophagy. Autophagy is a process conserved among all eukaryotic cells and is required for rapid degradation of large portions of the cytoplasm and organelles in the lysosomal lumen, occurring as a result of nutrient deprivation or normal developmental processes (Levine & Klionsky, 2004). Under nutrient-rich conditions, TOR blocks the initiation step of autophagy by facilitating dissociation of Autophagy-specific gene (Atg) 13 from Atg1, an essential factor required for the formation of an autophagic vesicle (autophagosome) in budding yeast (Kamada et al, 2000). Furthermore, the crucial roles of TOR and ATG1 in starvation- and development-dependent autophagy have been discovered in the fat body cells of Drosophila (Scott et al, 2004).
As cell growth and autophagy are opposite biological processes both regulated by TOR, we hypothesized that autophagy might be responsible for inhibiting cell growth under conditions of suppressed TOR signalling, such as starvation. To investigate the relationship between autophagy and cell growth regulation, we examined the interactions between the representative components of the two pathways, TOR/S6K and ATG1. Using biochemical and genetic approaches, we demonstrated that ATG1 negatively regulates S6K and, consequently, inhibits cell growth in both mammals and Drosophila.
Results And Discussion
Drosophila ATG1 and characterization of its mutants
The Drosophila CG10967 gene, which encodes a sole Drosophila orthologue of ATG1, is known as DmATG1 (supplementary Figs S1,S2A online). Previous studies demonstrated that homozygous EP3348 flies, in which a single P element is inserted into the 5′ untranslated region of DmATG1 (Fig 1A), show larval/pupal lethality (Spradling et al, 1999; Scott et al, 2004). However, after we cleared the genomic background of the EP3348 line by four backcrosses with w1118 flies, about 30% of homozygous mutants were found to develop to adults (Fig 2A). To confirm that the P-element insertion in EP3348 hampers transcription of DmATG1 (Scott et al, 2004), we carried out quantitative real-time reverse transcriptase–PCR (qRT–PCR), which showed highly reduced DmATG1 expression in the mutant (Fig 1B). Therefore, we named this cleared EP3348 allele as DmATG11.
Figure 1.
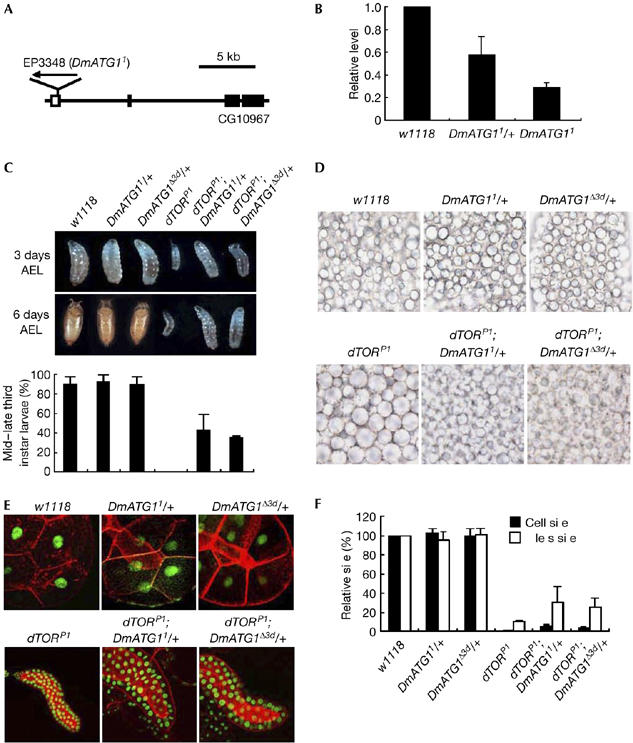
Involvement of Drosophila Autophagy-specific gene 1 in Drosophila Target of rapamycin-dependent cell growth and development. (A) Genomic structure of CG10967. The P-element insertion site of EP3348 (DmATG11) is denoted. (B) The transcriptional levels of DmATG1 in the third instar larvae were analysed by qRT–PCR. Ribosomal protein 49 (rp49) was used as an internal control; n=3. Bars indicate mean±s.d. (C) Images of the larvae of denoted genotypes at 3 days (top) and 6 days (middle) after egg-laying (AEL), and quantification of the larvae that developed into the mid–late third instar stage of each genotype (bottom); n=3. Bars indicate mean±s.d. Fifteen larvae of each genotype were examined in each experiment. (D) Suppression of lipid vesicle aggregation in the fat body of dTOR mutants by reduced gene dosage of DmATG1. The fat body images of the second/early third instar larvae of denoted genotypes under fully fed conditions. (E) Images of the larval salivary glands of denoted genotypes. Hoechst33342 (pseudo-coloured green) and phalloidin-TRITC (red) were used to visualize the nuclei and cell boundary of larval salivary gland cells, respectively. (F) Quantitative analysis of cell and nuclear sizes in Fig 1E; n=5. Bars indicate mean±s.d. DmATG1, Drosophila Autophagy-specific gene 1; dTOR, Drosophila TOR; qRT–PCR, quantitative real-time reverse transcriptase–PCR.
Figure 2.

Functional interaction between Drosophila Autophagy-specific gene 1 and Drosophila S6 kinase. (A) Quantification of adults that survived after eclosion for denoted genotypes; n=3. Bars indicate mean±s.d. Fifteen larvae of each genotype were examined in each experiment. (B,C) Activation of dS6K in DmATG1 mutants. Phosphorylation of dS6K was examined by immunoblot analyses using phosphospecific dS6K T398 antibody in the third instar larvae or pupae of denoted genotypes. Tubulin immunoblot was used as a loading control. Rheb expressing flies (hs>Rheb) were used as a positive control (C). hs>Rheb larvae were subjected to heat shock at 37°C for 1 h and then incubated at 30°C for 3 h before sample preparation to induce Rheb expression. For quantification, the levels of dS6K phosphorylation in each genotype were measured using Adobe Photoshop, and normalized to the tubulin levels. Results are expressed as a fold change compared with the wild-type controls (w1118). DmATG1, Drosophila Autophagy-specific gene 1; dS6k, Drosophila S6 kinase.
As Bettencourt-Dias et al (2004) have showed previously that CG10967 RNA intereference (RNAi) can induce mitotic abnormalities in Drosophila S2 cells, we first examined the mitotic phenotypes in the larval brain and imaginal discs of DmATG1 mutants, DmATG11 and DmATG1Δ3d (a null allele of DmATG1; Scott et al, 2004). However, we could not find any notable mitotic defects in the mutants compared with the wild-type control (w1118, supplementary Fig S3A,B online). Consistently, DmATG1 mutants had no gross chromosomal abnormalities in the neuroblast cells of third instar larval brains (supplementary Fig S3C online).
Next, we looked for any defects in autophagy in DmATG11 flies using toluidine blue-azure II staining and transmission electron microscopy (TEM) analyses. As expected, DmATG11 flies showed marked defects in the induction of autophagy under conditions of starvation (supplementary Fig S2B–D,F–H online), which is highly consistent with the previous study using DmATG1Δ3d (Scott et al, 2004). These results strongly supported that DmATG11 is a new hypomorphic mutant allele of DmATG1.
Functional interaction between ATG1 and TOR
To understand further the in vivo roles of ATG1 in Drosophila, we generated double-mutant lines for dTORP1 (a loss-of-function mutant allele of dTOR; Zhang et al, 2000) and DmATG1 mutants (DmATG11, DmATG1Δ3d and EP3348). As reported previously (Zhang et al, 2000), homozygous dTORP1 mutants showed growth arrest in the second/early third instar larval stage and markedly delayed growth. Surprisingly, homozygous dTORP1 larvae with a heterozygous genetic background of DmATG11 or DmATG1Δ3d not only grew faster than homozygous dTORP1, but also extended their developmental stage to the mid–late third instar larval stage (Fig 1C). Furthermore, the double-homozygous mutants between dTORP1 and various DmATG1 mutants survived up to the mid–late third instar larval stage (data not shown), which is inconsistent with the previous results (Scott et al, 2004).
In addition, we found that another phenotype of dTORP1 mutants, lipid vesicle aggregation in the fat body (Colombani et al, 2003), was also suppressed by a reduction of the gene dosage of DmATG1 (Fig 1D). These results implicated that ATG1 negatively mediates the developmental and physiological roles of TOR in Drosophila.
As dTOR regulates cell growth in a cell-autonomous manner (Oldham et al, 2000; Zhang et al, 2000), we decided to examine whether DmATG1 is also involved in this role of dTOR. Consistent with the previous report (Zhang et al, 2000), the cell and nuclear sizes of the salivary gland cells were markedly reduced in dTORP1 larvae (Fig 1E,F). Intriguingly, the heterozygous genetic background of DmATG11 or DmATG1Δ3d partly rescued the reduced cell and nuclear size phenotype of dTORP1 (Fig 1E,F), strongly implicating that DmATG1 mediates the crucial function of dTOR in cell growth regulation. This is further supported by recent results (Scott et al, 2007), that DmATG1-null cells have a relative growth advantage over wild-type cells when treated with rapamycin, a specific inhibitor of dTOR.
To examine the possibility that the suppression of dTORP1 phenotypes by DmATG1 mutation resulted from altered auto-phagic activities, we investigated the genetic interactions between dTOR and other autophagy-related genes, such as ATG6 (Beclin1) and UVRAG (UV radiation resistance associated gene), which are known to form a complex regulating autophagosome formation (Liang et al, 2006; supplementary Fig S4 online). As a result, ATG6 and UVRAG mutations did not suppress the developmental delay and cell growth defects of dTOR mutants (supplementary Fig S5 online), showing that the interaction between dTOR and DmATG1 is not caused indirectly by uncontrolled regulation of autophagy.
Functional interaction between ATG1 and S6K
To determine the functional interaction between ATG1 and S6K, a downstream effector of TOR, we generated double-mutant lines between DmATG11 and dS6K mutants—a hypomorphic allele, dS6K07084, and a null allele, dS6Kl−1 (Montagne et al, 1999). Using TEM analyses, we observed that a reduction of dS6K gene dosage did not rescue the defects in autophagosome formation in starved DmATG11 homozygous larvae (supplementary Fig S2E,I online). However, surprisingly, the reduced gene dosage of dS6K increased the eclosion rate of homozygous DmATG11 in a dS6K gene dosage-dependent manner (Fig 2A). Although we cannot completely exclude the possibility that S6K promotes autophagy as reported previously (Scott et al, 2004), these data indicate that dS6K has an important role in DmATG1-dependent developmental processes.
Next, we conducted a biochemical analysis to examine the effect of loss of DmATG1 on dS6K activation. As a result, we found that dS6K was markedly activated (∼threefold increase) in homozygous DmATG11 larvae and pupae, measured by the phosphorylation of dS6K at the Thr 398 site, compared with the wild-type controls (w1118, Fig 2B). Notably, the increased level of dS6K phosphorylation in DmATG11 mutants was about one-third of that in flies overexpressing Rheb (Fig 2C). Consistent with this, DmATG1 overexpression almost completely inhibited dS6K Thr 398 phosphorylation in Drosophila (Scott et al, 2007). These results strongly suggest an important role of ATG1 in the regulation of S6K.
Inhibition of S6K activity by ATG1
To extend our findings to the mammalian system and also to investigate further the molecular mechanism of the interaction between ATG1 and TOR/S6K, we examined the effect of ATG1 on the activity of S6K in mammalian cells. There are two isoforms of ATG1 in mammals, which are named UNC-51-like kinase (ULK) 1 and 2 (Kuroyanagi et al, 1998; Yan et al, 1998, 1999). However, according to the agreement on gene nomenclature made by researchers in the field of autophagy (Klionsky et al, 2003), we renamed them ATG1α and ATG1β, respectively. Nutrient deprivation of HEK293T cells abolished the phosphorylation of S6K at both Thr 229 and Thr 389 sites, which represents the activation status of S6K (Fig 3A, lanes 1–3). When nutrients including amino acids and glucose (DMEM) were added back to the cells, the phosphorylation of both sites in S6K was strongly induced (Fig 3A, lane 4). However, co-expression of wild-type mouse ATG1α (ATG1α WT) strongly inhibited S6K activity induced by DMEM (Fig 3A, lane 5). On the contrary, a kinase-dead form of ATG1α (ATG1α KI) was not able to block the nutrient-induced activation of S6K (Fig 3A, lane 6), showing that ATG1α inhibits S6K in a kinase activity-dependent manner. Consistently, epidermal growth factor (EGF)-stimulated S6K activation was also inhibited by ATG1α (Fig 3B). Furthermore, we showed that ATG1β, another isoform of ATG1, has the same inhibitory effect on S6K phosphorylation as ATG1α (supplementary Fig S6B online). These data strongly suggest that ATG1 regulates the activities of upstream kinases or phosphatases of S6K, which affect both Thr 229 and Thr 389 phosphorylation.
Figure 3.
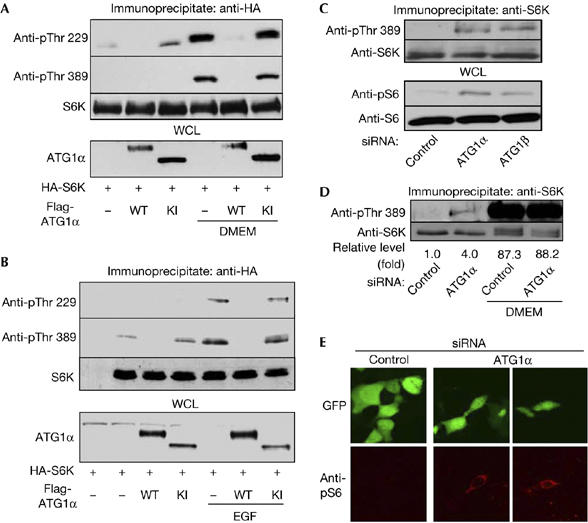
Inhibition of S6 kinase phosphorylation by Autophagy-specific gene 1. (A,B) HEK 293T cells transfected with denoted plasmids were (A) deprived of nutrients for 90 min, and then replenished with DMEM for 30 min or (B) stimulated with 50 ng/ml epidermal growth factor (EGF) for 30 min. The S6K immune complexes were analysed by immunoblot using appropriate antibodies (top three panels). Flag (ATG1α) blots were completed from the same cell lysates (bottom panel). (C) HEK 293T cells transfected with control siRNA, ATG1α siRNA or ATG1β siRNA were deprived of nutrients for 90 min, and then the phosphorylation and protein levels of endogenous S6K (top two panels) and S6 (bottom two panels) were determined. (D) HEK 293T cells transfected with control siRNA or ATG1α siRNA were deprived of nutrients for 90 min, and then replenished with DMEM for 30 min. The phosphorylation and protein levels of endogenous S6K were examined. For quantification, the levels of S6K Thr 389 phosphorylation were measured using Adobe Photoshop, and normalized to the S6K protein levels. Results are expressed as a fold change compared with the first indicated sample. (E) HEK293T cells were transfected with control siRNA or ATG1α siRNA with pEGFP-C1 (a transfection marker, green), and nutrient-starved for 90 min, fixed and stained with phosphospecific S6 antibody (red). ATG1, Autophagy-specific gene 1; HA, haemagglutinin; KI, ATG1α Lys 46 Ile (kinase-dead) mutant; siRNA, short interfering RNA; S6K, S6 kinase; WT, wild type.
As ATG1 is a crucial regulator of autophagy in yeast and Drosophila (Matsuura et al, 1997; Kamada et al, 2000; Levine & Klionsky, 2004; Scott et al, 2004), we tested whether overexpression of ATG1 can induce autophagy in mammalian cells. Nutrient deprivation was able to induce autophagy in MCF-7 cells (supplementary Fig S7A,B online), whereas overexpression of ATG1 did not induce autophagy in MCF-7 (supplementary Fig S7A,B online) and HEK 293T cells (supplementary Fig S7C online), indicating that the inhibition of TOR/S6K by ATG1 is not an indirect consequence of an ectopic induction of autophagy. This was further supported by the observation that overexpression of ATG6 and UVRAG did not inhibit the phosphorylation of S6K (supplementary Fig S7D online), which is also highly consistent with the above Drosophila data (supplementary Fig S5 online).
Then, we used short interfering RNA (siRNA) targeting for ATG1α and ATG1β messenger RNA to confirm that ATG1 inhibits S6K activity. The efficacy of siRNA was verified by qRT–PCR using ATG1-specific primers (supplementary Fig S8A,B online). Transfection of ATG1α and ATG1β siRNA to HEK 293T cells led to increased phosphorylation of S6K Thr 389 (Fig 3C, top panel) and S6 (the only proven in vivo substrate of S6K) Thr 235/236 (Fig 3C, lower middle panel). This result was further supported by immunocytochemistry by using phosphospecific S6 antibody; ATG1 siRNA transfection alone induced phosphospecific immunostaining of S6 in starved cells (Fig 3E). Taken together, these results clearly demonstrate that ATG1 inhibits S6K and S6 in vivo.
Notably, the level of S6K activation by ATG1 siRNA was about 5% of that by nutritional stimulation (Fig 3D). We think that this weak activation of S6K resulted from partial gene knockdown by RNAi (supplementary Fig S8) and genetic redundancy of ATG1 in mammals (Fig 3A; supplementary Fig S6B online). Consistent with this conclusion, we observed more pronounced activation of dS6K in DmATG1 mutants in Drosophila, which contains only a single orthologue of ATG1 (DmATG1; Fig 2B,C).
Modulation of S6K Thr 389 phosphorylation by ATG1
S6K is in the AGC kinase family, which also includes Akt and p90 ribosomal S6 kinase (RSK). These kinases are regulated by a similar mechanism in which both phosphorylation at their activation loop and a hydrophobic motif next to the kinase domain are required for full activation (Mora et al, 2004). 3-Phosphoinositide-dependent kinase 1 (PDK1) is a kinase responsible for phosphorylation at the activation loop of AGC kinases (Mora et al, 2004). In the case of S6K, PDK1 directly phosphorylates the Thr 229 residue at the activation loop of S6K, which is strictly dependent on the previous phosphorylation of Thr 389 at the hydrophobic motif (Collins et al, 2003; Mora et al, 2004). These motifs are well conserved among the family members in different species. Therefore, we examined whether ATG1α also affects the phosphorylation of Akt and RSK. Interestingly, the phosphorylation of Akt and RSK was not affected by ATG1α, with or without stimulation by insulin and EGF (Fig 4A, B, respectively). These data indicate that ATG1α specifically modulates S6K activity.
Figure 4.
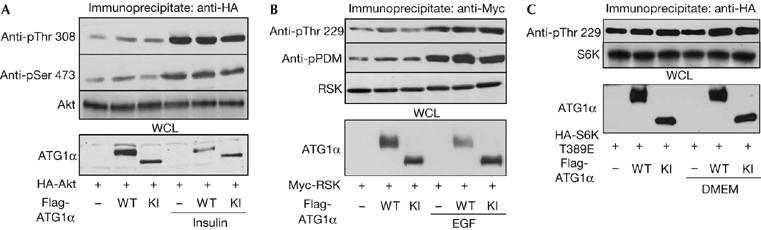
Inhibition of S6 kinase Thr 389 phosphorylation by Autophagy-specific gene 1α. (A,B) HEK 293T cells were transfected with HA–Akt (A) or Myc–RSK (B), and/or Flag–ATG1α WT or KI plasmids. The immunoprecipitated Akt and RSK proteins were analysed by immunoblot using appropriate antibodies (top three panels). Flag (ATG1α) blots were completed from the same cell lysates (bottom panel). (C) HEK 293T cells were transfected with HA–S6K Thr 389 Glu mutant and/or Flag–ATG1α WT or KI. The immunoprecipitated S6K mutants were analysed by immunoblot using anti-phosphospecific S6K Thr 229 (top panel) and HA (S6K) antibodies (middle panel). Flag (ATG1α) blot was completed from the same cell lysates (bottom panel). ATG1, Autophagy-specific gene 1; EGF, epidermal growth factor; HA, haemagglutinin; KI, ATG1α Lys 46 Ile (kinase-dead) mutant; RSK, ribosomal S6 kinase; WT, wild type.
Next, to understand the molecular mechanism of the specific regulation of S6K by ATG1, we investigated whether ATG1 affects the phosphorylation of Thr 229 in S6K by using an S6K mutant that specifically mimics the phosphorylated form of S6K, Thr 389 Glu. As a result, Thr 229 phosphorylation of the S6K Thr 389 Glu mutant was not affected by wild-type ATG1 (Fig 4C). Because Akt was not inhibited by ATG1 (Fig 4A), it is unlikely that ATG1 regulates Thr 389 phosphorylation of S6K by inhibiting the PDK1/Akt signalling module. Therefore, we believe that ATG1 modulates S6K activity by affecting S6K Thr 389-specific kinases or phosphatases.
In summary, under nutrient-rich conditions, activation of TOR leads to inhibition of ATG1, which facilitates S6K Thr 389 phosphorylation and the subsequent phosphorylation of Thr 229 by PDK1 to activate S6K fully. Consequently, activated S6K promotes cell growth. However, under conditions of starvation, TOR becomes quiescent and ATG1 can inhibit S6K by blocking Thr 389 phosphorylation. This nutrient-dependent signalling switch operated by TOR and ATG1 is highly consistent with that in yeast (Noda & Ohsumi, 1998; Kamada et al, 2000). The observations described in this study clearly show the presence of crosstalk between ATG1 and S6K signalling, in which ATG1 specifically inhibits S6K. We also showed that this is evolutionarily conserved between Drosophila and mammals. We believe that our biochemical data and fly system will be useful in future studies that address the detailed molecular mechanism of crosstalk between the two nutrition-dependent physiological processes—autophagy and cell growth.
Methods
Drosophila strains. The fly lines for hs-GAL4, dTORP1 and dS6K07084 were obtained from the Bloomington Stock Center (Indiana University, Bloomington, IN, USA). EP3348 was obtained from the Szeged Drosophila melanogaster P Insertion Mutant Stock Center (University of Szeged, Szeged, Hungary). The dS6Kl-1 fly line was a generous gift from Dr G. Thomas (University of Cincinnati Genome Research Institute, Cincinnati, OH, USA; Montagne et al, 1999). The DmATG1Δ3d fly strain was kindly provided by Dr T.P. Neufeld (University of Minnesota, Minneapolis, MN, USA; Scott et al, 2004). The UAS-Rheb fly line was provided by Dr F. Tamanoi (University of California, Los Angeles, USA; Patel et al, 2003).
Antibodies. Phosphospecific dS6K Thr 398, S6K Thr 389, S6 Ser 235/236, Akt Thr 308 and Akt Ser 473 antibodies were purchased from Cell Signalling Technology (Danvers, MA, USA). Phospho-(Ser/Thr) PDK1 Docking Motif (pPDM) antibody and S6 antibody were also purchased from Cell Signalling Technology. Phosphospecific S6K Thr 229 antibody was obtained from R&D Systems (Minneapolis, MN, USA). S6K antibody was generated as described previously (Chung et al, 1992). β-Tubulin mouse antibody (E7) was purchased from the Developmental Studies Hybridoma Bank (DSHB).
Supplementary information is available at EMBO reports online (http://www.emboreports.org)
Supplementary Material
supplementary Information
Acknowledgments
We thank Dr M. Muramatsu for mouse ATG1 cDNA, Dr J.U. Jung for LC3, ATG6 and UVRAG cDNA, and Dr G. Thomas, Dr T.P. Neufeld and Dr F. Tamanoi for fly stocks. The E7 monoclonal antibody was obtained from the Developmental Studies Hybridoma Bank (DSHB) maintained by the Department of Biological Sciences, University of Iowa.
References
- Bettencourt-Dias M et al. (2004) Genome-wide survey of protein kinases required for cell cycle progression. Nature 432: 980–987 [DOI] [PubMed] [Google Scholar]
- Brown EJ, Albers MW, Shin TB, Ichikawa K, Keith CT, Lane WS, Schreiber SL (1994) A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 369: 756–758 [DOI] [PubMed] [Google Scholar]
- Chung J, Kuo CJ, Crabtree GR, Blenis J (1992) Rapamycin-FKBP specifically blocks growth-dependent activation of and signaling by the 70 kd S6 protein kinases. Cell 69: 1227–1236 [DOI] [PubMed] [Google Scholar]
- Collins BJ, Deak M, Arthur JS, Armit LJ, Alessi DR (2003) In vivo role of the PIF-binding docking site of PDK1 defined by knock-in mutation. EMBO J 22: 4202–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombani J, Raisin S, Pantalacci S, Radimerski T, Montagne J, Leopold P (2003) A nutrient sensor mechanism controls Drosophila growth. Cell 114: 739–749 [DOI] [PubMed] [Google Scholar]
- Fingar DC, Salama S, Tsou C, Harlow E, Blenis J (2002) Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev 16: 1472–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18: 1926–1945 [DOI] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, Ohsumi Y (2000) Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J Cell Biol 150: 1507–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ et al. (2003) A unified nomenclature for yeast autophagy-related genes. Dev Cell 5: 539–545 [DOI] [PubMed] [Google Scholar]
- Kozma SC, Thomas G (1994) p70s6k/p85s6k: mechanism of activation and role in mitogenesis. Semin Cancer Biol 5: 255–260 [PubMed] [Google Scholar]
- Kuroyanagi H, Yan J, Seki N, Yamanouchi Y, Suzuki Y, Takano T, Muramatsu M, Shirasawa T (1998) Human ULK1, a novel serine/threonine kinase related to UNC-51 kinase of Caenorhabditis elegans: cDNA cloning, expression, and chromosomal assignment. Genomics 51: 76–85 [DOI] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6: 463–477 [DOI] [PubMed] [Google Scholar]
- Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, Jung JU (2006) Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 8: 688–699 [DOI] [PubMed] [Google Scholar]
- Matsuura A, Tsukada M, Wada Y, Ohsumi Y (1997) Apg1p, a novel protein kinase required for the autophagic process in Saccharomyces cerevisiae. Gene 192: 245–250 [DOI] [PubMed] [Google Scholar]
- Montagne J, Stewart MJ, Stocker H, Hafen E, Kozma SC, Thomas G (1999) Drosophila S6 kinase: a regulator of cell size. Science 285: 2126–2129 [DOI] [PubMed] [Google Scholar]
- Mora A, Komander D, van Aalten DM, Alessi DR (2004) PDK1, the master regulator of AGC kinase signal transduction. Semin Cell Dev Biol 15: 161–170 [DOI] [PubMed] [Google Scholar]
- Noda T, Ohsumi Y (1998) Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J Biol Chem 273: 3963–3966 [DOI] [PubMed] [Google Scholar]
- Oldham S, Montagne J, Radimerski T, Thomas G, Hafen E (2000) Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev 14: 2689–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel PH, Thapar N, Guo L, Martinez M, Maris J, Gau CL, Lengyel JA, Tamanoi F (2003) Drosophila Rheb GTPase is required for cell cycle progression and cell growth. J Cell Sci 116: 3601–3610 [DOI] [PubMed] [Google Scholar]
- Sabatini DM, Erdjument-Bromage H, Lui M, Tempst P, Snyder SH (1994) RAFT1: a mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 78: 35–43 [DOI] [PubMed] [Google Scholar]
- Scott RC, Schuldiner O, Neufeld TP (2004) Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev Cell 7: 167–178 [DOI] [PubMed] [Google Scholar]
- Scott RC, Juhasz G, Neufeld TP (2007) Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr Biol 17: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima H, Pende M, Chen Y, Fumagalli S, Thomas G, Kozma SC (1998) Disruption of the p70s6k/p85s6k gene reveals a small mouse phenotype and a new functional S6 kinase. EMBO J 17: 6649–6659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Stern D, Beaton A, Rhem EJ, Laverty T, Mozden N, Misra S, Rubin GM (1999) The Berkeley Drosophila Genome Project gene disruption project: single P-element insertions mutating 25% of vital Drosophila genes. Genetics 153: 135–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan J, Kuroyanagi H, Kuroiwa A, Matsuda Y, Tokumitsu H, Tomoda T, Shirasawa T, Muramatsu M (1998) Identification of mouse ULK1, a novel protein kinase structurally related to C. elegans UNC-51. Biochem Biophys Res Commun 246: 222–227 [DOI] [PubMed] [Google Scholar]
- Yan J et al. (1999) Mouse ULK2, a novel member of the UNC-51-like protein kinases: unique features of functional domains. Oncogene 18: 5850–5859 [DOI] [PubMed] [Google Scholar]
- Zhang H, Stallock JP, Ng JC, Reinhard C, Neufeld TP (2000) Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev 14: 2712–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary Information