

Abstract
Endocytosed antigens are proteolytically processed and small amounts of peptides captured by class II MHC molecules. The details of antigen proteolysis, peptide capture and how destruction of T-cell epitopes is avoided are incompletely understood. Using the tetanus toxin antigen, we show that the introduction of 3–6 cleavage sites is sufficient to trigger a partially unfolded conformation able to bind to class II MHC molecules. The known locations of T-cell epitopes and protease cleavage sites predict that large domains of processed antigen (8–35 kDa) are captured under these conditions. Remarkably, when antigen is bound to the B-cell antigen receptor (BCR), processing can trigger a concerted ‘hand-over' reaction whereby BCR-associated processed antigen is captured by neighbouring class II MHC molecules. Early capture of minimally processed antigen and confinement of the processing and class II MHC loading reaction to the membrane plane may improve the likelihood of T-cell epitope survival in the class II MHC pathway and may help explain the reciprocal relationships observed between B- and T-cell epitopes in many protein antigens and autoantigens.
Keywords: antigen processing, B-cell antigen receptor (BCR), class II MHC
Introduction
Before T lymphocytes can recognise antigens, limited antigen proteolysis is required to generate antigen peptides, which then bind to either class I or class II MHC molecules. The T-cell antigen receptor then recognises and responds to specific peptide/MHC complexes. For example, antigen processing by B lymphocytes following antigen capture via their B-cell antigen receptors (BCRs) permits B cells to present MHC class II-associated peptides to CD4 T cells, a key recognition event that ultimately allows B cells to become antibody-producing cells (Lanzavecchia, 1990; Watts, 2004). Similarly, presentation of viral peptides on class I MHC molecules identifies virally infected cells as targets for killing by cytotoxic CD8 T cells (Pamer and Cresswell, 1998; Shastri et al, 2002; Rock et al, 2004). Antigen processing and peptide capture by MHC molecules has been extensively studied, but some significant mechanistic questions have been difficult to address.
Whereas class I MHC molecules display peptides of narrowly defined size (8–10 residues), the peptides eluted from class II MHC molecules are considerably larger (∼15–20 residues) and have heterogeneous N and C termini (Engelhard, 1994). It is the open-ended class II MHC peptide binding groove that permits such extensions beyond the ‘core' sequence required for T-cell receptor engagement, but the upstream events that lead to the final output of this heterogeneous set of peptides are not known. Two models have been generally considered. On the one hand, the final set of bound peptides might be that generated by antigen processing and available for class II MHC binding in the loading compartment. On the other hand, longer precursors may bind first and then become trimmed. Such a ‘bind first trim later' scenario might offset the tendency of T-cell epitopes to be destroyed in a compartment populated with several broad-specificity proteolytic enzymes (Sercarz and Maverakis, 2003; Watts, 2004).
Detailed analysis of peptide sets eluted from class II MHC molecules tends to support the notion that processing and trimming reactions can continue after class II MHC binding of longer precursors (Nelson et al, 1997; Lippolis et al, 2002). In addition, there is evidence that class II MHC molecules can bind long peptides or even unfolded proteins both in vitro (Sette et al, 1989) and in vivo following antigen pulsing (Lee et al, 1988; Davidson et al, 1991; Lindner and Unanue, 1996; Castellino et al, 1998) or during class II MHC maturation (Villadangos et al, 2000). Although class II MHC binding to unfolded (Lee et al, 1988; Sette et al, 1989) or partially unfolded (Lindner and Unanue, 1996) antigens has been described, most antigens require processing events before class II MHC binding. However, the number and variety of processing steps necessary to generate a form of antigen competent to bind to class II MHC molecules is unclear. Also, what is the physical state of such antigen and how susceptible is it to additional proteolytic cleavages? Bound antibody (e.g. the BCR) can alter the course of antigen processing, stabilising distinct antigen fragments that differ depending on the epitope specificity of the immunoglobulin (Ig) (Davidson and Watts, 1989). This Ig-directed processing can influence eventual T-cell epitope display either positively or negatively (Watts and Lanzavecchia, 1993; Simitsek et al, 1995), but exactly how Ig-modulated processing affects class II MHC peptide capture is unclear. Answers to these and related questions should be facilitated when the relevant processing enzymes and cleavage sites for an antigen are known. An example of an antigen whose initial processing events are well characterised is the tetanus toxin C fragment (TTCF). Optimal presentation requires limited cleavage by the asparagine-specific endopeptidase AEP. AEP introduces limited cleavages into TTCF, and mutation of these sites or chemical inhibition of AEP slows presentation of T-cell epitopes from this antigen (Manoury et al, 1998; Antoniou et al, 2000; Loak et al, 2003). We proposed earlier that AEP action on TTCF might ‘unlock' the folded native antigen and initiate the downstream events leading to peptide capture (Watts, 2001). Our results demonstrated the necessity of AEP processing for optimal TTCF presentation but not whether this processing was sufficient. Here, we ask whether limited AEP cleavage is sufficient to generate a form of antigen able to bind to class II MHC molecules. We investigate the physical state of AEP processed antigen and attempt to reconstruct the events of antigen processing and class II MHC capture that occur when TTCF is processed as a complex with a specific BCR. Our results provide direct evidence for the capture by class II MHC of large antigen fragments following limited antigen processing and show that these complexes are recognised by T cells. Further, we demonstrate a plausible pathway for T-cell epitope capture following antigen uptake by the BCR.
Results
Physical state of AEP processed TTCF antigen
Processing of the TTCF antigen by recombinant human AEP introduces cleavages after asparagine residues 873, 1184 and 1219, as seen previously when TTCF was digested with lysosomal fractions isolated from the B lymphoblastoid cell lines EDR (Manoury et al, 1998) or Pala (Figure 1). Digestion of TTCF with higher levels of AEP leads to additional cleavages at asparagine residues 944, 1171 and 1230 (Figure 1A and B). Thus, two different levels of AEP lead to processed forms of the TTCF antigen, with approximately three or six cleavage sites introduced. We investigated the properties of these processed antigen forms, which we refer to as TTCF-L and TTCF-H, respectively. We used several different assays to assess the physical state of processed TTCF. Intrinsic protein fluorescence (IPF) can be used to assess the folded state of a protein or the ease with which it can be unfolded by denaturing agents. IPF detects primarily the fluorescence of tryptophan residues whose emission maximum varies depending on whether the amino acid is buried in the hydrophobic core (max ∼330 nm) or is exposed on the protein surface (max ∼350 nm). Following digestion with different levels of AEP, we exposed TTCF to increasing concentrations of urea and measured IPF at 330 and 350 nm following excitation at 280 nm. As expected, at high levels of urea (>4.5 M), undigested TTCF became unfolded, as shown by the shift in the IPF 330/350 ratio from 1.4 to 0.85 (Figure 2A). Whereas digestion at the lower level of AEP marginally increased the susceptibility of TTCF to unfolding in urea, digestion at the higher level of AEP induced a shift in the IPF 330/350 ratio even at non-denaturing levels of urea. The most likely explanation is that AEP digestion resulted in a limited degree of TTCF unfolding and aqueous exposure of Trp residues. As a further test of the conformational integrity of AEP digested TTCF, we assessed its binding to the conformation-specific anti-TTCF monoclonal antibody 15D1 (Antoniou et al, 2000). As shown in Figure 2B, binding of this antibody to plate-bound TTCF was competed by increasing concentrations of soluble TTCF. TTCF-L competed equally well, whereas TTCF-H competed less well, indicating unfolding and partial loss of the 15D1 epitope.
Figure 1.
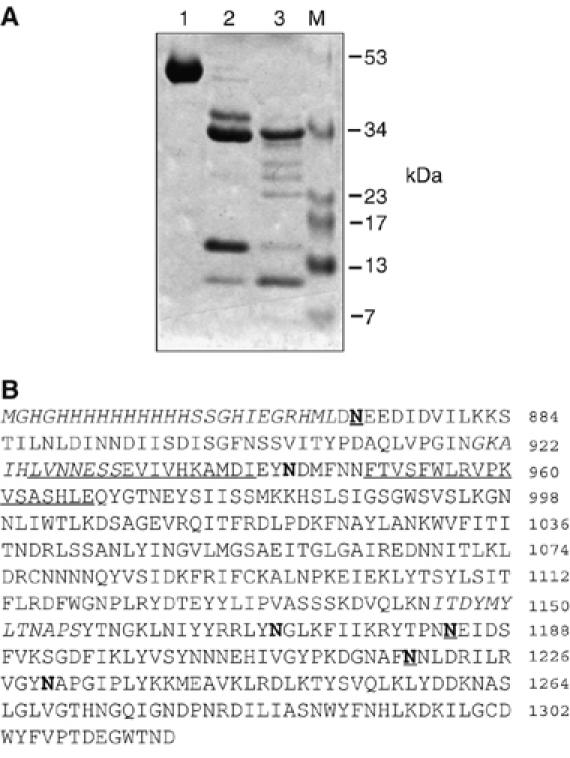
TTCF is cleaved by AEP at several Asn residues. (A) SDS–PAGE analysis of digestion products from AEP cleavage of TTCF. TTCF (lane 1) was digested for 4 h at 37°C with AEP at either 1 μg/ml, generating TTCF-L (lane 2), or 10 μg/ml, generating TTCF-H (lane 3). (B) Asparagine residues targeted by AEP were identified by N-terminal sequencing. TTCF was digested with 1 or 10 μg/ml AEP, transferred to PVDF membrane, the bands excised and subjected to Edman sequencing. AEP-cleaved Asn residues are in bold and Asn residues that are cleaved at the lower level of AEP are underlined. Epitopes recognised by T-cell clones/hybridomas used in later figures are either in italics (AK5, 1145–1156, AK116, 920–931) or underlined (4A8 LVNN… and 4E4; FTVS…). Note that AK116 and 4A8 epitopes are partially overlapping. Residues are numbered according to the 1315-residue whole tetanus toxin molecule.
Figure 2.
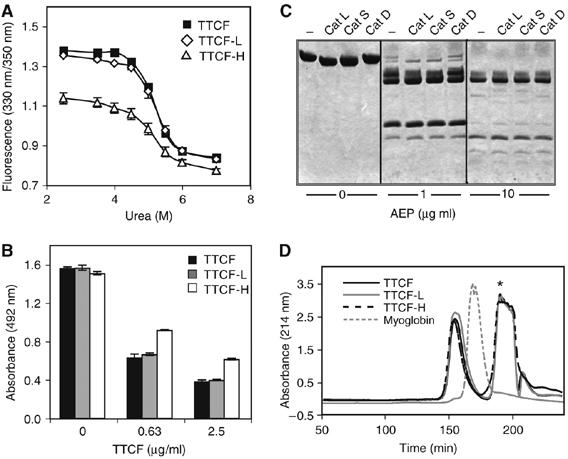
TTCF becomes partially denatured after cleavage by AEP. (A) Intrinsic tryptophan fluorescence of TTCF. The ratio of fluorescence at 330/350 nm was calculated for TTCF TTCF-L and TTCF-H as indicated following exposure to urea. (B) Binding of a conformation-dependent monoclonal antibody to TTCF. Immobilised TTCF was competed with TTCF, TTCF-L and TTCF-H for binding to the monoclonal antibody 15D1. The binding of 15D1 is shown as absorbance at 492 nm. (C) Digestion of TTCF with purified cathepsins after treatment with AEP. TTCF was digested with 1 or 10 μg/ml AEP, AEP activity was blocked with the tetrapeptide AENK and AEP-digested TTCF was further incubated with cathepsin L, S or D. (D) Size-exclusion chromatography of TTCF before and after digestion with AEP. TTCF, TTCF-L and TTCF-H comigrate and elute ahead of myoglobin. Asterisk denotes an OD214 nm absorbing buffer peak eluting at the column volume.
We next assessed the susceptibility of AEP-digested TTCF to the lysosomal proteases cathepsin L, S and D. Significant unfolding would be expected to render TTCF sensitive to these enzymes, which have broader specificity than AEP. Interestingly, digestion even at the higher level of AEP did not dramatically increase the sensitivity of TTCF to processing by these enzymes. Cathepsins L and S were clearly active in this assay as they reduced the size of the major AEP processing product owing to a cleavage close to the N terminus (Figure 2C) and cathepsin D cleaved the substrate myoglobin (Moss et al, 2005; data not shown). This result suggested that in spite of the introduction of a number of AEP cleavage sites, the degree of unfolding of TTCF was quite limited and not sufficient to expose many cleavage sites for other enzymes. To assess the extent of fragmentation further, we subjected digested and undigested TTCF to size fractionation by size-exclusion chromatography. As shown in Figure 2D, undigested and AEP-digested TTCF eluted at the same point, whereas myoglobin (17 kDa) was included in the gel and eluted much later. Taken together, these results suggest that following AEP processing, TTCF, although unfolded to some extent, still retains considerable conformational integrity.
Processing of TTCF by AEP is sufficient for class II MHC binding and presentation to T cells
We next assessed whether AEP-digested TTCF was able to bind to class II MHC molecules in solution and on the surface of antigen-presenting cells. We adapted an assay used to measure binding of synthetic peptides to soluble DR4 molecules (B1*0401), which assesses their ability to displace a biotinylated indicator peptide derived from the CLIP (class II MHC-associated invariant chain peptide) region of the invariant chain (Astill et al, 2003). The CLIP binds promiscuously to class II MHC molecules (Malcherek et al, 1995), and under physiological conditions is replaced by self or antigen-derived peptides in a reaction catalysed by the DM protein (Denzin and Cresswell, 1995). We incubated purified soluble DR4 molecules with biotinylated CLIP peptide (PKPPKPVSKMRMATPLLMQA) in the presence of increasing concentrations of either TTCF-L or TTCF-H. DR4 molecules were then recovered on anti-DR-coated plates and the amount of biotinylated peptide measured by time-resolved fluorescence. As shown previously (Astill et al, 2003) and in Figure 3A, unlabelled CLIP peptide was able to displace biotinylated CLIP at low micromolar concentrations. Although undigested TTCF was not able to displace biotinylated CLIP, AEP digestion of TTCF clearly induced CLIP displacing activity, particularly in TTCF-H, which measurably displaced the biotinylated CLIP peptide at 10 μM (Figure 3A).
Figure 3.
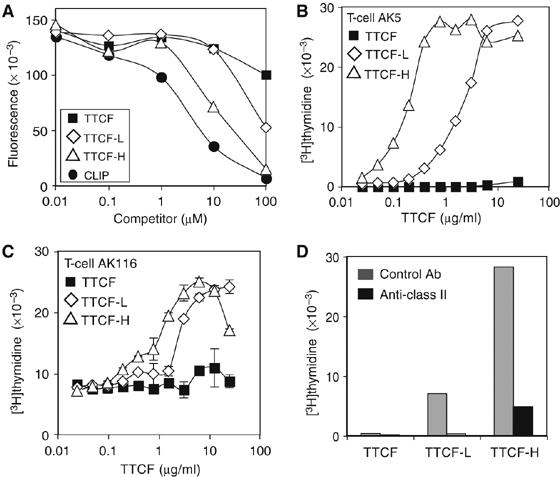
Cleavage by AEP promotes binding of TTCF to class II MHC. (A) A reporter CLIP peptide was competed with TTCF, TTCF-L, TTCF-H or a control CLIP peptide, as indicated, for binding to the class II MHC molecule DR4. (B, C) Fixed cells present AEP-digested TTCF. TTCF, TTCF-L and TTCF-H were added at the indicated concentrations to fixed EBV-B cells for presentation to T-cell clones AK5 (B) or AK116 (C). Proliferation was measured by [3H]thymidine incorporation. Results are representative of three experiments. (D) Presentation of predigested TTCF is blocked with an antibody against class II MHC (black bars). Fixed cells were incubated for 1 h with antibody DA6.231 at 25 μg/ml before inclusion in the antigen presentation assay as above. Inhibition was observed at all concentrations of TTCF tested. The results for 0.78 μg/ml TTCF (for AK5) are shown.
We next tested whether TTCF-L and TTCF-H could become bound to cellular MHC molecules and be presented to T cells. For this, we used cells lightly fixed with aldehyde, which cannot internalise and process antigen but can often present preprocessed or peptide antigen, presumably via a limited number of unoccupied cell surface MHC molecules (Shimonkevitz et al, 1983). Aldehyde-fixed EBV-transformed B lymphoblastoid cells were incubated with unprocessed TTCF, TTCF-L or TTCF-H. The B cells were then cocultured with different TTCF-specific T-cell clones from the same donor and T-cell proliferation was measured 48 h later. Although unprocessed TTCF could not trigger proliferation of T-cell clones AK5 or AK116, TTCF processed with AEP was presented by fixed EBV-B cells, as indicated by dose-dependent T-cell proliferation (Figure 3B and C). Although TTCF-H was more potent, TTCF-L, which contains on average only three cleavages, was nonetheless able to stimulate a T-cell response in T-cell clones AK5 and AK116 recognising residues 1145–1156 and 920–931, respectively. Other T-cell clones tested recognising different epitopes also responded (data not shown). Inspection of the locations of the AK5 and AK116 T-cell epitopes in relation to the mapped AEP processing sites (Figure 1) indicates that the AK5 and AK116 epitopes are likely to be on large fragments of TTCF (26.2 and 7.8 kDa for TTCF-H and 35 kDa for TTCF-L). Incubation with an anti-human class II monoclonal antibody (DA6.231) substantially inhibited recognition of TTCF-L and TTCF-H by AK5 and other T-cell clones (not shown), demonstrating that T-cell activation was indeed due to triggering by class II MHC/processed antigen (Figure 3D).
Single protease classes frequently dominate antigen processing and yield T-cell epitopes
The results thus far show that a limited processing by a single enzyme (AEP) converts the TTCF antigen to a form competent for class II MHC-restricted presentation. Before proceeding further, we wanted to test other protein substrates in the lysosomal digestion assay to ask (1) if single enzymes dominate digestion frequently or rarely and (2) where T-cell reagents were available, can a presentable form of antigen be generated by a single enzyme? Various proteins were exposed to human EBV-B-cell-derived or murine dendritic cell-derived lysosomes in the presence and absence of different protease inhibitors. As shown in Figure 4A and B, when lysosomes from human EBV-transformed cells were used, AEP clearly made a substantial contribution to the in vitro processing reaction for several though not all substrates. For example, the AEP inhibitor AENK blocked processing of ovalbumin, lactate dehydrogenase and the Neisseria meningitidis membrane protein Por A (Figure 4A and B), whereas digestion of myoglobin and the hepatitis B surface Ag (HBsAg) was more effectively blocked by pepstatin (Moss et al, 2005; Figure 4B). Other substrates including transferrin and to some extent the adr subtype of HBsAg appeared to be processed partly by leupeptin or E64-sensitive enzymes (Figure 4A and B). In some cases, for example, PorA, the dominant processing activity varied depending on whether lysosomes from DCs or EBV-B cells were used (data not shown). Where T-cell clones were available, for example, in the case of ovalbumin and myoglobin, purified AEP and cathepsin D were able to release T-cell epitopes from their respective antigen targets (Figure 4C; Moss et al, 2005). Thus, limited proteolysis by a single protease class acting on native antigen substrates may be generally sufficient to expose T-cell epitopes for class II MHC binding.
Figure 4.
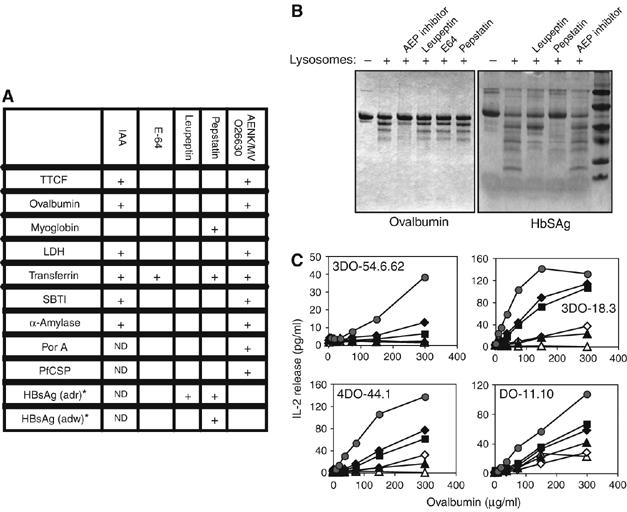
Single enzymes frequently dominate antigen processing and generate presentable antigen. (A) Pure proteins were digested with EBV-B cell-derived lysosomes in the presence of cysteine protease inhibitors iodoacetamide (IAA), E64 or leupeptin, the aspartyl protease inhibitor pepstatin or the AEP inhibitors AENK or MVO26630, and the products were analysed by SDS–PAGE. Inhibition of degradation is indicated (+). ND: not determined; SBTI, soybean trypsin inhibitor; LDH: lactate dehydrogenase; PorA: Neisseria meningitidis outer membrane protein; PfCSP: Plasmodium falciparum circumsporozoite peptide; HBsAg: hepatitis B surface antigen. *HBsAg was digested with lysosomes derived from murine spleen dendritic cells. (B) SDS–PAGE analysis of products from digestion of ovalbumin or HBsAg (adr sub-type) by human EBV-derived or murine DC-derived lysosomes, respectively. (C) AEP releases T-cell epitopes from ovalbumin. Ovalbumin was digested with AEP at 0.5 μg/ml (black triangles), 1 μg/ml (open diamonds), 5 μg/ml (black squares), 10 μg/ml (black diamonds), 20 μg/ml (grey circles) or no AEP (open triangles). A20 cells were fixed and the indicated concentration of predigested ovalbumin was added, together with ovalbumin-specific T-cell hybridomas DO-11.10, 3DO-54.6.62, 3DO-18.3 or 4DO-44.1.
Reconstructing physiological antigen processing and class II MHC binding
Compared with the true physiological situation, the experimental conditions for binding of soluble AEP processed TTCF antigen to fixed cells (Figure 3B–D) are likely to be less favourable, as most cell surface class II MHC is already peptide occupied. On the other hand, the amounts of processed antigen offered to fixed cells may be considerably higher than those found inside class II MHC processing compartments. We designed a novel experimental protocol that limits the level of antigen undergoing processing to that which can bind to specific cell surface BCRs. This also allowed us to test an earlier proposal that antigen processing in specific B cells and class II MHC peptide binding might be linked events confined to the membrane surface (Davidson and Watts, 1989; Watts et al, 1989).
We first took a soluble form of the BCR from one such clone (11.3; Lanzavecchia, 1985), mixed it with 125I-labelled TTCF and then performed an AEP digest to (i) assess whether the bound 11.3 Ig changed the pattern of processing and (ii) to confirm that, following digestion, TTCF fragments remain bound to the 11.3 Ig. As a control, we tested an Ig from a different human tetanus toxin-specific B cell (FC7) that recognises the B fragment domain rather than TTCF. As shown in Figure 5A, the initial pattern of AEP processing was essentially the same, but after 2 h, a 16 kDa 125I-labelled fragment (asterisked) was degraded more slowly in the presence of the binding 11.3 Ig compared with the non-binding FC7 Ig, indicating protection by the 11.3 Ig. Consistent with this, the more persistent fragment, but not lower-molecular-weight (M.Wt) fragments, was recovered when the 11.3 Ig was reisolated from the digestion (Figure 5A). As expected, no fragments were recovered on the FC7 antibody. A higher M.Wt processing product was also recovered on the 11.3 Ig, consistent with our finding that the different TTCF fragments remain associated following AEP digestion. The latter fragment corresponds to the N-terminal region of the molecule whereas the lower M.Wt protected fragment is a C-terminal fragment starting after the Asn 1184 cleavage site and extending to the C terminus (Simitsek et al, 1995; Manoury et al, 1998), its destruction in the absence of 11.3 Ig binding being due to eventual cleavage at the two downstream Asn residues identified in Figure 1.
Figure 5.
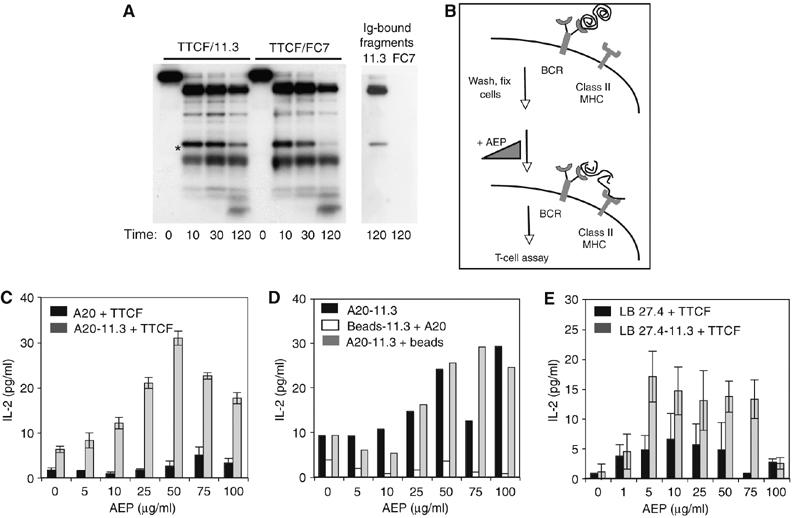
In situ processing and class II MHC capture. (A) Left panel: time course of AEP-catalysed digestion of 125I-TTCF complexed with 11.3 antibody (Ig) or control Ab, FC7. Right panel: TTCF fragments recovered on 11.3 Ab or control FC7 Ig. (B) Schematic of the cell surface processing and handover experiment. (C) Cell surface BCR-bound TTCF is presented to T cells after in situ cleavage by AEP. A20 cells transfected with 11.3 BCR (clone A20.8K; grey bars) or untransfected A20 (black bars) were loaded with TTCF on ice, washed, lightly fixed, digested with AEP as indicated and cocultured with T-cell hybridoma 4A8. (D) The source of TTCF must be on the same surface as MHC-II. Presentation to T-cell hybridoma 4A8 was tested under conditions where BCR-bound TTCF was bound and AEP digested on the surface of A20.8K without (black bars) or with Sepharose beads (grey bars). Digestion of TTCF bound to Sepharose beads linked to 11.3 Ig is not sufficient to load class II MHC on co-added A20 cells (white bars). (E) Presentation of BCR-bound TTCF to T-cell hybridoma 4E4 by 11.3 BCR-transfected LB27.4 cells (clone 4BB3; grey bars) or untransfected LB27.4 cells (black bars).
We next performed similar AEP processing of the TTCF/11.3 Ig complex, but this time when the human 11.3 Ig was expressed on the cell surface of A20 B cells (Knight et al, 1997). Our aim was to reconstruct the events of antigen processing and potentially class II MHC peptide capture but to use the cell surface as a surrogate ‘class II MHC compartment' to permit a level of control not technically possible otherwise. A20 B cells expressing the 11.3 BCR (clone A20.8K) were loaded with TTCF at 4°C, washed to remove unbound antigen and then lightly fixed to prevent antigen internalisation and processing. The cells were then exposed to graded amounts of AEP protease at 37°C after which the cells were washed again and cocultured with TTCF-specific T-cell hybridomas (Figure 5B). Under these conditions, the amount of TTCF in a 0.15 ml incubation reaction was ∼10 000 times lower (assuming ∼105 BCR per cell) than when fixed cells were incubated with 1 μg/ml of AEP-digested antigen. Remarkably, AEP processing of surface BCR-bound antigen induced the formation of class II MHC/processed antigen complexes able to trigger the H-2d-restricted TTCF-specific hybridoma 4A8, which recognises TTCF residues 925–941 (Figure 5C). T-cell stimulation was dramatically reduced when untransfected A20 cells were incubated with TTCF. Moreover, stimulation of the T-cell hybridoma was proportionate to the amount of AEP protease added but only up to certain level (∼50 mU/ml) above which the response was diminished (Figure 5C). This result raised the intriguing possibility that processed TTCF, generated while complexed to the 11.3 BCR, was being captured by class II MHC molecules on the surface of the same cell. In other words, there might be a concerted ‘handover' of processed antigen from the BCR to neighbouring class II MHC. However, it was also possible (although unlikely, given that AEP processed TTCF fragments seem to remain associated; see Figure 2) that processed TTCF diffused away from the membrane surface and was then recaptured by class II MHC molecules on the same or a different cell. To rule this out we immobilised soluble 11.3 Ig on inert (protein G) beads, loaded TTCF onto them and digested the bead-bound antigen with graded levels of AEP. The amount of bead-bound TTCF closely matched the level of TTCF bound by the A20.8K transfectants. Non BCR-transfected A20 cells were included as a source of suitable H-2d bearing class II MHC molecules and the reaction was cocultured with the 4A8 hybridoma as before. Under these conditions, no stimulation of the T-cell hybridoma was observed (Figure 5D). This was not because the beads were inhibitory, as inclusion of the same number of beads with TTCF-loaded A20.8K cells did not inhibit the processing and capture reaction (Figure 5D). It seems therefore that class II MHC molecules on the same cell are most likely involved in binding AEP-digested BCR-bound TTCF. This processing and class II MHC loading was reproducible when a different 11.3 BCR-transfected B cell (LB27.4) with a different MHC haplotype (H-2b) was used in the ‘handover' assay (Figure 5E). Moreover, two different T-cell specificities were stimulated, 4E4 (Figure 5E) and 1H7 (data not shown), recognising TTCF residues 950–966 and 1025–1041, respectively. As before, the T-cell response was largely dependent on both the presence of a TTCF-specific BCR and AEP digestion.
A role for DM in the cell surface processing and capture reaction
Within the intracellular compartments that harbour class II MHC, peptide loading is catalysed by the chaperone DM, which stabilises empty class II MHC molecules and acts as a peptide editor. DM has also been shown to be present on the surface of some human B cells and on immature human and murine DC and to catalyse peptide loading/editing at this site (Santambrogio et al, 1999; Arndt et al, 2000). We asked if DM might be playing a role in the BCR/TTCF processing and handover reaction. As shown in Figure 6A, A20 B cells, but not a DMα chain-negative variant 3A5 (Russell et al, 1999), expressed low but detectable levels of DM on their surface. We therefore performed further processing experiments in the presence of an antiserum specific for murine DM or as a control, the corresponding preimmune serum. As shown in Figure 6B, there was significant inhibition of the T-cell response by the specific serum but not by the preimmune control, indicating a role for cell surface DM in the capture of processed TTCF by class II MHC molecules. At this point, we cannot be certain if the residual class II MHC loading observed is due to partial blockade of DM function by the antibody or because of a DM-independent component of the handover reaction.
Figure 6.
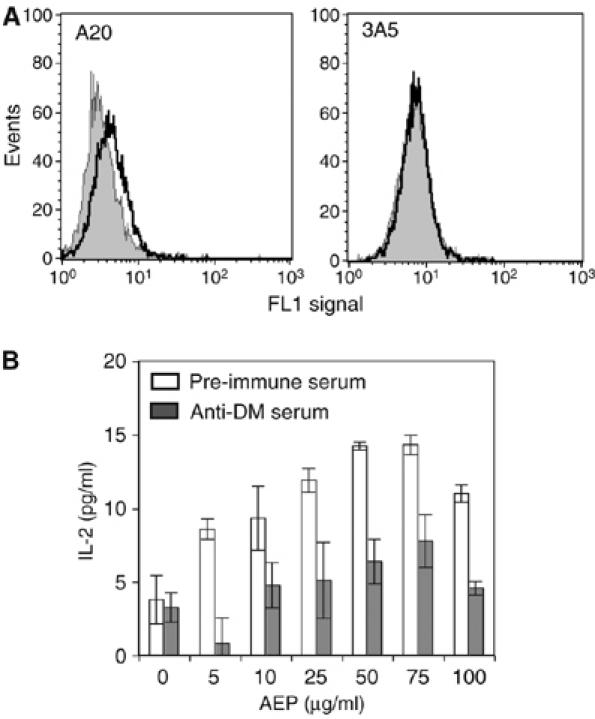
Involvement of H-2M/DM in the cell surface antigen processing and handover reaction. (A) Expression of H2-M at the cell surface. A20 cells or H2-M-negative cells 3A5 were stained with anti-H2-M serum (black) or preimmune serum (grey) and analysed by flow cytometry. (B) Blocking H2-M inhibits presentation of BCR-bound TTCF. Presentation of cell surface BCR-bound TTCF to 4A8 was carried out as in Figure 5C and E, except that A20.8K 11.3 transfectants were pretreated with anti-H2-M serum (grey bars) or preimmune serum (white bars).
Discussion
Antigen-presenting cells use the cytosolic and lysosomal proteolytic systems found in all cells and adapt them to ensure the display of a constellation of different MHC-associated peptides representative of recently encountered and recently synthesised proteins. In the class I MHC antigen processing pathway, a series of cytosolic processing, cytosol to ER transport and ER-based peptide trimming events provide ER-localised class I MHC molecules with a supply of suitable peptides (reviewed in Pamer and Cresswell, 1998; Shastri et al, 2002; Kloetzel, 2004; Rock et al, 2004; Groothuis and Neefjes, 2005). It is much less clear how antigen is prepared for class II MHC binding, even though several key processing enzymes and chaperones have been identified. The process often appears more haphazard than that for class I MHC involving largely random generation of processing products salvagable from complete destruction by fortuitous capture by class II MHC. Clear evidence that the class II MHC pathway is a hazardous environment for antigen has recently emerged. For example, the 85–99 MS-associated epitope in myelin basic protein is a target for AEP and this limits its presentation to T cells (Manoury et al, 2002). We recently showed that aspartyl protease activity released T-cell epitopes from myoglobin, but presentation actually improved when this activity was reduced in cells by elimination of the major aspartyl protease cathepsin D (Moss et al, 2005). In addition, when closely related forms of antigen that differ in their proteolytic susceptibility are compared (RNase and HRP), the more protease-resistant ones turn out to be better immunogens (Delamarre et al, 2006). More generally, dendritic cells seem to outperform macrophages as antigen-presenting cells at least in part because they express less processing enzyme activity, not more (Delamarre et al, 2005), and also because they have the capacity to attenuate protease activity by alkalinisation of phagosomal compartments (Savina et al, 2006).
There are likely to be additional mechanisms that increase the possibility of T-cell epitope binding. In contrast to class I MHC, the open-ended class II MHC binding groove can engage peptides much longer than those finally displayed on the cell surface. Early studies demonstrated the capacity of denatured but not native proteins to bind to isolated class II MHC molecules (Sette et al, 1989), and for certain proteins such as fibrinogen to be presented to T cells without processing (Lee et al, 1988). In the case of the tetanus toxin antigen, we found that processing products with apparent sizes (based on SDS–PAGE) up to 17 kDa could be eluted from class II MHC molecules following pulsing of TT-specific B cells with native 125I-labelled tetanus toxin (Davidson et al, 1991). Studies with hen egg lysozyme (HEL) illustrated the capacity of extended forms of HEL to bind to class II MHC molecules. In one study, cells pulsed with a partially denatured form of HEL could bind directly to recycling class II MHC without any processing (Lindner and Unanue, 1996), whereas in another, HEL fragment was isolated that was simultaneously bound to two different class II MHC molecules (Castellino et al, 1998). Thus, class II MHC molecules can bind extended antigen fragments, but linking the mechanics of this to antigen processing events has not previously been possible. Identification of AEP as the enzyme that initiates processing of the TTCF antigen allowed us to address this question and to test our earlier proposal that TTCF processing by AEP might be a specific example of an ‘unlocking' step that specific processing enzymes might trigger in antigens entering the class II MHC pathway (Antoniou et al, 2000). In addition, we were able to extend our earlier studies, which demonstrated that antibody-bound antigen is processed differently to free antigen and that this impacts on the presentation of T-cell epitopes (Simitsek et al, 1995; Watts et al, 1998).
In this study, we found that low levels of AEP were sufficient to generate a form of processed TTCF antigen able to bind to soluble as well as cell surface-expressed class II MHC molecules. Processing resulting in the cleavage of, on average, three principal sites, was sufficient, consistent with our earlier demonstration that one or two key cleavage sites were crucial for processing of this antigen, which indeed appears to be ‘unlocked' by these cleavages (Antoniou et al, 2000). More extensive processing by AEP, resulting in cleavage of approximately six sites, improved the ability of the antigen to engage class II MHC molecules. The limited action of AEP and the size spectrum of fragments generated illustrate the capacity of class II MHC to bind large products following processing. Interestingly, the extent of TTCF unfolding following AEP processing was more limited than we expected. IPF measurements after processing were consistent with antigen unfolding, but the cleavages introduced did not, at least in vitro, result in dissociation of the fragments or in greatly increased susceptibility to other proteases. Although initially surprising, this is consistent with the fact that when total lysosomal enzymes are used to digest TTCF, the other enzymes present do not degrade the antigen after the AEP cleavages have been introduced (Manoury et al, 1998). In contrast, complete denaturation of TTCF exposed a large number of cathepsin D and E processing sites (Hewitt et al, 1997). Clearly, further examples are needed, but these data are consistent with the possibility that class II MHC may be able to ‘probe' processed antigen conformations that still have considerable structural integrity and that are at least partially resistant to extensive processing. Although we did not analyse their physical state, two other antigens were able to bind to class II MHC and create a T-cell ligand following processing by single enzymes, AEP in the case of ovalbumin (Figure 4) and cathepsin D in the case of myoglobin (Moss et al, 2005).
To extend these studies to a more physiological setting, we revisited our earlier studies on TTCF processing in TTCF-specific B cells. In B cells with a high-affinity BCR, the substrate for processing is a BCR/TTCF complex, as the antigen does not dissociate from the BCR following endocytosis. Distinct radiolabelled antigen fragments accumulated in B cells with different epitope specificity owing to a protective or ‘footprinting' effect of the BCR Ig on its bound antigen (Davidson and Watts, 1989). In some situations, this Ig-influenced processing reaction had a striking impact on B-cell/T-cell relationships in the TTCF antigen, boosting some but inhibiting others (Watts and Lanzavecchia, 1993; Simitsek et al, 1995). Taking advantage of these earlier findings and our later demonstration that AEP initiates TTCF processing, we attempted to reconstruct processing and class II MHC capture of TTCF in as physiological a setting as possible. To gain control over the processing events, we performed this on the surface of fixed cells expressing a TTCF-specific BCR. Even though many of the conditions on the cell surface are probably much less favourable than in the class II MHC processing compartment, we found that processing by AEP of BCR-bound antigen led to the formation of antigen/class II MHC complexes. Formation of these complexes was dependent on a TTCF-specific BCR and on an optimal level of AEP. Antigen processed in situ by AEP was then ‘handed over' to neighbouring class II MHC molecules rather than diffusing away and then rebinding. Higher levels of AEP usually reduced the efficiency of generation of the antigen/MHC complex, perhaps because the fragment size spectrum was no longer optimal for the handover reaction. In the case of the 4A8 epitope, which contains asparagine residues, we cannot rule out the alternative possibility of destructive processing within the epitope, although those residues did not appear to be targeted by AEP (Figure 1).
It is worth noting that the BCR binding region and the ‘handed over' T-cell epitope need not be on a single continuous fragment. Given that the T-cell epitopes we used are quite distant from the C-terminally located 11.3 binding ‘footprint' and AEP processing sites lie in between in TTCF-H, non-contiguous fragments that remain associated non-covalently may well be involved. Thus far, the very low amounts of antigen involved in this cell surface processing and MHC capture reaction have thwarted our attempts to chemically characterise the relevant ‘handed-over' products.
It will be important to confirm that confinement of processing and class II MHC capture to the membrane plane can take place for other BCR/antigen combinations and ideally under conditions of normal antigen internalisation and intracellular processing. This currently presents a substantial technical challenge because of the very transient nature of the intermediates involved, their likely heterogeneity and the fact that BCR-internalised antigen may be recycled between the cell surface and internal compartments several times before being processed in B cells, that is, the process is very asynchronous (Davidson et al, 1990). Confining the processing and class II MHC capture reaction to the membrane surface is however attractive for a number of reasons. First, the kinetics of binding of processed antigen to suitable class II MHC molecules are likely to be faster as diffusion and collision events are confined to two rather than three dimensions. Second, these faster kinetics of binding and lack of mixing should reduce competition for class II MHC binding with peptides present in the bulk lumenal phase. Third, exposure to potentially destructive processing enzymes should be reduced. The model may also shed light on the longstanding issue of T-cell/B-cell epitope reciprocity in protein antigens (Berzofsky, 1983), that is, why B cells with a particular epitope specificity preferentially present certain T-cell epitopes. Effects of bound antibodies on T-cell epitope presentation have now been reported for autoantigens such as TPO (Quaratino et al, 2005), GAD (Jaume et al, 2002) and thyroglobulin (Dai et al, 1999), viral antigens such as gp120 (Chien et al, 2004) and HCV envelope protein (Shirai et al, 1999), bacterial antigens such as the P1 protein of Streptococcus mutans (Rhodin et al, 2004) and model antigens such as HEL (Guermonprez et al, 1999) and β-galactosidase (Manca et al, 1985). In some cases, antibody suppressed T-cell epitope presentation, but in others it was boosted, as we saw for the tetanus antigen (Simitsek et al, 1995). For example, HCV-infected individuals who did not show a T-cell response to hypervariable region 1 (HVR1) of the envelope protein did not make antibodies to this region either, although they showed both T and B responses to other regions of the same protein (Shirai et al, 1999). Conceivably, B cells specific for HVR1 are able to ‘handover' neighbouring T-cell epitopes efficiently during uptake and processing, whereas other B-cell specificities cannot.
Taken as a whole, our studies extend our understanding of the events that lie between the initiation of exogenous antigen processing and the final output of peptides on class II MHC molecules. They indicate that very limited processing may suffice to allow class II MHC binding and show that class II MHC can engage large fragments of processed antigen and present these to T cells. Moreover, where antigen is taken up via the BCR, the complete processing and class II loading reaction may be confined to the membrane surface. Our results provide direct evidence for a ‘bind first trim later' model of class II MHC peptide capture (Sercarz and Maverakis, 2003). Although shown here for the BCR, a similar mechanism might be operative for Fc receptors in other APC types that have engaged immune complexes. Finally, although we developed cell surface-restricted TTCF processing as an experimental tool and surrogate for the intracellular class II compartment, the cell surface may be a physiologically important location for class II peptide loading, as shown for dendritic cells, which have been shown to support an extracellular pathway for antigen processing and presentation (Santambrogio et al, 1999).
Materials and methods
Proteins, antibodies and other reagents
His-tagged TTCF was produced in Escherichia coli as described previously (Hewitt et al, 1997). Recombinant human AEP was produced in CHO cells and autoactivated as described (Li et al, 2003). Antibodies against TTCF have been described previously (Antoniou et al, 2000). Rabbit anti-H-2M serum (K557) and its preimmune counterpart were kindly provided by Lars Karlsson. Cathepsin L was a gift from AstraZeneca and cathepsin S a gift from Medivir UK Ltd. Recombinant human cathepsin D was purchased from Athens Research & Technology (Athens, GA, USA). Other reagents were purchased from Sigma.
Cell lines and culture conditions
The cell lines A20, LB27.4 and EBV (AK) were grown at 37°C with 5% CO2 in RPMI 1640 (Gibco) supplemented with 10% FCS, 100 μg/ml kanamycin, 2 mM glutamine, 1 mM pyruvate, non-essential amino acids and 50 μM β-mercaptoethanol. A20 cells transfected with the heavy and light chains of BCR 11.3 (A20.8K) were additionally maintained in 0.5 mg/ml G418 and 0.75 mg/ml hygromycin B and LB27.4 cells were maintained in selective medium as described (Antoniou et al, 2000). Human T-cell clones were maintained as described (Lanzavecchia, 1985). The TTCF-specific T-cell hybridomas used were 4A8, recognising residues 925–941 (Antoniou et al, 2000), and 4E4, recognising residues 950–966. Ovalbumin-specific T-cell hybridomas DO-11.10, 3DO54.6.62, 3DO18.3, 4DO-44.1 and the DMα-negative line 3A5 were kind gifts from P Marrack and K Rock, respectively.
In vitro digestions
Purified protein was digested at 0.5 mg/ml. AEP was typically added to a final concentration of 1 or 10 μg/ml, generating TTCF cleaved at an average of three sites or six sites, respectively. These two forms of cleaved TTCF are referred to as TTCF-L and TTCF-H. AEP was inactivated with 1 mM iodoacetamide and the sample dialysed extensively against PBS. Cathepsins D, S and L were used in digestions at 5 μg/ml. Total lysosomal extracts were prepared from Pala cells as described previously (Davidson et al, 1990) and typically used at 0.1 mg/ml in digestion reactions. The reaction buffer contained 50 mM sodium acetate at pH 4.5 and 5 mM DTT, and the reaction was allowed to proceed for 4 h at 37°C and the products analysed by SDS–PAGE. 125I-labelled TTCF was prepared as previously described (Davidson and Watts, 1989) to a specific activity of approximately 15 000 c.p.m./ng. The protein was mixed in PBS with a 10-fold molar excess of monoclonal anti-TTCF antibody 11.3 or control (TT B fragment specific) antibody FC7 purified from the supernatants of the 11.3 and FC7 B-cell lines. After 1–2 h, aliquots of the mixtures were diluted 10-fold in digestion buffer (0.1 M Na citrate, pH 5.0 and 0.01% CHAPS) containing 20 μg/ml AEP. At different times, the digestion was terminated by transferring to ice and analysed by electrophoresis on 15% Tris–tricine SDS gels. Labelled fragments remaining bound to 11.3 or FC7 Ig were recovered by precipitation with sheep anti-human light chain Ig and protein G–Sepharose.
Intrinsic tryptophan fluorescence and size-exclusion chromatography
After digestion with AEP, TTCF was analysed by intrinsic tryptophan fluorescence. AEP activity was quenched with 1 mM iodoacetamide and the samples diluted in 50 mM sodium phosphate pH 7.0, containing the indicated concentration of urea. Fluorescence was measured with an excitation wavelength of 280 nm and the emission between 300 and 400 nm was recorded. The ratio between 350 and 330 nm was calculated to give a measure of the degree of protein denaturation. Undigested and AEP-treated TTCF were analysed on a Superdex 200 size-exclusion column (Pharmacia) in 50 mM sodium phosphate pH 7.0 and 150 mM NaCl.
ELISAs
TTCF (5 μg/ml) in PBS was coated onto 96-well plates (Costar) overnight and then blocked in 10% FCS. TTCF-specific conformation-sensitive monoclonal antibody 15D1 (2 μg/ml) was mixed with TTCF, TTCF-L or TTCF-H at the indicated concentrations and this mixture was added to the TTCF-coated plates for 1.5 h. The binding of antibody to immobilised TTCF was detected using HRP-conjugated donkey anti-mouse antibody (Jackson).
HLA-DR4 binding assays
Binding affinities for intact and AEP-digested TTCF for HLA-DR4 were tested in vitro in a direct competition binding assay against a biotinylated indicator peptide comprising residues 98–117 of the MHC class II invariant chain. Briefly, TTCF digests or AEP alone were thoroughly dialysed against assay buffer (20 mM MES (2-[N-morpholino]ethanesulphonic acid) and 140 mM NaCl, pH 5.0) containing 1 mM iodoacetic acid. Competition assays were established by incubating 12 μg of immunoaffinity-purified HLA-DR4 with 2.5 μmol/l biotinylated indicator peptide and test protein or non-biotinylated indicator peptide at a range of concentrations (0.1–100 μmol/l in a final DMSO concentration of 20% in assay buffer supplemented with 1% w/v n-octylglucoside for 20 h at room temperature. Reaction mixtures were transferred to wells of a Maxisorp plate (Nalge Nunc, Hereford, UK) that had been precoated for 20 h at room temperature with 100 μl of anti-HLA-DR (L243) capture antibody (Astill et al, 2003). Plates were incubated for a further 1 h at room temperature and washed five times in TBST. Europium-conjugated streptavidin (Perkin Elmer Ltd, Hounslow, UK) was added at 1 μg/ml in dissociation-enhanced time-resolved fluoroimmunoassays (DELFIA) assay buffer (Wallac Oy, Turku, Finland) and incubated for 45 min at room temperature. Wells were washed a further five times in TBST and 100 μl DELFIA enhancement solution was added to each well. Fluorescent intensity was measured in a DELFIA fluorimeter. Binding assays were carried out in triplicate and all proteins were assayed simultaneously.
Antigen presentation
After digestion with AEP, TTCF was tested for its ability to bind MHC class II to the surface of EBV-B cells and stimulate autologous T-cell clones. EBV-B cells were fixed for 45 s with 0.05% glutaraldehyde, quenched in PBS, 0.1 M glycine, washed extensively and added at 1 × 105 cells/well in a round-bottomed 96-well plate together with 1 × 105 T cells and AEP-treated TTCF at the indicated concentrations. After 48 h, cells were pulsed with 1 μCi [3H]thymidine and harvested for scintillation counting 16 h later. AEP was also tested for its ability to convert ovalbumin into an MHC class II binding form. After digestion with varying concentrations of AEP, the digested ovalbumin (Worthington) was incubated at different concentrations with 1 × 105 A20 cells, fixed as above. Four different ovalbumin-specific T-cell hybridomas (1 × 105 cells) were used to assess antigen presentation. After 24 h, IL-2 release was measured by ELISA.
Presentation of BCR-bound TTCF
Presentation of cell surface BCR-bound TTCF was examined using either A20 or LB27.4 murine B cells transfected with the 11.3 TTCF-specific BCR (Antoniou et al, 2000). TTCF was incubated at 5 μg/ml with clones A20.8K or LB27.4 4BB3 (2 × 107 cells/ml) in 10% FCS on ice for 2 h. Unbound TTCF was removed by washing and the cells were fixed as above. Cells were resuspended in PBS pH 6.0 to 2 × 107 cells/ml and AEP wad added at different concentrations for 30 min at 37°C. Cells were washed extensively with RPMI containing 1% FCS and added at 2 × 105 cells/well together with 1 × 105 T-cell hybridomas. IL-2 release was measured after 24 h by ELISA.
In some cases, Sepharose beads were used as the source of TTCF. We measured the amount of biotinylated TTCF bound to 2 × 105 A20.8K cells to be 0.625 pmol (31.25 ng). Protein G beads (Sigma) carrying an equivalent amount of 11.3 BCR-bound TTCF were prepared and included as the source of TTCF in control experiments.
For H2-M blocking experiments, anti-H2-M serum (K557) or preimmune serum was added to BCR transfectants at 1/250 for 1 h on ice before addition of TTCF.
FACS analysis of surface H2-M expression
For FACS analysis, cells were washed in 1% FCS/PBS, stained with anti H2-M serum diluted in 10% FCS/PBS at 4°C and fixed for 10 min in 3% PFA. K557 serum and preimmune serum (kind gifts from Lars Karlsson) were used at 1/250, and donkey anti-rabbit FITC (Jackson) was used at 1/200 dilution.
Acknowledgments
We thank P Marrack, K Rock and A Lanzavecchia for gifts of cells, L Karlsson for anti H-2M sera, C van Els, J Reiman and G Corradin for Por A, HBsAg and PfCSP, respectively, D Campbell for protein sequencing and D Keane for technical assistance. This work was supported by a Wellcome Trust Travelling Fellowship to CXM and a Wellcome Trust Programme Grant to CW.
References
- Antoniou AN, Blackwood SL, Mazzeo D, Watts C (2000) Control of antigen presentation by a single protease cleavage site. Immunity 12: 391–398 [DOI] [PubMed] [Google Scholar]
- Arndt SO, Vogt AB, Markovic-Plese S, Martin R, Moldenhauer G, Wolpl A, Sun Y, Schadendorf D, Hammerling GJ, Kropshofer H (2000) Functional HLA-DM on the surface of B cells and immature dendritic cells. EMBO J 19: 1241–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astill TP, Ellis RJ, Arif S, Tree TI, Peakman M (2003) Promiscuous binding of proinsulin peptides to Type 1 diabetes-permissive and -protective HLA class II molecules. Diabetologia 46: 496–503 [DOI] [PubMed] [Google Scholar]
- Berzofsky JA (1983) T-B reciprocity. An Ia-restricted epitope-specific circuit regulating T cell–B cell interaction and antibody specificity. Surv Immunol Res 2: 223–229 [DOI] [PubMed] [Google Scholar]
- Castellino F, Zappacosta F, Coligan JE, Germain RN (1998) Large protein fragments as substrates for endocytic antigen capture by MHC class II molecules. J Immunol 161: 4048–4057 [PubMed] [Google Scholar]
- Chien PC Jr, Cohen S, Tuen M, Arthos J, Chen PD, Patel S, Hioe CE (2004) Human immunodeficiency virus type 1 evades T-helper responses by exploiting antibodies that suppress antigen processing. J Virol 78: 7645–7652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Carayanniotis KA, Eliades P, Lymberi P, Shepherd P, Kong Y, Carayanniotis G (1999) Enhancing or suppressive effects of antibodies on processing of a pathogenic T cell epitope in thyroglobulin. J Immunol 162: 6987–6992 [PubMed] [Google Scholar]
- Davidson HW, Reid PA, Lanzavecchia A, Watts C (1991) Processed antigen binds to newly synthesized MHC class II molecules in antigen-specific B lymphocytes. Cell 67: 105–116 [DOI] [PubMed] [Google Scholar]
- Davidson HW, Watts C (1989) Epitope-directed processing of specific antigen by B lymphocytes. J Cell Biol 109: 85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson HW, West MA, Watts C (1990) Endocytosis, intracellular trafficking, and processing of membrane IgG and monovalent antigen/membrane IgG complexes in B lymphocytes. J Immunol 144: 4101–4109 [PubMed] [Google Scholar]
- Delamarre L, Couture R, Mellman I, Trombetta ES (2006) Enhancing immunogenicity by limiting susceptibility to lysosomal proteolysis. J Exp Med 203: 2049–2055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES (2005) Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science 307: 1630–1634 [DOI] [PubMed] [Google Scholar]
- Denzin LK, Cresswell P (1995) HLA-DM induces CLIP dissociation from MHC class II alpha beta dimers and facilitates peptide loading. Cell 82: 155–165 [DOI] [PubMed] [Google Scholar]
- Engelhard VH (1994) Structure of peptides associated with class I and class II MHC molecules. Annu Rev Immunol 12: 181–207 [DOI] [PubMed] [Google Scholar]
- Groothuis TAM, Neefjes J (2005) The ins and outs of intracellular peptides and antigen presentation by class I MHC molecules. Curr Top Microbiol Immunol 300: 127–148 [DOI] [PubMed] [Google Scholar]
- Guermonprez P, Lo-Man R, Sedlik C, Rojas MJ, Poljak RJ, Leclerc C (1999) mAb against hen egg-white lysozyme regulate its presentation to CD4(+) T cells. Int Immunol 11: 1863–1872 [DOI] [PubMed] [Google Scholar]
- Hewitt EW, Treumann A, Morrice N, Tatnell PJ, Kay J, Watts C (1997) Natural processing sites for human cathepsin E and cathepsin D in tetanus toxin: implications for T cell epitope generation. J Immunol 159: 4693–4699 [PubMed] [Google Scholar]
- Jaume JC, Parry SL, Madec AM, Sonderstrup G, Baekkeskov S (2002) Suppressive effect of glutamic acid decarboxylase 65-specific autoimmune B lymphocytes on processing of T cell determinants located within the antibody epitope. J Immunol 169: 665–672 [DOI] [PubMed] [Google Scholar]
- Kloetzel P (2004) Generation of major histocompatibility complex class I antigens: functional interplay between proteasomes and TPPII. Nat Immunol 5: 661–669 [DOI] [PubMed] [Google Scholar]
- Knight AM, Lucocq JM, Prescott AR, Ponnambalam S, Watts C (1997) Antigen endocytosis and presentation mediated by human membrane IgG1 in the absence of the Igα/Igβ dimer. EMBO J 16: 3842–3850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzavecchia A (1985) Antigen-specific interaction between T and B cells. Nature 314: 537–539 [DOI] [PubMed] [Google Scholar]
- Lanzavecchia A (1990) Receptor-mediated antigen uptake and its effect on antigen presentation to class II-restricted T lymphocytes. Annu Rev Immunol 8: 773–793 [DOI] [PubMed] [Google Scholar]
- Lee P, Matsueda GR, Allen PM (1988) T cell recognition of fibrinogen. A determinant on the A alpha-chain does not require processing. J Immunol 140: 1063–1068 [PubMed] [Google Scholar]
- Li D, Matthews SP, Antoniou AN, Mazzeo D, Watts C (2003) Multistep autoactivation of asparaginyl endopeptidase in vivo and in vitro. J Biol Chem 278: 38980–38990 [DOI] [PubMed] [Google Scholar]
- Lindner R, Unanue ER (1996) Distinct antigen MHC class II complexes generated by separate processing pathways. EMBO J 15: 6910–6920 [PMC free article] [PubMed] [Google Scholar]
- Lippolis JD, White FM, Marto JA, Luckey CJ, Bullock TN, Shabanowitz J, Hunt DF, Engelhard VH (2002) Analysis of MHC class II antigen processing by quantitation of peptides that constitute nested sets. J Immunol 169: 5089–5097 [DOI] [PubMed] [Google Scholar]
- Loak K, Li DN, Manoury B, Billson J, Morton F, Hewitt E, Watts C (2003) Novel cell-permeable acyloxymethylketone inhibitors of asparaginyl endopeptidase. Biol Chem 384: 1239–1246 [DOI] [PubMed] [Google Scholar]
- Malcherek G, Gnau V, Jung G, Rammensee HG, Melms A (1995) Supermotifs enable natural invariant chain-derived peptides to interact with many major histocompatibility complex-class II molecules. J Exp Med 181: 527–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manca F, Kunkl A, Fenoglio D, Fowler A, Sercarz E, Celada F (1985) Constraints in T-B cooperation related to epitope topology on E. coli b-galactosidase I. The fine specificity of T cells dictates the fine specificity of antibodies directed to conformation-dependent determinants. Eur J Immunol 15: 345–350 [DOI] [PubMed] [Google Scholar]
- Manoury B, Hewitt EW, Morrice N, Dando PM, Barrett AJ, Watts C (1998) An asparaginyl endopeptidase processes a microbial antigen for class II MHC presentation [see comments]. Nature 396: 695–699 [DOI] [PubMed] [Google Scholar]
- Manoury B, Mazzeo D, Fugger L, Viner N, Ponsford M, Streeter H, Mazza G, Wraith DC, Watts C (2002) Destructive processing by asparagine endopeptidase limits presentation of a dominant T cell epitope in MBP. Nat Immunol 3: 169–174 [DOI] [PubMed] [Google Scholar]
- Moss CX, Villadangos JA, Watts C (2005) Destructive potential of the aspartyl protease cathepsin D in MHC class II-restricted antigen processing. Eur J Immunol 35: 3442–3451 [DOI] [PubMed] [Google Scholar]
- Nelson CA, Vidavsky I, Viner NJ, Gross ML, Unanue ER (1997) Amino-terminal trimming of peptides for presentation on major histocompatibility complex class II molecules. Proc Natl Acad Sci USA 94: 628–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamer E, Cresswell P (1998) Mechanisms of MHC class I--restricted antigen processing. Annu Rev Immunol 16: 323–358 [DOI] [PubMed] [Google Scholar]
- Quaratino S, Ruf J, Osman M, Guo J, McLachlan S, Rapoport B, Londei M (2005) Human autoantibodies modulate the T cell epitope repertoire but fail to unmask a pathogenic cryptic epitope. J Immunol 174: 557–563 [DOI] [PubMed] [Google Scholar]
- Rhodin NR, Van Tilburg ML, Oli MW, McArthur WP, Brady LJ (2004) Further characterization of immunomodulation by a monoclonal antibody against Streptococcus mutans antigen P1. Infect Immun 72: 13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock KL, York IA, Goldberg AL (2004) Post-proteasomal antigen processing for major histocompatibility complex class I presentation. Nat Immunol 5: 670–677 [DOI] [PubMed] [Google Scholar]
- Russell HI, York IA, Rock KL, Monaco JJ (1999) Class II antigen processing defects in two H2d mouse cell lines are caused by point mutations in the H2-DMa gene. Eur J Immunol 29: 905–911 [DOI] [PubMed] [Google Scholar]
- Santambrogio L, Sato AK, Carven GJ, Belyanskaya SL, Strominger JL, Stern LJ (1999) Extracellular antigen processing and presentation by immature dendritic cells. Proc Natl Acad Sci USA 96: 15056–15061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, Lennon-Dumenil AM, Seabra MC, Raposo G, Amigorena S (2006) NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell 126: 205–218 [DOI] [PubMed] [Google Scholar]
- Sercarz EE, Maverakis E (2003) MHC-guided processing: binding of large antigen fragments. Nat Rev Immunol 3: 621–629 [DOI] [PubMed] [Google Scholar]
- Sette A, Adorini L, Colon SM, Buus S, Grey HM (1989) Capacity of intact proteins to bind to MHC class II molecules. J Immunol 143: 1265–1267 [PubMed] [Google Scholar]
- Shastri N, Schwab S, Serwold T (2002) PRODUCING NATURE'S GENE-CHIPS: the generation of peptides for display by MHC class I molecules. Annu Rev Immunol 20: 463–493 [DOI] [PubMed] [Google Scholar]
- Shimonkevitz R, Kappler J, Marrack P, Grey H (1983) Antigen recognition by H-2-restricted T cells. I. Cell-free antigen processing. J Exp Med 158: 303–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai M, Arichi T, Chen M, Nishioka M, Ikeda K, Takahashi H, Enomoto N, Saito T, Major ME, Nakazawa T, Akatsuka T, Feinstone SM, Berzofsky JA (1999) T cell recognition of hypervariable region-1 from hepatitis C virus envelope protein with multiple class II MHC molecules in mice and humans: preferential help for induction of antibodies to the hypervariable region. J Immunol 162: 568–576 [PubMed] [Google Scholar]
- Simitsek PD, Campbell DG, Lanzavecchia A, Fairweather N, Watts C (1995) Modulation of antigen processing by bound antibodies can boost or suppress class II major histocompatibility complex presentation of different T cell determinants [see comments]. J Exp Med 181: 1957–1963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villadangos JA, Driessen C, Shi G-P, Chapman HA, Ploegh HL (2000) Early endosomal maturation of MHC class II molecules independently of cysteine proteases and H-2DM. EMBO J 19: 882–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts C (2001) Antigen processing in the endocytic compartment. Curr Opin Immunol 13: 26–31 [DOI] [PubMed] [Google Scholar]
- Watts C (2004) The exogenous pathway for antigen presentation on major histocompatibility complex class II and CD1 molecules. Nat Immunol 5: 685–692 [DOI] [PubMed] [Google Scholar]
- Watts C, Antoniou A, Manoury B, Hewitt EW, McKay LM, Grayson L, Fairweather NF, Emsley P, Isaacs N, Simitsek PD (1998) Modulation by epitope-specific antibodies of class II MHC-restricted presentation of the tetanus toxin antigen. Immunol Rev 164: 11–16 [DOI] [PubMed] [Google Scholar]
- Watts C, Lanzavecchia A (1993) Suppressive effect of antibody on processing of T cell epitopes. J Exp Med 178: 1459–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts C, West MA, Reid PA, Davidson HW (1989) Processing of immunoglobulin-associated antigen in B lymphocytes. Cold Spring Harb Symp Quant Biol 1: 345–352 [DOI] [PubMed] [Google Scholar]