

Abstract
Somatotrophs are the only pituitary cells that express Ret, GFRα1 and GDNF. This study investigated the effects of Ret in a somatotroph cell line, in primary pituitary cultures and in Ret KO mice. Ret regulates somatotroph numbers by inducing Pit-1 overexpression, leading to increased p53 expression and apoptosis, both of which can be prevented with Ret or Pit-1 siRNA. The Pit-1 overexpression is mediated by sustained activation of PKCδ, JNK, c/EBPα and CREB induced by a complex of Ret, caspase 3 and PKCδ. In the presence of GDNF, Akt is activated, and the Pit-1 overexpression and resulting apoptosis are blocked. The adenopituitary of Ret KO mice is larger than normal, showing Pit-1 and somatotroph hyperplasia. In normal animals, activation of the Ret/Pit-1/p53 pathway by retroviral introduction of Ret blocked tumor growth in vivo. Thus, somatotrophs have an intrinsic mechanism for controlling Pit-1/GH production through an apoptotic/survival pathway. Ret might be of value for treatment of pituitary adenomas.
Keywords: caspase-3, c/EBPa, GDNF, pituitary, PKCd
Introduction
Final adult height and body composition are maintained in a constant interval for each given species. Somatototrophs, the pituitary cells secreting growth hormone (GH), are specialized secretory cells. However, they are not terminally differentiated: the pituitary retains some plasticity throughout life, enabling appropriate responses to events such as puberty and pregnancy (Melmed, 2003). Somatotrophs play a key role in the control of growth during infancy and puberty, although they continue to secrete GH throughout adult life. Somatotroph number and function are tightly regulated, as GH hypersecretion leads to excessive growth in the form of either gigantism or acromegaly. Genetic factors leading to somatotroph hypoplasia are well known, but little is known about the mechanisms controlling physiological adjustments in somatotroph number. In this connection, the incidence of pituitary somatotroph adenomas is surprisingly low (about 3–4 cases per 106 per year) compared with adenomas of other endocrine glands (Holdaway and Rajasoorya, 1999). More puzzling still is the consistent benignity of these tumors, which never metastasize (Melmed, 2003; Kaltsas et al, 2005).
We have recently demonstrated that somatotrophs are the only pituitary secretory cells expressing Ret, GDNF and GFRα1, both in humans (Japon et al, 2002) and in rats (Urbano et al, 2000). This expression pattern is maintained in all somatotroph adenomas. Ret is a transmembrane receptor well known for its differentiation- and survival-controlling functions in epithelial cells, neurons and neuroendocrine cells. Alternative splicing produces two main isoforms: Ret long (Ret-L or Ret51), 1072 amino acids (aa) long, and Ret short (Ret-S or Ret9), 1114 aa long, with different sequences in the C-terminal region (51 aa long in Ret-L and 9 aa long in Ret-S) (Tahira et al, 1990). Ret has four coreceptors (GFRα1, 2, 3 and 4), with ligands GDNF, NTN, ART and PSP, respectively. Extensive research on Ret function in mammals indicates that it has two main functions. First, it actively promotes survival, growth and extension/migration of cells such as neurons (neurite and axonal sprouting) and epithelial renal cells (branching). In these roles, Ret binds to its ligand GDNF and its GPI-anchored extracellular coreceptor GFRα1, and this binding induces the tyrosine kinase activity of Ret, triggering various signal transduction pathways (Bredesen et al, 2004; Porter and Dhakshinamoorthy, 2004; Arighi et al, 2005). Second, Ret appears to induce apoptosis, but only in the absence of GDNF; to date, however, this effect has only been demonstrated in transient transfection of HEK293T and neuroblastoma cell lines (Bordeaux et al, 2000). In line with this, Ret has been included in the family of ‘dependence receptors', together with p75NTR, DCC, UNC5H, PTC1 and the androgen receptor. In humans, mutations that activate Ret lead to multiple endocrine neoplasia type 2 (MEN 2), whereas inactivating mutations lead to Hirschprung's disease (Santoro et al, 2004; Arighi et al, 2005).
Here, we report a study of Ret functions in a somatotroph cell line and in primary pituitary cultures. We uncover a pathway in which Ret, in the absence of GDNF, induces Pit-1 overexpression, leading to increased p53 expression and apoptosis. We identify the kinases involved in the induction of Pit-1 promoter overexpression by Ret. The cytoplasmic Ret portion (IC-Ret) complexes with caspase 3 and the kinase PKC delta, all becoming cleaved during the process. We also present results indicating that newborn Ret KO mice pituitaries are larger than normal, showing Pit-1-expressing cell and somatotroph hyperplasia. Finally, the Ret/Pit-1/p53 apoptotic pathway acts in an in vivo model of pituitary hyperplasia. In this model, retroviral Ret delivery suppressed estrogen-induced hyperplasia but did not alter normal pituitary function.
Results
Ret-induced apoptosis is associated with Pit-1 expression
To investigate the role of Ret in somatotrophs, we used a pituitary cell line, GH4C1, that does not express some of the characteristic receptors of normal somatotrophs (e.g. GHRH-R), but that nonetheless expresses the ghrelin receptor (NRL unpublished) and small amounts of Pit-1, and secretes some GH. This cell line does not express Ret but expresses both GFRα1 and GDNF mRNA (data not shown) and the corresponding proteins (Figure 1A, left). We tried to produce transfectants stably expressing human Ret-L or Ret-S; colonies were obtained in various transfections, but in no case was Ret expression detected (Figure 1A). While transient transfection of GH4C1 or human embryonic kidney (HEK293) cells led to Ret expression (Figure 1A), Ret stable transfection had a deleterious effect on colony number after selection with neomycin (G418) (Supplementary Figure 1). In a previous study using the HEK293T cell line, transient transfection with Ret induced apoptosis that could be blocked by GDNF (Bordeaux et al, 2000). Similarly, in our somatotroph cells, transfection with Ret (either isoform) potently induced apoptosis within 24 h, whereas addition of GDNF (50 ng/ml) completely blocked this response (Figure 1B, left). zVAD, a broad caspase inhibitor, likewise blocked the apoptosis, suggesting an involvement of caspase activity (Figure 1B, right).
Figure 1.
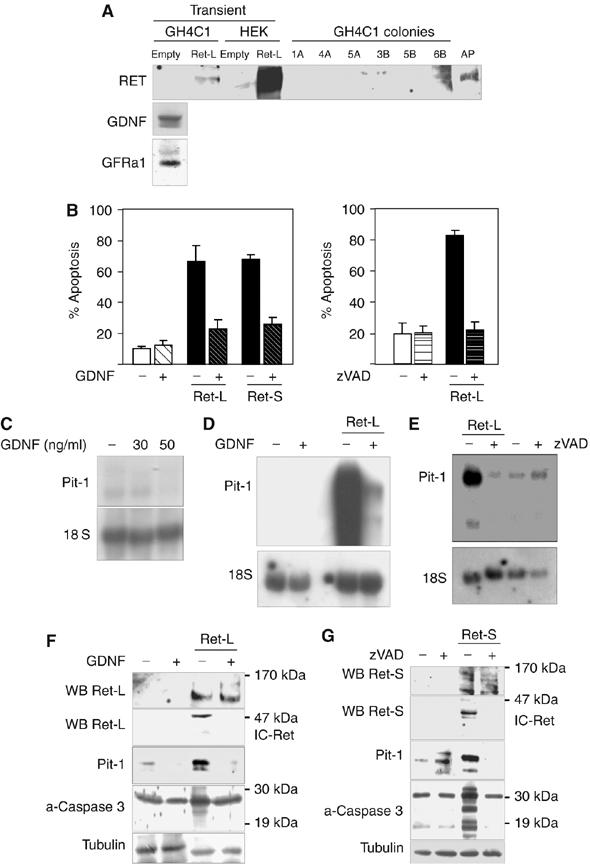
Both the Ret-L and Ret-S isoforms strongly induced apoptosis and Pit-1 expression in the GH4C1 somatotroph line. (A) GH4C1 cells expressed GDNF and GFRα1 but not Ret. No stable Ret-expressing transfectants could be obtained (results shown only for Ret-L): the few colonies obtained were all Ret-negative, except colony 6B, which died after the second passage. In transient transfections, full-length Ret (150–170 kDa) was expressed strongly in HEK293 cells and weakly in GH4C1 cells. (B) Forty-eight hours after transient transfection of GH4C1 cells with either Ret-L or Ret-S, marked apoptosis was detected. Apoptosis could be prevented by treatment with GDNF or the broad caspase inhibitor zVAD. (C) In the absence of Ret, GDNF had a weak inhibitory effect on Pit-1 mRNA expression in GH4C1 cells. (D) Transfection with Ret induced a marked increase in Pit-1 mRNA expression, which was blocked by GDNF. (E) The broad caspase inhibitor zVAD blocked the effect of Ret on Pit-1 mRNA expression. (F) Ret transfection likewise induced a marked increase in Pit-1 protein expression, again blocked by GDNF (results shown only for Ret-L). Note that detection of full-length Ret required immunoprecipitation or loading more than 150 μg proteins/lane, whereas the processed intracellular fragment (IC-Ret) could be detected easily with 50 μg proteins/lane. Both IC-Ret and activated caspase 3 (19 kDa) were induced by transfection with Ret when GDNF was absent but not when GDNF was present. (G) zVAD also reduced the Ret-induced increase in Pit-1 protein expression (results shown only for Ret-S) and blocked IC-Ret formation and activation of caspase 3.
We next investigated the effects of Ret transfection and GDNF on the expression of specific pituitary somatotroph genes. In the absence of Ret, GDNF had no consistent effect on the hormones GH or PRL (data not shown), but had a small dose-dependent inhibitory effect on Pit-1 mRNA expression (Figure 1C). Transfection with Ret induced a marked increase in Pit-1 mRNA, which was blocked by incubation with GDNF (Figure 1D). The caspase inhibitor zVAD, which blocked Ret-induced apoptosis, also blocked the effect of Ret on Pit-1 mRNA expression (Figure 1E). Ret likewise induced an increase in Pit-1 protein levels, which were again reduced by GDNF and zVAD (Figure 1F and G). Ret has been classed as a ‘dependence receptor': in HEK and neuroblastoma cell lines, it actively induces apoptosis in the absence of its ligand GDNF, whereas the presence of the ligand allows cell survival (Bordeaux et al, 2000; Bredesen et al, 2004; Porter and Dhakshinamoorthy, 2004). The Ret protein is processed by caspase 3 at two intracytoplasmic sites, releasing a fragment of about 40–50 kDa (IC-Ret) that has been proposed to induce apoptosis. IC-Ret was readily detected by Western blotting in our GH4C1 cells after transient transfection with either Ret-L or Ret-S. IC-Ret was not detectable in cells treated with zVAD, and levels were significantly reduced by GDNF (Figure 1F and G), in contrast with the previous findings in HEK cells. Basal levels of activated caspase 3 were present in the GH4C1 cells, and caspase-3 activity was strongly induced by Ret transfection and reduced by GDNF or zVAD; that is, the effects on caspase 3 paralleled the effects on apoptosis, IC-Ret presence and Pit-1 level (Figure 1F, G and B, E).
In the presence of GDNF, Ret forms a heterodimeric complex with GDNF and GFRα1, activating Ret's tyrosine kinase activity (so that it phosphorylates susceptible cytoplasmic substrates). We therefore investigated whether the tyrosine kinase activity of Ret is necessary for induction of apoptosis and/or of Pit-1 expression. The kinase-dead mutants of Ret-L (Ret-LKR, K758R substitution) and Ret-S (Ret-SKR, K758R substitution) were transiently transfected into GH4C1 cells. The transfected cells did not show increased tyrosine kinase activity in the presence of GDNF (unlike cells transfected with Ret-L or Ret-S) (Figure 2B), although Ret was expressed to similar levels (Figure 2C). While all Ret isoforms had apoptotic effects, the protective effects of GDNF were not seen in the cells transfected with the kinase-dead mutants (Figure 2D). Interestingly, both kinase-dead Ret isoforms strongly induced Pit-1 mRNA and protein expression (like the normal isoforms), but, unlike in cells transfected with the normal isoforms, GDNF did not inhibit this response (Figure 2E and F); again, this is in line with a relationship between Ret's apoptotic activity and its induction of Pit-1 expression.
Figure 2.
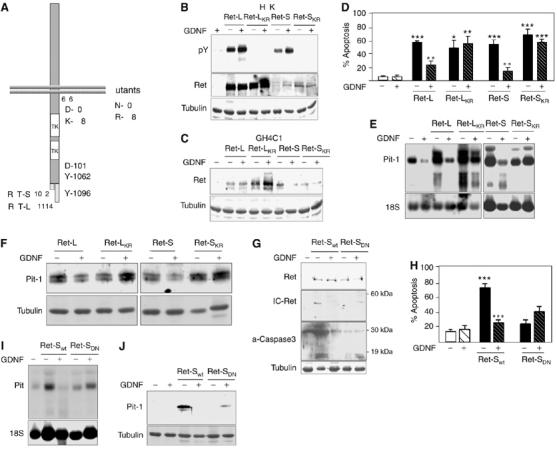
Ret-induced apoptosis and Pit-1 expression are independent of Ret's tyrosine kinase activity, but dependent on activation of caspase 3. (A) Cartoon showing the wild-type and mutant receptors used in these experiments. Ret-LKR is a kinase-dead mutant (K758R) of Ret-L; Ret-SKR is a kinase-dead mutant (K758R) of Ret-S; Ret-SDN, cloned in the pBR expression vector, has a substitution in the proximal caspase-3-processing consensus site of the Ret-S isoform (control: Ret-Swt in the same vector). (B) When expressed in HEK293 cells, neither kinase-dead mutant showed tyrosine kinase activity. As HEK293 cells secrete GDNF, a basal phosphorylation of the Ret receptor is seen in the untreated lanes. (C) The kinase-dead mutants were expressed in GH4C1 somatotrophs and (D) strongly induced apoptosis that could not be blocked by GDNF, like the wild-type isoforms. The kinase-dead mutants likewise markedly increased Pit-1 mRNA (E) and protein (F) expression; however, these increases were not blocked by GDNF. (G) In cells transfected with the Ret-SDN mutant, neither IC-Ret (the processed intracellular form) nor activated caspase 3 could be detected. In line with this, Ret-SDN did not induce either apoptosis (H) or Pit-1 mRNA (I), or protein (J) expression (*P<0.05; **P<0.01; ***P<0.001).
Given our results showing that Ret's apoptotic activity is associated with caspase-3 processing and the appearance of the intracytoplasmic fragment IC-Ret, we transfected cells with Ret-SDN, a Ret-S mutant with a D707N substitution affecting the first caspase-3 consensus site in the cytoplasmic tail of the receptor (Figure 2A). As previously demonstrated (Bordeaux et al, 2000), transfected Ret-SDN could not be processed by caspase 3 in vivo, and no IC-Ret band was detected in Western blotting (Figure 2G). Ret-SDN did not induce either apoptosis (Figure 2H) or Pit-1 mRNA or protein expression (Figure 2I and J).
Ret-induced apoptosis is mediated by Pit-1, and Pit-1 overexpression increases p53 levels
The data shown in Figures 1 and 2 suggested a direct relationship between caspase-3-processed intracytoplasmic Ret, Pit-1 expression and apoptosis. The time course of caspase-3 activity in extracts of transfected cells showed that the maximal activity was reached immediately after initiating the Ret transfection (2 h) (Figure 3A). Coincidentally, the induction of Pit-1 expression in somatotrophs was a very early event, seen as soon as 2 h after Ret transfection (Figure 3B), and maintained throughout the experiment (Figure 3C). Ret also induced a marked increase in p53 at 24 h after transfection (Figure 3D) before the induction of apoptosis, which peaked around 48 h after transfection (see Figures 1 and 2). Incubation with zVAD (which blocked IC-Ret generation, induction of Pit-1 expression and apoptosis; Figures 1B, E and G) also blocked the p53 increase (Figure 3E).
Figure 3.
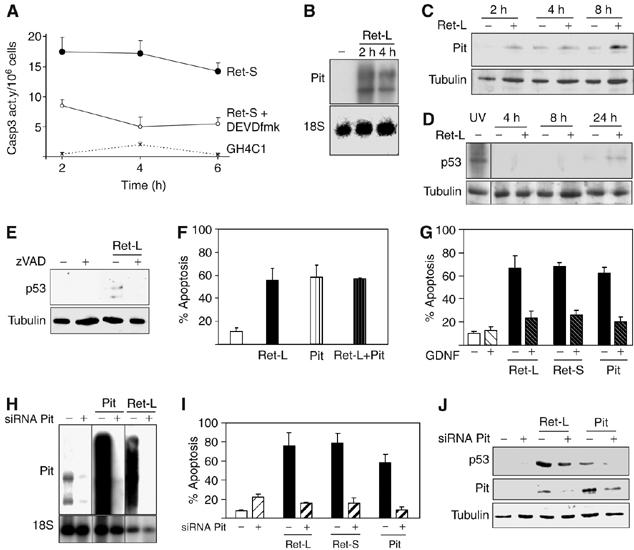
Ret-induced caspase-3 activation and Pit-1 overexpression in somatotrophs are early events causing a later p53 increase and apoptosis. (A) Time-course assay of caspase-3 activity after initiation of Ret-S transfection compared with empty-vector transfected cells or in the presence of the caspase-3 inhibitor DEVDfmk. The highest activity was seen at the first time point, only 2 h after the initiation of Ret transfection. (B) The Ret-induced increase in Pit-1 mRNA expression was already detectable within 2 h of transfection, suggesting a direct effect on the Pit-1 promoter that increased Pit-1 protein (C) with time after transfection. (D) An increase in p53 protein expression was only detectable 24 h after transfection. (E) Caspase inhibition with zVAD (which inhibited caspase-3 activity, Ret processing, apoptosis and Pit-1 overexpression; Figures 1 and 2) blocked the Ret-induced p53 increase. (F) Transfection with Pit-1 induced similar levels of apoptosis in somatotrophs as transfection with Ret; the two effects were not additive. (G) Intriguingly, Pit-1-induced apoptosis was also inhibited by GDNF (Supplementary Figure 2A). (H) Pit-1 siRNA blocked Pit-1 mRNA expression, whether basal or induced by Ret or Pit-1 transfection. For each Pit-1 siRNA treatment (+) and its control (−, GFP siRNA), lanes are shown with different exposure times. Pit-1 siRNA blocked apoptosis induced by Ret-L, Ret-S and Pit-1 (I) and the Ret-induced increase in Pit-1 and p53 protein expression (J).
This sequence of events suggested that Ret-induced apoptosis in somatotrophs is mediated by Pit-1 induction. Transfection of a Pit-1 expression construct induced similar levels of apoptosis to Ret transfection, and the two treatments were not additive (Figure 3F). However, Pit-1-induced apoptosis was blocked by GDNF (Figure 3G), simultaneously with the inhibition of transfected Pit-1 expression (Supplementary Figure 2A). To confirm that Pit-1 expression is essential for Ret-induced apoptosis, we used Pit-1 siRNA to block basal and Ret-induced Pit-1 expression (Figure 3H). The presence of Pit-1 siRNA entirely blocked the apoptosis induced by transfection with Ret-L, Ret-S or Pit-1 (Figure 3I), and also blocked p53 induction (Figure 3J). These results suggest that caspase-3-processed IC-Ret induces Pit-1 expression, which in turn increases p53 expression and causes apoptosis.
Ret-induced c/EBPα binding to the Pit-1 promoter depends on PKCδ, JNK and CREB
Next, we performed experiments designed to identify kinases involved in the induction of Pit-1 expression by Ret. Sustained p-JNK and p-CREB expressions were detected in somatotrophs transfected with all Ret isoforms and mutants that induced apoptosis, but not in somatotrophs transfected with the non-apoptosis-inducing mutant Ret-SDN (Figure 4A). As previously described (Chiariello et al, 1998; Feng et al, 1999; Poteryaev et al, 1999; Trupp et al, 1999; Hayashi et al, 2000; Pezeshki et al, 2001), short-term (15 min) treatment with GDNF also induced p-JNK or p-CREB activation (Figure 4B) that returned to basal levels after 30 min (Supplementary Figure 2B) and was not altered by Ret expression. However, long-term (24 h) treatment with GDNF, which blocks Ret-induced apoptosis, also blocked the Ret-induced p-JNK and p-CREB sustained expression (Figure 4C). Likewise, we investigated whether PKCδ is activated by Ret. PKCδ has been implicated in apoptosis induced by DNA-damaging agents or Fas ligand: after its induction, PKCδ is phosphorylated (p-PKCδ) and processed, and the free catalytic domain migrates to the nucleus inducing apoptosis (DeVries et al, 2002; Mecklenbrauker et al, 2002, 2004), although the mechanism remains unknown. p-PKCδ was detected in somatotrophs transfected with the apoptosis-inducing Ret forms, but not in cells transfected with Ret-SDN (Figure 4D). In contrast to the effects on JNK or CREB, short-term treatment (15 min) with GDNF reduced p-PKCδ levels in cells transfected with Ret-L and Ret-S but not in cells transfected with the kinase-dead mutant Ret-LKR (Figure 4D). This reduction in p-PKCδ levels by GDNF was maintained after 24 h of treatment, coinciding with the reduction in p-CREB and p-JNK levels (Figure 4C). Ret-induced p-PKCδ production was also blocked by the caspase inhibitor zVAD (Figure 4E). These results suggested that Ret may induce Pit-1 and apoptosis through activation of PKCδ. Rottlerin, a PKCδ inhibitor, blocked PKCδ phosphorylation by all the apoptosis-inducing Ret forms (Figure 4F). Rottlerin also blocked Ret-induced p-JNK expression but not Ret-induced p-CREB expression (Figure 4G). Rottlerin strongly reduced Ret-dependent Pit-1 expression, and in line with this reduced Ret-induced apoptosis; however, it did not affect Pit-induced apoptosis (Figure 4H and I). Finally, killer CREB, a dominant-negative form of CREB, significantly reduced Ret-induced apoptosis (Figure 4H). These data suggest a two-branched pathway for Ret-induced Pit-1 expression, one branch dependent on activation of PKCδ/JNK and the other dependent on activation of CREB. Pit-1 mRNA induction by this pathway was very rapid, suggesting a transcriptional effect. The most important consensus binding sites on the rat Pit-1 promoter are shown in Figure 4J. Both CRE elements are essential for GHRH- and ghrelin-induced Pit-1 expression (Soto et al, 1995; Garcia et al, 2001). In somatotrophs cotransfected with Ret and promoter/Luc constructs with decreasing promoter length, luciferase activity was detected with the −2500, −959 and −358 promoter fragments (strongest activity with the −358 fragment), whereas further shortening to −231 abolished the Ret-dependent activity (Figure 4J). Luciferase activity was also completely abolished by treatment with GDNF or by cotransfection with Ret-SDN instead of Ret-S (Figure 4K). Luciferase activity in cells cotransfected with Ret-S and the −358 promoter fragment was markedly reduced by rottlerin and by killer CREB, and completely abolished by adding both together, confirming the additive effects of the PKCδ/JNK and CREB pathways (Figure 4L). These results indicate that the Ret-responsive region of the rat Pit-1 promoter lies between this protein's positions −358 and −231. A consensus binding site for the Pit-1 transcription factor c/EBPα appears near the first CRE, just before −231. Interestingly, the human Pit-1 promoter has a similar conserved site located upstream to the TATA region (Pfaffle et al, 1992), and initial results corroborate a Ret-dependent regulation of the hPit-1 promoter (data not shown). To confirm that Ret induces binding of c/EBPα to the rat Pit-1 promoter, we used chromatin immunoprecipitation (ChIP) assay with c/EBPα-specific antibodies, followed by PCR using primers for a small region of the Pit-1 promoter, including the c/EBPα binding site. A basal level of c/EBPα binding was detected in untransfected cells, and an increase in this binding in Ret-transfected cells was detected within only 4 h of transfection. The increase in c/EBPα binding was completely abolished by GDNF and significantly reduced by rottlerin or cotransfection with killer CREB (Figure 4M).
Figure 4.
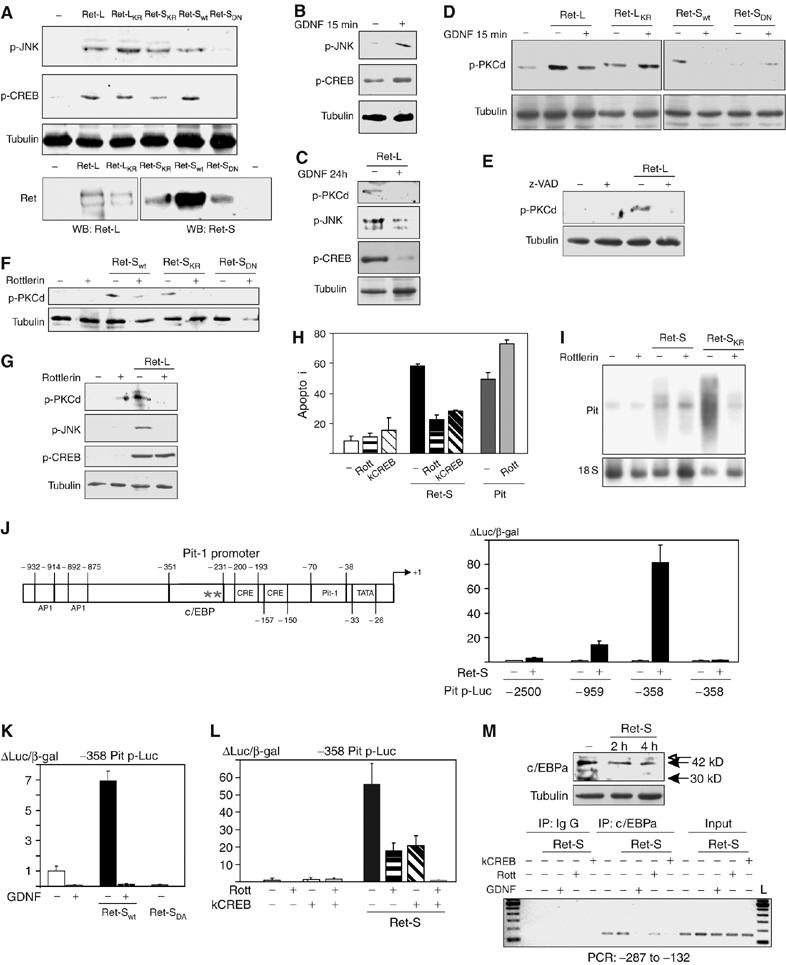
Ret-induced sustained activation of PKCδ, JNK and CREB leads to binding of c/EBPα to the Pit-1 promoter. (A) Twenty-four hours after transfection of GH4C1 cells with apoptosis-inducing Ret forms, but not after transfection with Ret-SDN, p-JNK and p-CREB levels showed a sustained increase. (B) We could not study the short-term effects of GDNF on p-JNK or p-CREB levels, as GDNF induced short-term phosphorylation of both kinases in the absence of Ret (peak at 15 min, return to basal at 30 min; Supplementary Figure 2B). (C) Long-term treatment with GDNF (24 h) blocked the Ret-induced phosphorylation of both kinases. (D) Transfection of GH4C1 cells with the apoptosis-inducing Ret forms, but not Ret-SDN, induced a sustained increase in levels of p-PKCδ. Brief treatment with GDNF (15 min) inhibited this PKCδ phosphorylation, which was maintained after long-term (24 h) GDNF treatment (C). (E) zVAD treatment blocked the Ret-induced phosphorylation of PKCδ. (F) Rottlerin (Rott), a specific PKCδ inhibitor, effectively blocked the Ret-induced phosphorylation of PKCδ and (G) JNK, but not of CREB, suggesting that PKCδ/JNK and CREB form part of two parallel and additive branches of the pathway. (H) Both rottlerin and the dominant-negative form of CREB (killer CREB, kCREB) blocked Ret- but not Pit-induced apoptosis. (I) Rottlerin blocked Ret-induced Pit-1 mRNA expression. (J) Schematic representation of the rat Pit-1 promoter, with known (Carneiro et al, 1998; Garcia et al, 2001) (inside boxes) and putative (outside boxes) response elements. Cells were cotransfected with Ret plus Pit-1 promoter-luciferase constructs with increasing deletions of the promoter. In the presence of Ret-S, Pit-1 promoter deleted to −2500 showed a three-fold induction in luciferase activity that was enhanced with further deletions, especially to −358, but that was lost with further deletion to −231. (K) The −358 bp region appears to be very important for the response to Ret, as the response was blocked by GDNF treatment and could not be induced by the Ret-SDN. (L) Both rottlerin and kCREB reduced Pit-1 promoter induction by Ret, although complete abolishment could only be achieved with both inhibitors together. (M) A consensus c/EBPα site was present in this small region of the promoter. Specific binding of c/EBPα to the Pit-1 promoter could be demonstrated by ChIP only 4 h after transfection with Ret. This was abolished by GDNF treatment and reduced by treatment with rottlerin or kCREB. c/EBPα Western blotting showed both the 42 and 30-kDa isoforms present in pituitary cell lines (Schaufele, 1996).
The intracytoplasmic portion of Ret forms a complex with caspase 3 and PKCδ
Our data suggested a strong relationship between the intracellular Ret portion, caspase 3, PKCδ, Pit-1 expression and apoptosis. It seemed that blockage of Ret or caspase-3 processing also inhibited Pit-1 expression. In fact, caspase-3 activity was detected as early as 2 h after transfection of either Ret-S, the kinase-dead mutant Ret-SKR or the intracellular portion of Ret (IC-Ret707–1017). However, there was no caspase-3 activity after transfection with Ret-SDN, a mutant that could not be processed by caspase-3 (Figure 5A, upper panel). GDNF was able to block Ret-induced caspase-3 activity, Pit-1 and apoptosis, but could not stop the apoptosis induced by IC-Ret707–1017, which correspondingly induced Pit-1 expression (Figure 5A, lower panels). We have found another caspase-3-processed protein, PKCδ (Voss et al, 2005), implicated in the pathway from Ret to the Pit-1 promoter. As both the catalytic fragment of PKCδ and caspase 3 have been located in the nucleus, we investigated the location of the processed intracellular fragment of Ret. Both the full-length Ret-S and the fragment IC-Ret707–1017 transfected in GH4C1 showed cytoplasmic location (Figure 5B and Supplementary Figure 2D).
Figure 5.
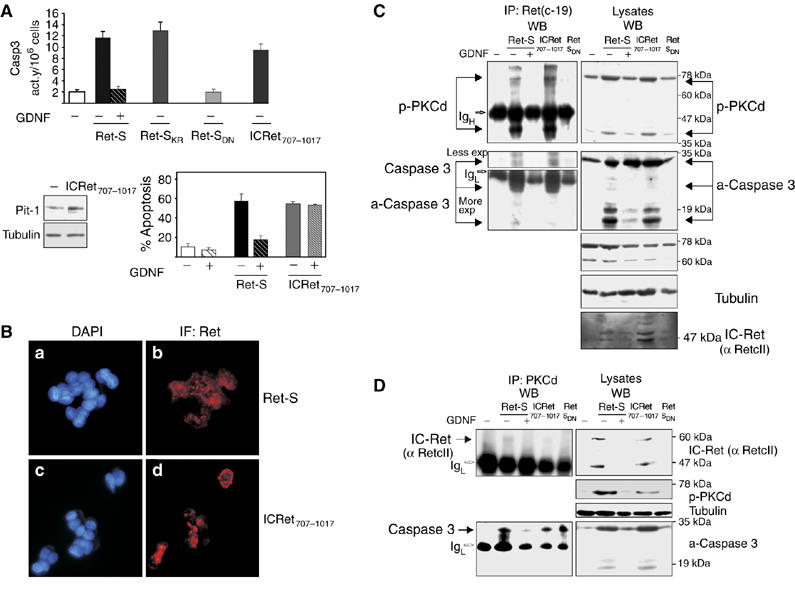
The intracellular portion of Ret forms a complex with caspase 3 and PKCδ in which all three become cleaved and activated. (A) Transfection of the intracellular portion of Ret, IC-Ret707–1017, induced an early (2 h) caspase-3 activity similar to full-length Ret-S or the kinase-dead Ret-SKR but not Ret-SDN. IC-Ret707–1017 induced Pit-1 expression and apoptosis not inhibited by GDNF. (B) Ret immunofluorescence in cells transfected with full-length Ret-S (b) or ICRet707−1017 (d) showed peripheral cytoplasmic location. DAPI staining was used as a nuclear marker (a, c). As it is a transient transfection, not all the cells expressed Ret (see Supplementary Figure 2E). (C) Left: immunoblotting with anti-p-PKCδ and anti-caspase 3 antibodies of anti-Ret immunoprecipitates indicated specific binding of Ret either to the cleaved 40 kDa p-PKCδ (more) and 25–19 kDa caspase 3 (less), or to the full-length PKCδ (70 kDa, less) and caspase 3 (30 kDa, more). The complex was prevented by GDNF treatment and was not present after transfection of the Ret-SDN mutant. Right: levels of proteins in the lysates. (D) Similar anti-PKCδ immunoprecipitation revealed binding of PKCδ to full-length caspase 3 in the presence of any isoform of the Ret receptor, even the inactive Ret-SDN; thus the binding was independent of caspase-3 activation. However, PKCδ bound to IC-Ret exclusively in the presence of Ret-S or IC-Ret707–1017, but not in the presence of Ret-SDN. Again, GDNF treatment prevented the formation of both the IC-Ret–PKCδ complex and the Ret-induced PKCδ–caspase-3 complex.
As it has been demonstrated that PKCδ binds to and phosphorylates caspase 3 in monocytes (Voss et al, 2005), we investigated whether IC-Ret binds either of these proteins. Initial experiments showed the presence of PKCδ in Ret immunoprecipitates, with a weak band around 80 kDa and a stronger band around 40 kDa (data not shown); straight Western blotting against PKCδ showed the 80-kDa band and a shorter (unphosphorylated) band around 60 kDa (Figure 5C, right). We thought that the smaller 40-kDa band that appeared in the Ret immunoprecipitates could be the processed/phosphorylated catalytic fragment of PKCδ, absent in the 60-kDa band. Therefore, we performed IP-Ret IP followed by Western blotting against phosphorylated PKCδ (Figure 5C, upper left). Full-length Ret-S and IC-Ret707–1017 both co-immunoprecipitated with the p-PKCδ; the strongest band was the processed 40-kDa band of p-PKCδ and the weakest band was the full-length 80-kDa p-PKCδ. Formation of this Ret/p-PKCδ complex was prevented in the presence of the ligand GDNF. The Ret-SDN mutant was not able to associate with p-PKCδ, in line with its inability to induce Pit-1 expression or apoptosis. In the absence of GDNF, full-length Ret-S and IC-Ret707–1017 also co-immunoprecipitated with unprocessed (32 kDa) or processed (20/19 kDa) caspase 3 (Figure 5C, lower left). However, Ret-SDN associated very weakly with unprocessed caspase 3, in keeping with its inability to induce caspase-3 processing/activation. To further study this complex, we immunoprecipitated PKCδ and assessed its association with either caspase 3 or the intracellular portion of Ret. The presence of any Ret construct, even Ret-SDN, induced the formation of the PKCδ/full-length caspase-3 complex, albeit in residual amounts in the presence of GDNF (Figure 5D, lower left). However, IC-Ret was found to be associated with PKCδ only in the presence of Ret-S or IC-Ret707–1017 (Figure 5D, upper left), that is, the same samples in which PKCδ was associated with Ret (compare upper panels of Figure 5C and D). These results indicated that the intracellular portion of Ret induces the formation of a complex with both full-length caspase 3 and PKCδ that results in cleavage (and activation) of the trio, and suggested that the complex was unstable when Ret could not be processed (Ret SDN), or when GDNF induced Ret dimerization and tyrosine kinase activation.
GDNF prevents Ret-induced Pit-1 expression and apoptosis through Akt activation in both the somatotroph line and in primary pituitary cultures
Many pathways induced by GDNF have been described, both in the presence (Chiariello et al, 1998; Feng et al, 1999; Hayashi et al, 2000; Santoro et al, 2004; Arighi et al, 2005) and absence (Poteryaev et al, 1999; Trupp et al, 1999; Pezeshki et al, 2001) of Ret. In neurons, the blocking of Ret-induced apoptosis by GDNF is mediated by the kinase Akt: specifically, a GDNF/GFRα1/Ret complex induces PI3K activation, leading to Akt phosphorylation and activation (Besset et al, 2000; De Vita et al, 2000; Hayashi et al, 2000). We therefore investigated whether this same pathway acts in somatotrophs. GDNF strongly induced Akt phosphorylation in cells transfected with wild-type Ret, but not in cells transfected with the kinase-dead mutants Ret-LKR or Ret-SKR (Supplementary Figure 2C). The PI3K inhibitor LY294002 partially reduced the GDNF-induced phosphorylation of Akt (Figure 6A). Likewise, the blocking of Ret-induced apoptosis (Figure 6B) and Pit-1 expression (Figure 6C) by GDNF was abolished by coincubation with LY294002.
Figure 6.
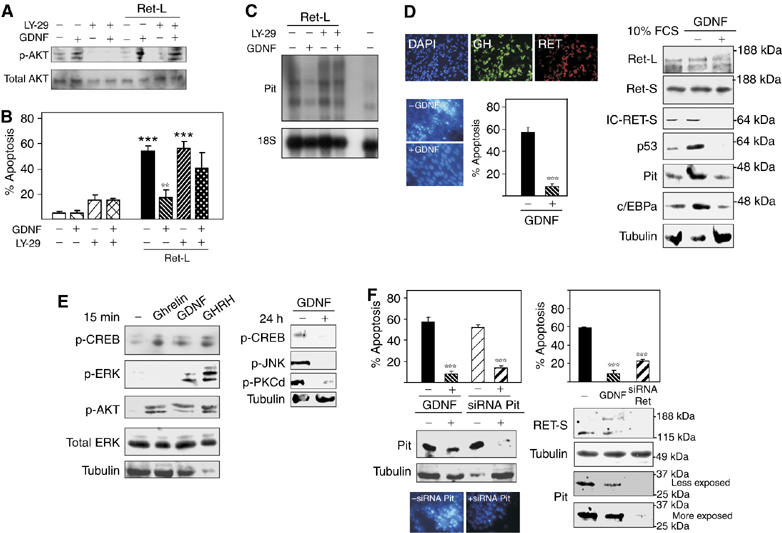
GDNF prevents Ret-induced p-PKCδ, p-JNK and apoptosis in both GH4C1 cells and primary pituitary cultures by phosphorylating Akt and thus blocking the Ret-induced increase in Pit-1. Both Pit-1 and Ret siRNA prevent apoptosis and Pit-1 expression. (A) GDNF induced Akt phosphorylation in Ret-transfected cells, and the PI3K inhibitor LY294002 (LY-29) reduced this phosphorylation. In the presence of LY-29, Ret induced apoptosis (B) and Pit-1 mRNA overexpression (C) despite the presence of GDNF. (D) The percentage of GH+/Ret+ cells in primary cultures correlated with that in the whole pituitary (more than 40%). In these cultures, change from DMEM+10% FCS to serum-deprived medium (DMEM+0.5% FCS) induced marked apoptosis and IC-Ret, c/EBPa, Pit-1 and p53 expression. All of them could be rescued by GDNF. (E) Short-term treatment (15 min) of primary cultures with Pit-1 regulators (ghrelin, GDNF or GHRH) induced phosphorylation of CREB and Akt, whereas phosphorylation of ERK was induced only by GHRH (Pombo et al, 2000). Ghrelin and GHRH (Soto et al, 1995; Garcia et al, 2001) induced increased Pit-1 mRNA expression over a period of about 2 h, whereas GDNF reduced Ret-induced Pit-1 mRNA expression. As in the GH4C1 cell line, prolonged treatment with GDNF in primary cultures blocked the expression of p-CREB, p-JNK and p-PKCδ. (F) Both Pit-1 and Ret siRNA abolished Ret-induced Pit-1 protein expression after serodeprivation in primary cultures, in parallel with prevention of apoptosis (**P<0.01; ***P<0.001).
We next investigated whether the results obtained in the GH4C1 somatotroph line were reproduced in primary pituitary cultures expressing endogenous Ret receptors. Pituitary cultures were grown in collagen-IV-coated dishes. Around 30% of cells were somatotrophs, in line with the high proportion of somatotrophs seen in vivo, and 99.9% were GH and Ret positive (Figure 6D); note that somatotrophs are the only cells expressing Ret and GFRα1 (Urbano et al, 2000; Japon et al, 2002; and Supplementary Figure 3) in the rat and human pituitary. After 2 days, the original medium (containing 10% FCS) was changed to serum-deprived medium (only 0.5% FCS). Serum deprivation induced massive apoptosis and c/EBPα, Pit-1 and p53 overexpression that could be blocked by addition of GDNF, which blocked also IC-Ret expression (Figure 6D). In line with our findings in GH4C1, short-term GDNF treatment induced phosphorylation of Akt and CREB, but not of other kinases such as ERK. Treatment with the somatotroph-specific factors GHRH or ghrelin (both known to induce a short-term increase in Pit-1 expression; Soto et al, 1995; Garcia et al, 2001) likewise induced phosphorylation of Akt (Figure 6E). These results indicate that Akt is a key mediator in Pit-1 induction to prevent apoptosis, controlling its time course. Long-term treatment of the cultures with GDNF reduced p-CREB, p-JNK and p-PKCδ expression (Figure 6E). Either Pit-1 or Ret siRNA transfection of the cultures completely blocked the apoptosis or the Pit-1 expression (Figure 6F), indicating a role of Ret in maintenance of the somatotroph phenotype in adult pituitary.
Activation of the Ret/Pit-1/p53/apoptosis pathway in vivo prevents tumor growth
The above results indicated that the apoptotic pathway in primary pituitary cultures is the same as in the GH4C1 line. To investigate whether this pathway acts in vivo, we analyzed the pituitaries of 20 d.p.c. Ret KO mice in comparison with their wild-type littermates. Ret KO mice die around the first day of life owing to the absence of kidney (Schuchardt et al, 1994). Coronal and sagittal (data not shown) sections of the heads were obtained every 5 μm. The first section in which the pituitary appeared was called level 0. From level 0 to the end, 5-μm sections were obtained every 25 μm, H&E-stained and used to measure the three pituitary lobes, that is, the adenopituitary (AP), intermediate lobe (IL) and neuropituitary (NP), and to reconstruct their volume (μm3) using ImageJ software (NIH) (Figure 7A, B and D). Areas of the pituitary lobes (μm2) in the three largest sections were also calculated (Figure 7A, E and F). AP volume in Ret KO mice was significantly larger than in the wild type (P=0.007; Figure 7D), as was AP area (P=2.4 × 10−5; Figure 7E), whereas IL and NP volumes and areas were similar to those of the wild type. To rule out bias in the different sections, we calculated the ratios of AP to IL area and NP to IL area for each section. The AP/IL ratio was 1.4 times greater in the Ret KO mice (P=0.009), whereas the NP/IL ratio showed no difference from the wild type (P=0.38; Figure 7F).
Figure 7.
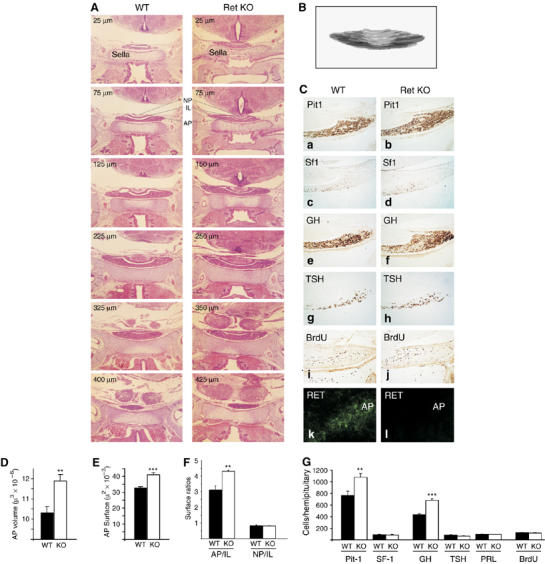
The Ret/Pit-1/p53 pathway acts in vivo: Ret KO mice present somatotroph hyperplasia. (A) H&E pituitary series of wild-type and KO mice (selected sections, × 40). The first section in which the pituitary appeared on top of the sella turcica (Sella) was labelled as 0 μm. The Ret KO mice showed a longer pituitary. (B) From level 0 μm to the end, 5-μm sections were taken every 25 μm and used to measure the three pituitary lobes, adenopituitary (AP), intermediate lobe (IL) and neuropituitary (NP), and to reconstruct their volume. (C) The central sections were used to study Pit-1-, Sf-1-, GH-, TSH-, PRL- (not shown) and BrdU-positive cells (hemipituitaries, × 200). As a control, Ret immunofluorescence of pituitary sections is shown in the bottom panel. (D) Ret KO pituitaries had significantly higher volume and AP area (E) than wild-type littermates. (F) Area ratios between the three pituitary lobes in each section were calculated. The AP/IL ratio was higher in the Ret KO mice, although no difference could be detected in the NP/IL ratio in the same sections. (G) Ret KO mice showed Pit-1 hyperplasia but no alteration in SF-1 cells (corticotrophs). Of the three Pit-1-positive cell types, only GH were more abundant; no difference was detected in TSH or PRL abundance. Likewise, we did not detect any difference in the abundance of BrdU-positive cells (**P<0.01; ***P<0.001).
There are five different types of secretory cell in the adenopituitary: somatotrophs (producing GH), lactotrophs (PRL) and thyrotrophs (TSH) are Pit-1-expressing cells derived from a common precursor, whereas gonadotrophs (LH and FSH) and corticotrophs (ACTH) are Pit-1 negative. Corticotrophs (ACTH) also express a specific nuclear transcription factor called SF-1 (Morohashi and Omura, 1996). Whereas Pit-1-positive nuclei showed increased abundance in Ret KO mice (P=0.0097), the number of SF-1-positive nuclei was similar in the two groups of mice (P=0.98; Figure 7G). We counted the different Pit-1-positive lineages. Ret KO mice presented more GH-producing cells per hemipituitary (P=9.8 × 10−6), whereas the number of TSH-producing cells was slightly, although not significantly, decreased (P=0.09), and the number of PRL-producing cells was similar to that in wild type (P=0.92). The total number of GH-, TSH- and PRL-producing cells in the wild-type pituitary equalled the number of Pit-1-positive cells. In the Ret KO mice, however, a subset of Pit-1-positive cells were not stained for any hormone marker, suggesting a delay in differentiation. Therefore, as GH cells are the only cells expressing Ret in the normal pituitary, somatotroph hyperplasia seems to have been the main cause of the larger size of the Ret KO pituitaries. There was no difference between the two groups in proliferative index in the pituitary, as measured by BrdU staining (Figure 7C and H); similar results were found by Ki-67 staining (data not shown). We conclude that somatotroph hyperplasia in Ret KO mice is likely due to enhanced survival through the differentiation process.
To investigate whether the most important feature of the pathway in vivo is Pit-1 expression or GH phenotype, we engineered the overexpression of Ret in Ret-negative/Pit-1-expressing pituitary lactotrophs, and then looked for increased Pit-1 expression, increased p53 expression and apoptosis. We used a retrovirus bearing the human Ret-S cDNA (LPC-Ret) or the empty construct (LPC). For retroviral expression, we needed actively dividing cells, and so we used estrogen-induced lactotroph hyperplasia (Tsukahara et al, 1994). The LPC or LPC-Ret retrovirus was delivered to estrogen-injected female rats by stereotaxic injection. One week later, the pituitaries were dissected out, weighed and processed. Estrogens induced a strongly significant increase in pituitary weight in LPC-infected rats by comparison with non-treated controls (P=0.00019). Infection with the Ret-expressing retrovirus prevented this estrogen-induced hyperplasia (LPC+E2 versus LPC-Ret+E2 P=0.00077, LPC-Ret+E2 versus control P=0.34) (Figure 8A and Supplementary Figure 4C). In line with this, PRL immunostaining was much weaker in LPC-Ret-infected pituitaries than in LPC-infected pituitaries; however, the small areas that showed detectable PRL staining in LPC-Ret-infected pituitaries also showed strong Ret costaining (Figure 8B). The estrogen-treated LPC-Ret-infected pituitaries contained detectable levels of IC-Ret and increased amounts of Pit-1, p53, p-CREB and p-JNK by comparison with both estrogen-treated LPC-infected pituitaries and untreated pituitaries. Cleaved PARP (80 kDa) was only present in LPC-RET-infected pituitaries (Figure 8C). Some pituitaries were dissected out just 24 h after retroviral delivery, when no marked alteration in lactotroph numbers was expected. The proportion of Ret-expressing cells was 37.8±9.2% in LPC-infected pituitary sections versus 61.8±7.3% in LPC-Ret-infected pituitary, whereas the opposite pattern was seen in PRL-expressing cells (62.2±9.2% in LPC-infected pituitary versus 38.1±7.4% in LPC-Ret-infected pituitary). More interestingly, as in normal pituitary (Urbano et al, 2000), the proportion of double labelled Ret/PRL cells was only 0.66±0.73% in LPC-infected pituitary, versus 10.7±1.8% in LPC-Ret-infected pituitary (P=0.0011) (see Supplementary Figure 3A). p-PKCδ, IC-Ret, Pit-1 and p53 levels were increased within 24 h of LPC-RET infection. Phosphorylated c/EBPα levels were increased in LPC-Ret-infected pituitary, whereas active caspase-3 levels, reduced in LPC-infected pituitary, returned to control levels (Supplementary Figure 3B). Taken together, these results demonstrate that Ret expression blocks estrogen-induced lactotroph hyperplasia in vivo by the same mechanism as revealed in the GH4C1 somatotroph cell line and in primary pituitary cultures, that is, via Pit-1 overexpression leading to increased p53 expression and thus apoptosis.
Figure 8.
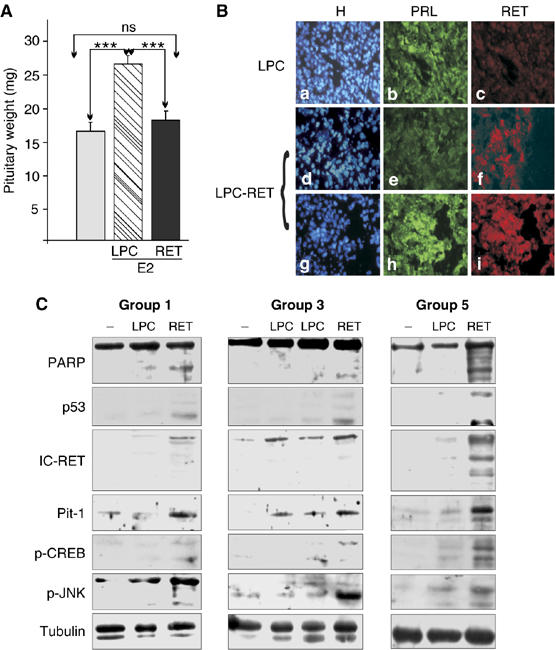
The Ret/Pit-1/p53 pathway acts in vivo: retroviral RET delivery in rat pituitary prevents tumor growth. (A) Mean (±s.e.m.) pituitary weights in estrogen- (E2) and vehicle-treated (C) rats, 7 days after the stereotaxic delivery of empty (LPC) or Ret-containing (LPC-RET) retrovirus. LPC-RET significantly reduced the E2-induced pituitary hyperplasia, returning pituitary weight to basal levels (for individual pituitary weights, see Supplementary Figure 3C). (B) The LPC-RET-infected pituitaries showed a marked decrease in PRL immunostaining. Areas still positive for PRL were also Ret-positive, in contrast with the absence of any PRL+/Ret+ areas in LPC-transfected pituitaries. (C) LPC-RET-infected pituitaries showed increased IC-Ret, Pit-1, p53, p-CREB and p-JNK expression 7 days after transfection. Cleaved PARP (80 kDa) was only present in LPC-RET-infected pituitaries. Data for groups 1, 3 and 5 are shown. Similar increases were already detectable 24 h after transfection (Supplementary Figure 4) (***P<0.001).
Discussion
Dependence receptors like Ret only induce apoptosis in the absence of a particular ligand, so that cell survival depends on the presence of the ligand. Besides, it seems likely that these proteins also play key roles in cell differentiation (Bredesen et al, 2004). Our present results rule out the possibility that Ret is necessary for differentiation to the somatotroph fate, and establish that Ret's role in the pituitary is to regulate Pit-1 expression and control survival of differentiated somatotrophs (Figures 6F and 7A–C). We might speculate that this acts as a mechanism to prevent excessive body growth.
For apoptotic effect, dependence receptors need to be intracellularly processed by proteases, releasing a fragment with apoptotic properties. It remains unclear which specific protease processes each receptor (Bredesen et al, 2004). We found processing of the Ret receptor to be caspase-dependent, both in cell cultures and in vivo. It has been postulated that initial processing of Ret takes place under low levels of caspase-3 activation, which induces greater caspase activation in an activating loop (Bredesen et al, 2004). In somatotrophs, the maximal caspase-3 activity was detected immediately after Ret transfection (Figures 3 and 5), with the caspase-3 processing Ret intracellularly (to IC-Ret) in the absence of GDNF, and inducing sustained activation of PKCδ, JNK, c/EBPα and CREB, leading to strong and sustained overexpression of Pit-1, which in turn led to increased p53 expression and apoptosis. By contrast, GDNF activated Ret's tyrosine kinase activity, leading to Akt activation, and thus reduced Pit-1 expression, promoting survival (Figure 6B). Long-term phosphorylation of either CREB or JNK seems to be the key to the Ret-induced process, as GDNF inducing short-term phosphorylation of both has opposite effects (Chiariello et al, 1998; Feng et al, 1999; Poteryaev et al, 1999; Trupp et al, 1999; Hayashi et al, 2000; Pezeshki et al, 2001). Recently, the UNC5H2 dependence receptor has been found to bind and activate DAP kinase to induce apoptosis (Llambi et al, 2005). We provide evidence that the intracellular portion of Ret forms a complex with PKCδ and caspase 3 (Figure 5) and binding of caspase 3 and PKCd has been described previously (Voss et al, 2005). Ternary complex formation was dependent solely on the presence of Ret, even an inactive form (Figure 5D), but was not enough to initiate the activation of the complex. Our data suggest that after binding to the complex, IC-Ret initiates the processing and activation of both PKCδ and caspase 3. A certain basal level of activated caspase 3 could initiate Ret cleavage and thus trigger an activating loop (Llambi et al, 2005); alternatively, the presence of the monomeric Ret cytoplasmic tail may elicit formation of the ternary complex in which caspase 3 would become stabilized and activated. We favor this latter model, as total caspase-3 levels are increased by Ret transfection. In contrast to previous findings in HEK cells (Bordeaux et al, 2000), in somatotrophs, GDNF was able to suppress Ret processing by dimerization of the Ret receptors and Akt recruiting, disrupting the IC-Ret/PKCδ/caspase-3 complex.
The PKCδ catalytic fragment migrates to the nucleus (DeVries et al, 2002), but its targets remain unknown. In many systems, PKCδ is implicated in apoptosis downstream of p53 but upstream of JNK (Humphries et al, 2006; Liu et al, 2006; Miyamoto et al, 2002). PKCδ-dependent CREB phosphorylation has been described for the B-cell receptor (Blois et al, 2004). However, in our cells, inhibition of PKCδ, although it blocked JNK activation, did not abolish CREB phosphorylation. Our results indicate a cooperative action of c/EBPα and CREB on the Pit-1 promoter. Similar cooperation has been described in human monocyte differentiation (Brenner et al, 2003). Taken together, our data suggest a model in which, once the ternary complex (IC-Ret/PKCδ/caspase 3) is formed and processed, PKCδ either alone or bound to caspase-3 activates JNK. It remains to be determined whether JNK activation occurs in the cytosol or in the nucleus, and how CREB phosphorylation is initiated and maintained independent of PKCδ activation.
Previous reports have already suggested a role of c/EBPα in the pituitary (Schaufele, 1996). Pit-1 establishes somatotroph (GH), lactotroph (PRL) and thyrotroph (TSH) phenotypes during pituitary development. Based on in vitro experiments in lactotroph cell lines, it is currently accepted that Pit-1 has two functions, regulating both differentiation and proliferation (Castrillo et al, 1991). However, there is no substantial in vivo evidence in rodents or humans to support this latter possibility: the Pit-1 gene is not altered in adenomas, and Pit-1 expression appears to be more closely related to GH secretion than to tumorigenesis (Delhase et al, 1993; Hoggard et al, 1993; Pellegrini et al, 1994). Moreover, Pit-1 mutations have been found in human hypopituitarism without pituitary hypoplasia (Pfaffle et al, 1992), whereas no increase in Pit-1 expression is required for in vivo estrogen-induced lactotroph hyperplasia in rats (Tsukahara et al, 1994). Our data show that too much Pit-1 is deleterious for the somatotroph; conversely, knocking down Pit-1 prevents apoptosis. The apoptosis induced by dependence receptors appears to be independent of both the intrinsic (caspase 9/Apaf1) and the extrinsic (FADD/caspase 8) pathways (Porter and Dhakshinamoorthy, 2004). The induction of p53 expression by Pit-1 links Ret-induced apoptosis with p53. To date, the only reported relationship of dependence receptors with p53 is via netrin-1, a ligand that blocks p53-dependent apoptosis by binding to the UNC5B dependence receptor (Tanikawa et al, 2003).
In the presence of the PI3K inhibitor LY-294002, Ret induced Pit-1 overexpression and apoptosis despite the presence of GDNF, although Akt phosphorylation was not completely abolished. GDNF induces Akt activation through binding of a Gab–p85PI3K complex to the phosphorylated Y1062 of activated Ret, thus preventing apoptosis (Besset et al, 2000; De Vita et al, 2000; Hayashi et al, 2000). In somatotrophs, the PI3K–Akt pathway seems to be essential for Pit-1 downregulation and cell survival. This pathway could act at various levels: by downregulating Ret processing by caspase inhibition, by decreasing c/EBPα phosphorylation through PP2A activation (Wang et al, 2004) or by regulating p53-dependent apoptosis. The fact that GDNF is not able to block Pit-1 induction/apoptosis induced by transfection of the IC-Ret707–1017 fragment, but decreases Pit-1 induction of exogenous transfected Pit-1 and consequent apoptosis, suggests that the GDNF action is before the Pit-1 action, and rules out any direct effect on p53; it seems probable that IC-Ret707–1017 sequesters PI3K/Akt, preventing its activation near the membrane. GDNF (via Akt induction) and siRNA Pit-1 were also able to prevent apoptosis in the primary pituitary culture. GHRH, ghrelin and GDNF share the ability to activate Akt and regulate Pit-1 transcription. GHRH and ghrelin cause very short-lived Pit-1 induction (2 h) (Soto et al, 1995; Garcia et al, 2001), whereas GDNF reduces Pit-1 transcription. We propose that Akt activation by GHRH and ghrelin switches off the Pit-1 promoter, confining the induction to this short period. In fact, GDNF is able to decrease Pit-1 levels after transfection of exogenous Pit-1 expression vector; in this case, the positive feedback of exogenous Pit-1 activating the endogenous Pit-1 promoter is disrupted by Akt. A role for Akt in pituitary tumorigenesis is suggested by the finding of increased p-Akt levels in pituitary tumors (Musat et al, 2005).
Our results support the notion of dependence receptors being tumor suppressors (Mazelin et al, 2004), where Ret, like the other dependence receptors, would be a two-sided tumor regulator: a tumor suppressor because of its dependence receptor activity and an oncogene because of its tyrosine kinase activity. We further show that activation of the Ret/Pit-1/p53 pathway was able to block tumor growth in vivo, probably by the same mechanisms described in cultures. These results raise the possibility of Ret-based therapy as a treatment for conventional Pit-1-expressing chemotherapy-resistant adenomas.
The Ret/Pit-1/p53 pathway, conserved in mouse, rat and human somatotrophs, appears to be a mechanism to prevent oversized individuals, exerting a strong pressure on somatotrophs to stay in the pituitary, and limiting their numbers (Supplementary Figure 5). Disrupting Ret expression during development provokes somatotroph hyperplasia (Figure 7); the final result in the adult animal might perhaps be gigantism. If a precursor cell is to become a somatotroph, it needs Pit-1, and to maintain sufficient Pit-1 expression in a somatotroph after birth, Ret expression is required (Urbano et al, 2000; Japon et al, 2002). But if a somatotroph produces too much Pit-1 or migrates to areas in which GDNF is present at much lower concentrations, Ret-induced Pit-1 overexpression will kill it. This would further explain why all GH-secreting adenomas maintain normal Ret/GFRα1 and GDNF expression, and why GH-secreting adenomas are unable to metastasize.
Materials and methods
A more detailed list of methods is given as Supplementary data.
Cell culture and apoptosis detection
Rat GH4C1 pituitary cells and HEK293 cells were cultured in DMEM plus 10% FCS and apoptosis was detected by Hoechst staining (Bravo et al, 2003). zVAD (Sigma) was dissolved in DMSO and used at 50 μM. Rottlerin (Sigma) was dissolved in DMSO and used at 20 nM. LY294002 (Sigma) was dissolved in DMSO and used at 10 μM.
Constructs and transfections
Cells were transfected with Fugene (Roche) or JetSi (PolyPlus). A detailed list of the constructs and siRNA sequences used in this work is provided as Supplementary data.
Primary adenopituitary culture
Pituitary cells were cultured by a modification of our previously described procedure (Coya et al, 1999; Garcia et al, 2001; Supplementary data). Cells were plated in Ikawi culture dishes coated with type-IV collagen (Sigma) and incubated for 4 days. To induce apoptosis, the medium was changed to DMEM plus 0.5% FCS for 36 h. Apoptosis was detected by Hoechst staining.
Northern blotting
RNA was extracted with RNAeasy (Qiagen).
Caspase-3 activity
We used the Caspase 3 Apoalert kit (Clontech) following the manufacturer's instructions.
Immunodetection
Cell extracts were obtained with Triton lysis buffer as described (Carneiro et al, 1998; Garcia et al, 2001). A detailed list of the antibodies used is provided as Supplementary data.
Immunocytochemistry
To estimate the proportion of secretory cells in pituitary cultures and cryosections, we used a double immunofluorescence procedure, with the two immunostainings performed sequentially. See Supplementary data for the list of antibodies and procedures.
Chromatin immunoprecipitation assay
ChIP assay was performed with a kit from Upstate, using c/EBPα (Santa Cruz Biotechnology) and rabbit IgG as control. PCR was performed at 59°C for 35 cycles with forward primer 5′-CCAAAAGA CGCCTATTTTTCA-3′ and reverse primer 5′-GGAGAAACCTGGTTGT GACGTA-3′.
Mouse pituitary analysis
Full-term pregnant mothers (20 days) were injected with 100 μg/g BrdU (Sigma) i.p. 1 h before kiling. Pups' heads were fixed in formalin for 24 h followed by immersion in 70% ethanol until analysis by standard paraffin inclusion. The heads were completely sectioned (5 μm) sagittally (initial group, n=4/genotype) or coronally (group 2, n=3/genotype; and group 3, n=4/genotype), and every 25 μm a hematoxilin–eosin staining was performed for estimation of pituitary lobe volume (n=4/genotype) and area (n=7/genotype) (ImageJ, http://rsb.info.nih.gov/ij/). Pit-1, Sf-1 (n=4/genotype), and GH, TSH and PRL (n=7/genotype) immunohistochemistry in the pituitaries from littermates were performed by standard techniques. A detailed procedure is provided as Supplementary data. ImageJ was also used to count the stained cells.
In vivo stereotaxic retroviral delivery
Ret-expressing or empty retrovirus were delivered after stereotaxic injection to estrogen-treated rats. See Supplementary data for a more detailed description.
Statistical analysis
Results were compared by non-parametric t-tests. (*P<0.05; **P<0.01; ***P<0.001).
Supplementary Material
Supplementary Figure 1
{kind=link}
Supplementary Figure 2
{kind=link}
Supplementary Figure 3
Supplementary Figure 4
{kind=link}
Supplementary Figure 5
{kind=link}
Supplementary Data
Acknowledgments
We thank Dr JL Labandeira for the stereotaxic device, Dr C Ibañez for the initial contribution of pREP-Ret-L, Dr P Mehlen for the Ret mutants, Dr A Bell for the kCREB vector and Dr K Morohashi for the SF-1 antibody. We also thank M Blanco for helpful support. This work was funded by CICYT-MEC and Xunta de Galicia grants to CVA, CD, CS and MJ.
References
- Arighi E, Borrello MG, Sariola H (2005) RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev 16: 441–467 [DOI] [PubMed] [Google Scholar]
- Besset V, Scott RP, Ibanez CF (2000) Signaling complexes and protein–protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J Biol Chem 275: 39159–39166 [DOI] [PubMed] [Google Scholar]
- Blois JT, Mataraza JM, Mecklenbrauker I, Tarakhovsky A, Chiles TC (2004) B cell receptor-induced cAMP-response element-binding protein activation in B lymphocytes requires novel protein kinase Cdelta. J Biol Chem 279: 30123–30132 [DOI] [PubMed] [Google Scholar]
- Bordeaux MC, Forcet C, Granger L, Corset V, Bidaud C, Billaud M, Bredesen DE, Edery P, Mehlen P (2000) The RET proto-oncogene induces apoptosis: a novel mechanism for Hirschsprung disease. EMBO J 19: 4056–4063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo SB, Pampin S, Cameselle-Teijeiro J, Carneiro C, Dominguez F, Barreiro F, Alvarez CV (2003) TGF-beta-induced apoptosis in human thyrocytes is mediated by p27kip1 reduction and is overridden in neoplastic thyrocytes by NF-kappaB activation. Oncogene 22: 7819–7830 [DOI] [PubMed] [Google Scholar]
- Bredesen DE, Mehlen P, Rabizadeh S (2004) Apoptosis and dependence receptors: a molecular basis for cellular addiction. Physiol Rev 84: 411–430 [DOI] [PubMed] [Google Scholar]
- Brenner S, Prosch S, Schenke-Layland K, Riese U, Gausmann U, Platzer C (2003) cAMP-induced Interleukin-10 promoter activation depends on CCAAT/enhancer-binding protein expression and monocytic differentiation. J Biol Chem 278: 5597–5604 [DOI] [PubMed] [Google Scholar]
- Carneiro C, Alvarez CV, Zalvide J, Vidal A, Dominguez F (1998) TGF-beta1 actions on FRTL-5 cells provide a model for the physiological regulation of thyroid growth. Oncogene 16: 1455–1465 [DOI] [PubMed] [Google Scholar]
- Castrillo JL, Theill LE, Karin M (1991) Function of the homeodomain protein GHF1 in pituitary cell proliferation. Science 253: 197–199 [DOI] [PubMed] [Google Scholar]
- Chiariello M, Visconti R, Carlomagno F, Melillo RM, Bucci C, de Franciscis V, Fox GM, Jing S, Coso OA, Gutkind JS, Fusco A, Santoro M (1998) Signalling of the Ret receptor tyrosine kinase through the c-Jun NH2-terminal protein kinases (JNKS): evidence for a divergence of the ERKs and JNKs pathways induced by Ret. Oncogene 16: 2435–2445 [DOI] [PubMed] [Google Scholar]
- Coya R, Alvarez CV, Perez F, Gianzo C, Dieguez C (1999) Effects of TGF-beta1 on prolactin synthesis and secretion: an in-vitro study. J Neuroendocrinol 11: 351–360 [DOI] [PubMed] [Google Scholar]
- De Vita G, Melillo RM, Carlomagno F, Visconti R, Castellone MD, Bellacosa A, Billaud M, Fusco A, Tsichlis PN, Santoro M (2000) Tyrosine 1062 of RET-MEN2A mediates activation of Akt (protein kinase B) and mitogen-activated protein kinase pathways leading to PC12 cell survival. Cancer Res 60: 3727–3731 [PubMed] [Google Scholar]
- Delhase M, Vergani P, Malur A, Velkeniers B, Teugels E, Trouillas J, Hooghe-Peters EL (1993) Pit-1/GHF-1 expression in pituitary adenomas: further analogy between human adenomas and rat SMtTW tumours. J Mol Endocrinol 11: 129–139 [DOI] [PubMed] [Google Scholar]
- DeVries TA, Neville MC, Reyland ME (2002) Nuclear import of PKCdelta is required for apoptosis: identification of a novel nuclear import sequence. EMBO J 21: 6050–6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng L, Wang CY, Jiang H, Oho C, Dugich-Djordjevic M, Mei L, Lu B (1999) Differential signaling of glial cell line-derived neurothrophic factor and brain-derived neurotrophic factor in cultured ventral mesencephalic neurons. Neuroscience 93: 265–273 [DOI] [PubMed] [Google Scholar]
- Garcia A, Alvarez CV, Smith RG, Dieguez C (2001) Regulation of Pit-1 expression by ghrelin and GHRP-6 through the GH secretagogue receptor. Mol Endocrinol 15: 1484–1495 [DOI] [PubMed] [Google Scholar]
- Hayashi H, Ichihara M, Iwashita T, Murakami H, Shimono Y, Kawai K, Kurokawa K, Murakumo Y, Imai T, Funahashi H, Nakao A, Takahashi M (2000) Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene 19: 4469–4475 [DOI] [PubMed] [Google Scholar]
- Hoggard N, Callaghan K, Levy A, Davis JR (1993) Expression of Pit-1 and related proteins in diverse human pituitary adenomas. J Mol Endocrinol 11: 283–290 [DOI] [PubMed] [Google Scholar]
- Holdaway IM, Rajasoorya C (1999) Epidemiology of acromegaly. Pituitary 2: 29–41 [DOI] [PubMed] [Google Scholar]
- Humphries MJ, Limesand KH, Schneider JC, Nakayama KI, Anderson SM, Reyland ME (2006) Suppression of apoptosis in the protein kinase Cdelta null mouse in vivo. J Biol Chem 281: 9728–9737 [DOI] [PubMed] [Google Scholar]
- Japon MA, Urbano AG, Saez C, Segura DI, Cerro AL, Dieguez C, Alvarez CV (2002) Glial-derived neurotropic factor and RET gene expression in normal human anterior pituitary cell types and in pituitary tumors. J Clin Endocrinol Metab 87: 1879–1884 [DOI] [PubMed] [Google Scholar]
- Kaltsas GA, Nomikos P, Kontogeorgos G, Buchfelder M, Grossman AB (2005) Clinical review: diagnosis and management of pituitary carcinomas. J Clin Endocrinol Metab 90: 3089–3099 [DOI] [PubMed] [Google Scholar]
- Liu J, Yang D, Minemoto Y, Leitges M, Rosner MR, Lin A (2006) NF-kappaB is required for UV-induced JNK activation via induction of PKCdelta. Mol Cell 21: 467–480 [DOI] [PubMed] [Google Scholar]
- Llambi F, Lourenco FC, Gozuacik D, Guix C, Pays L, Del Rio G, Kimchi A, Mehlen P (2005) The dependence receptor UNC5H2 mediates apoptosis through DAP-kinase. EMBO J 24: 1192–1201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazelin L, Bernet A, Bonod-Bidaud C, Pays L, Arnaud S, Gespach C, Bredesen DE, Scoazec JY, Mehlen P (2004) Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature 431: 80–84 [DOI] [PubMed] [Google Scholar]
- Mecklenbrauker I, Kalled SL, Leitges M, Mackay F, Tarakhovsky A (2004) Regulation of B-cell survival by BAFF-dependent PKCdelta-mediated nuclear signalling. Nature 431: 456–461 [DOI] [PubMed] [Google Scholar]
- Mecklenbrauker I, Saijo K, Zheng NY, Leitges M, Tarakhovsky A (2002) Protein kinase Cdelta controls self-antigen-induced B-cell tolerance. Nature 416: 860–865 [DOI] [PubMed] [Google Scholar]
- Melmed S (2003) Mechanisms for pituitary tumorigenesis: the plastic pituitary. J Clin Invest 112: 1603–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto A, Nakayama K, Imaki H, Hirose S, Jiang Y, Abe M, Tsukiyama T, Nagahama H, Ohno S, Hatakeyama S, Nakayama KI (2002) Increased proliferation of B cells and auto-immunity in mice lacking protein kinase Cdelta. Nature 416: 865–869 [DOI] [PubMed] [Google Scholar]
- Morohashi KI, Omura T (1996) Ad4BP/SF-1, a transcription factor essential for the transcription of steroidogenic cytochrome P450 genes and for the establishment of the reproductive function. FASEB J 10: 1569–1577 [DOI] [PubMed] [Google Scholar]
- Musat M, Korbonits M, Kola B, Borboli N, Hanson MR, Nanzer AM, Grigson J, Jordan S, Morris DG, Gueorguiev M, Coculescu M, Basu S, Grossman AB (2005) Enhanced protein kinase B/Akt signalling in pituitary tumours. Endocr Relat Cancer 12: 423–433 [DOI] [PubMed] [Google Scholar]
- Pellegrini I, Barlier A, Gunz G, Figarella-Branger D, Enjalbert A, Grisoli F, Jaquet P (1994) Pit-1 gene expression in the human pituitary and pituitary adenomas. J Clin Endocrinol Metab 79: 189–196 [DOI] [PubMed] [Google Scholar]
- Pezeshki G, Franke B, Engele J (2001) Evidence for a ligand-specific signaling through GFRalpha-1, but not GFRalpha-2, in the absence of Ret. J Neurosci Res 66: 390–395 [DOI] [PubMed] [Google Scholar]
- Pfaffle RW, DiMattia GE, Parks JS, Brown MR, Wit JM, Jansen M, Van der Nat H, Van den Brande JL, Rosenfeld MG, Ingraham HA (1992) Mutation of the POU-specific domain of Pit-1 and hypopituitarism without pituitary hypoplasia. Science 257: 1118–1121 [DOI] [PubMed] [Google Scholar]
- Pombo CM, Zalvide J, Gaylinn BD, Dieguez C (2000) Growth hormone-releasing hormone stimulates mitogen-activated protein kinase. Endocrinology 141: 2113–2119 [DOI] [PubMed] [Google Scholar]
- Porter AG, Dhakshinamoorthy S (2004) Apoptosis initiated by dependence receptors: a new paradigm for cell death? Bioessays 26: 656–664 [DOI] [PubMed] [Google Scholar]
- Poteryaev D, Titievsky A, Sun YF, Thomas-Crusells J, Lindahl M, Billaud M, Arumae U, Saarma M (1999) GDNF triggers a novel ret-independent Src kinase family-coupled signaling via a GPI-linked GDNF receptor alpha1. FEBS Lett 463: 63–66 [DOI] [PubMed] [Google Scholar]
- Santoro M, Melillo RM, Carlomagno F, Vecchio G, Fusco A (2004) Minireview: RET: normal and abnormal functions. Endocrinology 145: 5448–5451 [DOI] [PubMed] [Google Scholar]
- Schaufele F (1996) CCAAT/enhancer-binding protein alpha activation of the rat growth hormone promoter in pituitary progenitor GHFT1-5 cells. J Biol Chem 271: 21484–21489 [DOI] [PubMed] [Google Scholar]
- Schuchardt A, D'Agati V, Larsson-Blomberg L, Costantini F, Pachnis V (1994) Defects in the kidney and enteric nervous system of mice lacking the tyrosine kinase receptor Ret. Nature 367: 319–320 [DOI] [PubMed] [Google Scholar]
- Soto JL, Castrillo JL, Dominguez F, Dieguez C (1995) Regulation of the pituitary-specific transcription factor GHF-1/Pit-1 messenger ribonucleic acid levels by growth hormone-secretagogues in rat anterior pituitary cells in monolayer culture. Endocrinology 136: 3863–3870 [DOI] [PubMed] [Google Scholar]
- Tahira T, Ishizaka Y, Itoh F, Sugimura T, Nagao M (1990) Characterization of ret proto-oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma cell line. Oncogene 5: 97–102 [PubMed] [Google Scholar]
- Tanikawa C, Matsuda K, Fukuda S, Nakamura Y, Arakawa H (2003) p53RDL1 regulates p53-dependent apoptosis. Nat Cell Biol 5: 216–223 [DOI] [PubMed] [Google Scholar]
- Trupp M, Scott R, Whittemore SR, Ibanez CF (1999) Ret-dependent and -independent mechanisms of glial cell line-derived neurotrophic factor signaling in neuronal cells. J Biol Chem 274: 20885–20894 [DOI] [PubMed] [Google Scholar]
- Tsukahara S, Kambe F, Suganuma N, Tomoda Y, Seo H (1994) Increase in Pit-1 mRNA is not required for the estrogen-induced expression of prolactin gene and lactotroph proliferation. Endocr J 41: 579–584 [DOI] [PubMed] [Google Scholar]
- Urbano AG, Suarez-Penaranda JM, Dieguez C, Alvarez CV (2000) GDNF and RET-gene expression in anterior pituitary-cell types. Endocrinology 141: 1893–1896 [DOI] [PubMed] [Google Scholar]
- Voss OH, Kim S, Wewers MD, Doseff AI (2005) Regulation of monocyte apoptosis by the protein kinase Cdelta-dependent phosphorylation of caspase-3. J Biol Chem 280: 17371–17379 [DOI] [PubMed] [Google Scholar]
- Wang GL, Iakova P, Wilde M, Awad S, Timchenko NA (2004) Liver tumors escape negative control of proliferation via PI3K/Akt-mediated block of C/EBP alpha growth inhibitory activity. Genes Dev 18: 912–925 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary Figure 5
Supplementary Data