

Abstract
The Fanconi anemia (FA) core complex plays a central role in the DNA damage response network involving breast cancer susceptibility gene products, BRCA1 and BRCA2. The complex consists of eight FA proteins, including a ubiquitin ligase (FANCL) and a DNA translocase (FANCM), and is essential for monoubiquitination of FANCD2 in response to DNA damage. Here, we report a novel component of this complex, termed FAAP100, which is essential for the stability of the core complex and directly interacts with FANCB and FANCL to form a stable subcomplex. Formation of this subcomplex protects each component from proteolytic degradation and also allows their coregulation by FANCA and FANCM during nuclear localization. Using siRNA depletion and gene knockout techniques, we show that FAAP100-deficient cells display hallmark features of FA cells, including defective FANCD2 monoubiquitination, hypersensitivity to DNA crosslinking agents, and genomic instability. Our study identifies FAAP100 as a new critical component of the FA-BRCA DNA damage response network.
Keywords: FAAP100, FANCB, FANCD2, FANCL, Fanconi anemia
Introduction
Fanconi anemia (FA) is a rare genetic disease characterized by congenital defects, bone marrow failure, chromosomal instability, and cancer susceptibility (Joenje and Patel, 2001; Kennedy and D'Andrea, 2005). A hallmark feature of this disease is that cells derived from FA patients are highly sensitive to DNA crosslinking agents such as mitomycin C (MMC) and cisplatin. For this reason, the disease has been considered as a useful model for studying the repair mechanism of DNA interstrand crosslinks (ICLs).
To date, 13 FA complementation groups (FANC-A, B, C, D1, D2, E, F, G, I, J, L, M, and N) have been described, and 12 of the corresponding disease genes have been identified (see reviews Niedernhofer et al, 2005; Thompson, 2005; Patel, 2007). The only group whose corresponding gene has not been identified is FA-I. The FA proteins participate in a DNA damage response pathway, termed the FA pathway. A key step in this pathway is the monoubiquitination of FANCD2, an event that occurs either during S-phase of the cell cycle or following a variety of DNA damages, including ICLs, double-stranded DNA breaks, and blocked replication forks (Garcia-Higuera et al, 2001; Taniguchi and D'Andrea, 2002). At least nine FA gene products, FANC-A, B, C, E, F, G, L, M, and I, act upstream of this reaction, because cells deficient in any one of these proteins are defective in FANCD2 monoubiquitination (Garcia-Higuera et al, 2001; Taniguchi and D'Andrea, 2002; Meetei et al, 2003a, 2004, 2005; Levitus et al, 2004). Conversely, three FA proteins, FANCD1, FANCN, and FANCJ, are dispensable for FANCD2 monoubiquitination and are thought to function either downstream or in parallel pathways.
Recent evidence suggests that the FA proteins may not act in a simple linear pathway, but rather, form an interactive network (Venkitaraman, 2004). Importantly, this network includes the breast cancer susceptibility gene products, BRCA1 and BRCA2. One FA gene, FANCD1, is identical to BRCA2 (Howlett et al, 2002), whereas another FA gene, FANCJ, is identical to BRIP1 (also called BACH1) (Levitus et al, 2005; Levran et al, 2005; Litman et al, 2005), a DNA helicase that interacts with BRCA1(Cantor et al, 2004). Most recently, a third FA gene, FANCN, is found to be PALB2 (Reid et al, 2007; Xia et al, 2007), which interacts with and is essential for the function of BRCA2 (Xia et al, 2006). The connection between the FA and BRCA proteins argues that defects in FA proteins not only play a role in the pathophysiology of FA, but also in genome instability and tumorigenesis in non-FA patients. Indeed, inactivation of FA genes, by either mutations or epigenetic silencing, has been reported in cancers derived from non-FA patients (Taniguchi et al, 2003; van der Heijden et al, 2003). Thus, investigation of the FA-associated protein function may provide novel insight into the general mechanism of genome maintenance and cancer prevention.
We have previously purified the FA core complex with an FANCA antibody (Meetei et al, 2003a, 2003b). This complex includes not only the five known FA proteins (FANC-A, C, E, F, and G), but also four new polypeptides, which are named FAAPs for FANCA-associated polypeptides. Three of the four FAAPs, FAAP43, FAAP95, and FAAP250, have been shown to be integral components of the FA core complex and essential for FANCD2 monoubiquitination. Moreover, the genes encoding these proteins are defective in FA complementation groups L, B, and M, respectively (Meetei et al, 2003a, 2004, 2005). Thus, eight of the nine components of the FA core complex are FA proteins (FANC-A, B, C, E, F, G, L, and M). Furthermore, two of the newly discovered FA proteins have enzymatic activities: FANCL is a ubiquitin ligase essential for FANCD2 monoubiquitination in vivo (Meetei et al, 2003a), whereas FANCM is an ortholog of archeal DNA repair protein Hef (Meetei et al, 2005; Mosedale et al, 2005), and has a DNA-stimulated ATPase and DNA translocase activity (Meetei et al, 2005). It was proposed that the entire FA core complex functions as an E3 ubiquitin ligase that monoubiquitinates FANCD2 in response to DNA damage (Meetei et al, 2005). Moreover, the core complex appears to have at least two additional functions that are also essential in repair of crosslinked DNA damage: one is to recruit monoubiquitinated FANCD2 to chromatin and the other is an uncharacterized step after FANCD2 binding to chromatin (Matsushita et al, 2005). How FA core complex accomplishes its multiple functions remains unclear.
Here, we describe a novel component of the FA core complex, termed FAAP100, which is essential for the stability and function of the complex. We demonstrate that cells depleted of FAAP100 by siRNA or cells in which the FAAP100 gene is deleted display hallmark features of FA cells. Our data suggest that FAAP100 plays an essential role in the FA DNA damage response network, and its gene could be mutated in FA patients of a novel complementation group.
Results
FAAP100 is an integral component of the FA core complex
FAAP100 is the 100 kDa polypeptide in the FA core complex purified with an FANCA antibody (Figure 1A) (Meetei et al, 2003a, 2003b). We identified this polypeptide by mass spectrometry as LOC80233, a hypothetical protein with unknown function (gene name: C17orf70; accession number: NP_079437). The LOC80233 sequence in the GenBank is incomplete. We identified a full-length cDNA for this gene, which encodes a protein of 881 amino-acid residues (accession number: DQ989324). An antibody against LOC80233 recognized the 100 kDa polypeptide in the FA complex purified by the FANCA antibody (Figure 1B), indicating that LOC80233 is FAAP100.
Figure 1.
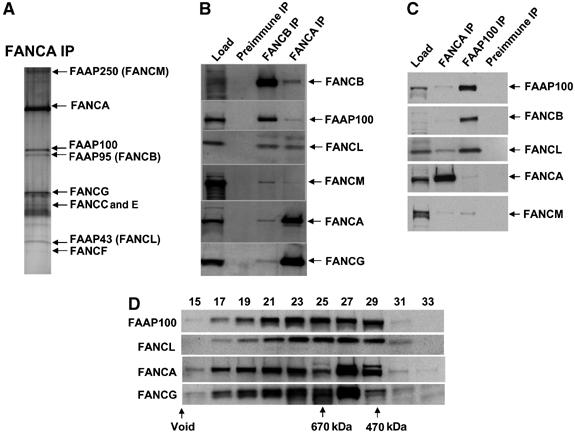
FAAP100 is a new intrinsic component of the FA core complex. (A) A silver-stained SDS gel showing the FA core complex purified by an FANCA antibody from HeLa nuclear extract (Meetei et al, 2003a). (B) Immunoblotting shows the presence of FAAP100 in the FANCA and FANCB immunoprecipitates. (C) Immunoblotting shows that the antibody against FAAP100 co-immunoprecipitated several FA core complex components. (D) Immunoblotting shows that the Superose 6 gel filtration profile of FAAP100 is coincidental with that of FANCL, and is somewhat different from that of FANCA and FANCG.
The following lines of evidence suggest that FAAP100 is an integral component of the FA core complex. First, FAAP100 was co-immunoprecipitated by antibodies against multiple core complex components, including FANCA and FANCB (Figure 1B). Second, reciprocal immunoprecipitation with the FAAP100 antibody obtained every FA core complex component that has been tested, including FANCA, FANCB, FANCL, and FANCM (Figure 1C). We noticed that the amounts of FANCB and FANCL isolated by the FAAP100 antibody are much higher than that of FANCM and FANCA, hinting that the interactions between FAAP100, FANCB, and/or FANCL could be more direct and thus stronger. Third, the gel filtration profile of FAAP100 significantly overlaps with that of several other FA proteins, indicating that these proteins may associate in one or more complexes (Figure 1D). Specifically, the profile of FAAP100 is coincidental with that of FANCL, suggesting that these two proteins are predominantly present in the same subcomplex(es) in vivo. However, the profiles of FANCA and FANCG are almost identical to each other, suggesting that the majority of these latter two proteins are present in the same complexes.
FAAP100 interacts with FANCB and FANCL
We investigated whether there is direct binding of FAAP100 to any of the other FA core complex members, using the mammalian two-hybrid assay (Medhurst et al, 2006). FAAP100 fused to the GAL4 DNA-binding domain (BD) was cotransfected with various FA proteins fused to the VP16 activation domain (AD). Only when BD-FAAP100 was coexpressed with AD-FANCB, was an induction of reporter gene activation over controls detected, indicating a direct interaction between this protein pair (Figure 2A).
Figure 2.
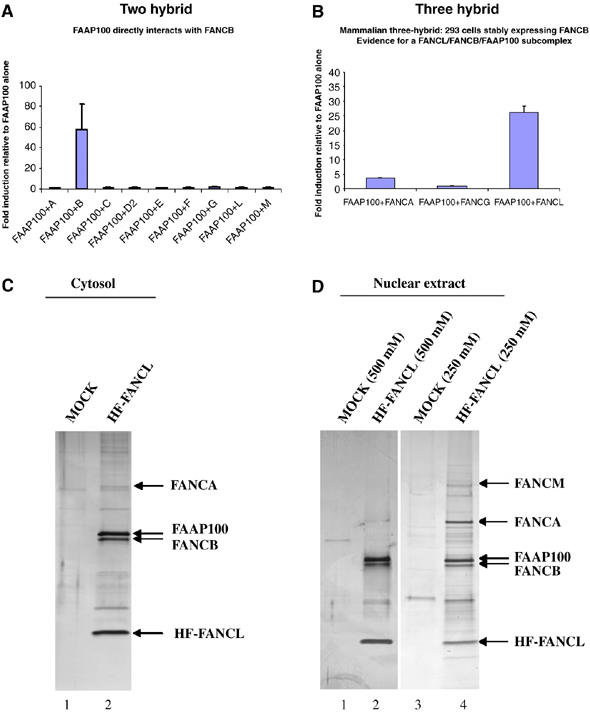
FAAP100 interacts with the FANCB and FANCL. (A) Mammalian two-hybrid assay shows direct interaction between FAAP100 and FANCB. BD-FAAP100 was cotransfected with AD-FA proteins and a luciferase reporter construct to test for direct interactions in HEK 293 cells. Fold induction of luciferase expression is relative to FAAP100 alone. Each experimental data set was obtained in triplicate. (B) Mammalian three-hybrid assay shows that the interaction between FAAP100 and FANCL depends on the presence of FANCB. HEK 293 cells stably expressing FLAG-FANCB were cotransfected with the indicated protein pairs and assayed for luciferase activation. Fold induction relative to reporter gene activation when full-length FAAP100 is expressed alone. Each experimental data set was obtained in triplicate. (C, D) Silver-stained SDS gels showing HF-FANCL-associated polypeptides purified from either cytosol (C), or nuclear extract (D), of HeLa cells by tandem-affinity purification. The major polypeptides identified by mass spectrometry are indicated by arrows. The ‘MOCK' lanes have control samples that were obtained by purification using HeLa cells that do not express HF-FANCL. The salt concentration of the washing buffer (250 or 500 mM) used in affinity purification was indicated.
We also performed three-hydrid analysis in human embryonic kidney (HEK) 293 cells that stably express FANCB. The rationale is that the interactions between FAAP100 and some other core complex components may depend on the presence of FANCB. Coexpression of BD-FAAP100 with AD-FANCA or AD-FANCG in these cells did not activate the reporter gene over background levels (Figure 2B). However, when BD-FAAP100 and AD-FANCL were coexpressed, there was a strong activation of the reporter gene (26-fold higher than controls). These data are consistent with the co-immunoprecipitation and fractionation data presented above (Figure 1), and suggest that FAAP100 may form a subcomplex with FANCB and FANCL through direct interaction.
A stable subcomplex containing FANCL, FANCB, and FAAP100 can be biochemically purified
The importance of FANCL as the crucial catalytic subunit of the FA core complex prompted us to purify its associated proteins with an unbiased biochemical approach. This should also test more directly the issue whether FANCL indeed forms a stable subcomplex with FAAP100 and FANCB. We generated a stable HeLa cell line expressing FANCL linked to two tags, a FLAG epitope and a 6-histidine tag, to allow its tandem-affinity purification with a FLAG antibody and a metal affinity column, respectively. This fusion protein, termed HF-FANCL, can complement an FA-L patient cell line (Supplementary Figure 1A and B), indicating that the tags do not interfere with the function of the protein. The majority of FAAP100 co-immunoprecipitated with HF-FANCL, as evidenced by significant depletion of FAAP100 after HF-FANCL precipitation (Supplementary Figure 1C), which is consistent with the notion that the majority of these two proteins coexist in vivo.
HF-FANCL-associated proteins from both cytosol and nuclear extracts of HeLa cells were isolated and analyzed by mass spectrometry. In the cytosol preparation, three major polypeptides were obtained, which were identified as HF-FANCL, FAAP100, and FANCB (Figure 2C). The levels of these three polypeptides are roughly stoichiometric, consistent with the three-hybrid data that suggested an FAAP100/FANCB/FANCL subcomplex. Below, we abbreviate this complex as L-B-P100. FANCA was the only other FA protein identified by mass spectrometry, but its level was considerably lower than that in L-B-P100. These data suggest that L-B-P100 could represent the most abundant form of FANCL in the cytosol, with only a small fraction in association with FANCA. The fact that FAAP100 can be purified independently by antibodies against two different FA core complex subunits (FANCA and FANCL) provides further evidence that FAAP100 is an integral component of the same complex.
In the nuclear preparation of HF-FANCL-associated proteins, three major polypeptides were also identified as HF-FANCL, FANCB, and FAAP100 (Figure 2D, lane 4). The stoichiometry of the three is indistinguishable from that of the cytosolic L-B-P100 complex, suggesting that they may have remained together as a unit when imported into the nucleus. Two other HF-FANCL-associated proteins were found to be FANCA and FANCM. The level of FANCA is roughly stoichiometric to proteins in L-B-P100, whereas that of FANCM appears to be substoichiometric. Other FA proteins may also be present but at levels too low to be detected by mass spectrometry (we detected the presence of FANCG by immunoblotting; data not shown).
When HF-FANCL-associated proteins were purified from nuclear extract under high-salt washing conditions, only FAAP100 and FANCB remained in stoichiometric association with HF-FANCL, whereas FANCA and FANCM were largely removed (Figure 2D, lane 2). These data provide biochemical evidence for existence of an L-B-P100 subcomplex module within the FA core complex, and suggest that the interactions within this module are stronger than those between this module and the other components of the core complex.
FAAP100 is conserved in vertebrates and contains a putative coiled-coil domain
Searching the genomic sequence databases with the BLAST algorithm revealed that FAAP100 protein is conserved only in vertebrates including mouse, Xenopus, and fish Tetraodon nigroviridis (Supplementary Figure 2), but not in invertebrates and yeast. This feature of FAAP100 is shared by several other components of the FA core complex, including FANC-A, B, C, E, F, and G, suggesting that the genes encoding these proteins may have arisen at about the same time during evolution.
Examination of FAAP100 sequence revealed the presence of a putative coiled-coil domain (also called leucine-zipper) (Supplementary Figure 2). This domain could mediate protein–protein interactions by either homodimerization (interacting with the same domain) or heterodimerization (interacting with a different coiled-coil domain). Potential coiled-coil domains are present in FANCA and FANCG (Lo Ten Foe et al, 1996; Demuth et al, 2000), which directly interact with each other (Garcia-Higuera et al, 1999; Waisfisz et al, 1999). We found that FANCB, the protein that directly interacts with FAAP100 in the two-hybrid assay (Figure 2A), may also contain a coiled-coil domain (Supplementary Figure 3). It remains to be determined whether the interaction between FAAP100 and FANCB is mediated through these domains.
FAAP100 is essential for the monoubiquitination of FANCD2 and the stability of the FA core complex
We depleted FAAP100 in HeLa cells by two different siRNA oligos and found that these cells not only have reduced levels of FAAP100 but also of other components of the FA core complex, including FANC-B, L, A, and G (Figure 3A). This feature mimics that of FANCB-depleted HeLa cells, which showed a reduced level of FANCL (Meetei et al, 2004). The data suggest that FAAP100, like FANCB, is required for stability of the FA core complex. Importantly, FAAP100-depleted cells display reduced levels of monoubiquitinated FANCD2 both in the absence and presence of DNA-damaging agents such as MMC, cisplatin, and hydroxyurea (Figure 3B and C). This feature is shared by cells depleted of other FA core complex components (Meetei et al, 2003a, 2004, 2005), indicating that FAAP100 is an essential part of the FA core complex required for FANCD2 monoubiquitination.
Figure 3.
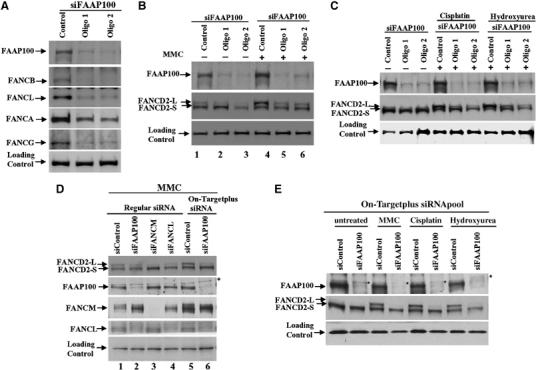
FAAP100 is essential for FANCD2 monoubiquitination and the stability of the FA core complex. (A) Immunoblotting shows that HeLa cells depleted of FAAP100 by two different siRNA oligos not only have reduced levels of FAAP100, but also of several other FA core complex components, including FANCB, FANCL, FANCA, and FANCG. HeLa cells transfected with a scrambled siRNA oligo are shown as control. The image of FAAP100 depletion (top panel) was reproduced from the lanes 1–3 of the top panel in (B), to allow easy comparison of the reduction of FAAP100 protein level with those of other FA proteins. (B, C) Immunoblotting shows that HeLa cells depleted of FAAP100 display reduced level of monoubiquitinated FANCD2 in cells that are either untreated (−), or treated with mitomycin C (MMC), cisplatin, or hydroxyurea (+). The monoubiquitinated and non-ubiquitinated FANCD2 proteins are indicated by FANCD2-L and FANCD2-S, respectively. (D) Immunoblotting shows that HeLa cells depleted of FAAP100 by On-Targetplus SMARTpool siRNAs display a level of monoubiquitinated FANCD2 comparable to cells depleted of FANCM or FANCL (compare the relative ratio between FANCD2-L and FANCD2-S in lane 6 with those in lanes 3 and 4). In addition, cells depleted by On-Target siRNAs have lower levels of FAAP100 and monoubiquitinated FANCD2 (compare lane 6 with lane 2). A nonspecific polypeptide was marked with an asterisk, which was resolved from FAAP100 using a 6% SDS gel. (E) Immunoblotting shows that HeLa cells depleted of FAAP100 by On-Targetplus SMARTpool siRNAs (siFAAP100) reduced monoubiquitination of FANCD2 in the presence of several DNA-damaging drugs. Note that the FAAP100 level in depleted cells is similar that of a nonspecific polypeptide (marked by an asterisk), indicating that its level is very low and close to the detection limit of the antibody. The cell lysis buffer in (A), (B) and (C) includes 8 M urea, which allows more efficient extraction of FANCD2-L and FANCB. The cell lysis buffer in (D) and (E), and those used in previous studies of FANCL, FANCB, and FANCM, do not include urea. Thus, the results in (D) and (E) should be more suitable for comparison with the previous studies.
We noticed that the level of monoubiquitinated FANCD2 in FAAP100-depleted cells is similar to that of cells depleted of FANCB (Meetei et al, 2004), but higher than those depleted of FANCL or FANCM (Meetei et al, 2003a, 2005). One possible explanation is that FAAP100 and FANCB are non-enzymatic components of the core complex, and are therefore less important for FANCD2 monoubiquitination than the enzymatic components, FANCL and FANCM. Another possibility is that both non-enzymatic and enzymatic subunits are of equal importance, but the former proteins are depleted less efficiently than the latter. The fact that cells from FA patients with mutations in the FANCB, FANCL, and FANCM genes all have complete absence of monoubiquitinated FANCD2 supports the second possibility (Meetei et al, 2003a, 2004, 2005). To distinguish further between these two possibilities, we utilized an improved siRNA technique, On-Targetplus SMARTpool siRNAs, to deplete FAAP100. These siRNAs are designed by better bioinformatics tools and chemically modified to reduce significantly the off-target effects of siRNA. The On-Targetplus siRNAs reduced FAAP100 protein level by about 90% (data not shown), which is more efficient than by regular siRNAs (Figure 3D, compare lane 6 to lane 2; and Figure 3E). Importantly, the new siRNAs decreased monoubiquitinated FANCD2 to levels comparable to cells depleted of FANCL or FANCM (Figure 3D, compare lane 6 with lanes 2 and 3; and E), which supports the hypothesis that FAAP100 is as important for FANCD2 monoubiquitination as FANCL and FANCM.
Consistent with results obtained by using regular siRNAs (Figure 3A), the On-Targetplus siRNAs also reduced the levels of several subunits of the FA core complex, in particular FANCL and FANCB (Supplementary Figure 4A and B). The findings support the notion that FAAP100 is essential for stability of the FA core complex.
In contrast to cells depleted of FANCL or FANCM (Meetei et al, 2003a, 2005), HeLa cells depleted of FAAP100 by either regular or On-Targetplus siRNA have no significant difference in MMC-induced chromosome breakage levels and MMC-sensitivity compared with cells treated with control siRNAs (data not shown). This could be due to the fact that the siRNA depletion is incomplete, so that the FAAP100 protein level fails to reach the critical threshold to disrupt its normal function in the latter two processes. In support of this possibility, we noticed that there was still a residual level of FAAP100 in FAAP100-silenced cells that co-immunoprecipitated with FANCB (Supplementary Figure 4B). Moreover, several studies of the FA pathway have shown that siRNA-depleted cells often do not show full FA phenotypes. For example, cells depleted of several FA genes show reduced monoubiquitinated FANCD2 levels and about 2–3-fold increase in MMC sensitivity, whereas cells with mutations in FA genes show complete absence of monoubiquitinated FANCD2 and about 10-fold increase in MMC sensitivity (Meetei et al, 2003a, 2004, 2005; Xia et al, 2006, 2007). To define unambiguously the role of FAAP100, its complete inactivation by either pathogenic mutations in FA patients or gene knockout is required. The data described below show that FAAP100 knockout cells do have full FA phenotypes indistinguishable from cells inactivated of other FA genes. Therefore, lack of the full FA phenotype in FAAP100-depleted cells is most likely due to the technical limitation of siRNA that fails to completely inactivate the protein.
The stability of FAAP100 depends on FANCB and FANCL
During the screening of FA patients for FAAP100 mutations, we found that the FAAP100 protein level is generally lower in lymphoblastoid cells derived from FA-B patients (Supplementary Figure 5 and data not shown). We examined specifically cells derived from two FA-B patients, HSC230 and EUFA178, and found that the FAAP100 protein level is lower in both the nuclear and cytoplasmic extracts compared to those from a wild type lymphoblastoid cell line (Figure 4A). The results indicate that the stability of FAAP100 depends on FANCB.
Figure 4.
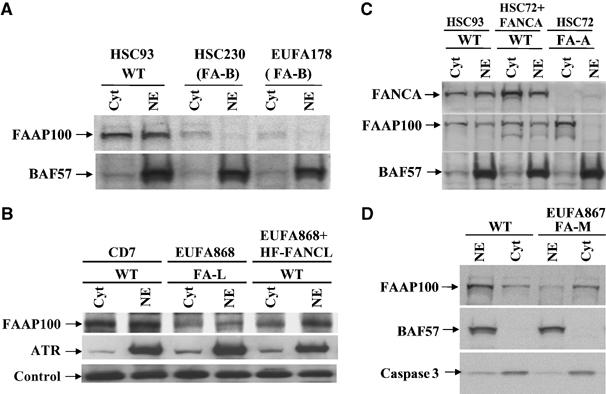
FAAP100 is unstable in FA patient cells deficient of FANCB or FANCL, and its nuclear localization is defective in cells deficient of FANCA or FANCM. (A) Immunoblotting shows that the FAAP100 protein level is reduced in nuclear (NE) and cytoplasmic (cyt) extracts of two different FA patient cell lines deficient of FANCB (FA-B). A cell line from a normal individual, HSC93, was included as a wild-type control (WT). BAF57, a component of SWI/SNF chromatin remodeling complex, was used as a loading control. (B) Immunoblotting shows that the FAAP100 protein level is reduced in the FA patient cell line deficient of FANCL (FA-L). Its level is increased when the same cell line was complemented by reintroduction of HF-FANCL (‘WT') (Supplementary Figure 1). ATR was used as a control for nuclear extract and a crossreactive band was used as a loading control. (C) Immunoblotting shows that the level of FAAP100 is strongly reduced in the nuclear extract of a FA patient cell line (HSC72) deficient of FANCA (FA-A). Importantly, in the same cell line complemented with expression of exogenous FANCA (HSC72+FANCA), the level of FAAP100 in nuclear extract becomes normal. (D) Immunoblotting reveals that the level of FAAP100 is reduced in the nuclear extract of a FA patient cell line (EUFA867) deficient of FANCM. BAF57 and caspase 3 are loading controls for nuclear and cytosolic extracts, respectively.
We found that the FAAP100 protein level is also low in the FA-L patient-derived cell line, EUFA868, and that this level became higher when the same cells were complemented by HF-FANCL (Figure 4B and Supplementary Figure 1D). These results suggest that the stability of FAAP100 depends on both its partners in the L-B-P100 subcomplex. Combined with previous findings that the level of FANCL is reduced in FA-B cells (Meetei et al, 2004), and the siRNA data showing reduced levels of FANCL and FANCB in FAAP100-depleted cells (Figure 3 and Supplementary Figure 4), our results suggest that assembly of the L-B-P100 subcomplex stabilizes each component and protects them against degradation.
The nuclear localization of FAAP100 depends on FANCA and FANCM
We found that the nuclear localization of FAAP100 is deficient in a patient cell line with mutations in FANCA (FA-A), and that this deficiency is corrected when the same cell line is complemented with exogenous FANCA (Figure 4C). This property mimics that of FANCB and FANCL, whose nuclear localization also depends on FANCA (Meetei et al, 2003a, 2004). These data imply that the entire L-B-P100 subcomplex could be coregulated by FANCA during their import into the nucleus or the assembly of the nuclear FA core complex.
The level of FAAP100 is also reduced in the nuclear extract of a FA patient cell line deficient of FANCM (Figure 4D). This feature resembles that of FANCL and FANCA, both of which show reduced levels in the nuclear extract of FA-M cells (Meetei et al, 2005), suggesting that FANCM may regulate nuclear localization of both L-B-P100 and FANCA.
DT40 cells inactivated of FAAP100 display hallmark features of FA cells
Recently, some results based on siRNA were found to be artifacts due to off-target effects (Ma et al, 2006). To exclude the possibility of making a similar mistake, and also to overcome the inability of the siRNA to produce a full FA phenotype in HeLa cells, we disrupted FAAP100 in the chicken DT40 cells. This B cell line has been used extensively for functional analysis of the FA genes (Niedzwiedz et al, 2004; Matsushita et al, 2005), and even played a role in the discovery of FANCJ (Bridge et al, 2005; Levitus et al, 2005).
We cloned chicken FAAP100 cDNA (DDBJ accession number AB270761) (Supplementary Figure 2) and genomic DNA. We made a targeting vector (Supplementary Figure 6A) and inactivated both alleles of the gene, as evidenced by Southern blot and RT–PCR analysis (Supplementary Figure 6B and C).
Molecular analyses of the FAAP100-null cells revealed that they display all hallmark features of FA cells. First, these cells have no detectable level of monoubiquitinated FANCD2 in response to a DNA crosslinking drug, MMC (Figure 5A). This result is in accord with the siRNA data in HeLa cells, and further supports the notion that FAAP100 is an essential component of the core complex and necessary for FANCD2 monoubiquitination. Second, FAAP100-null cells are extremely sensitive to DNA crosslinking drugs cisplatin and MMC (Figure 5B and C). Third, FAAP100-deficient cells show increased levels of MMC-induced chromosomal aberrations, while spontaneous chromosomal aberrations were only slightly elevated (0.03 versus 0.1 per cell for wild-type and FAAP100-deficient cells, respectively) (Figure 5D). Collectively, these findings clearly show that a defect in FAAP100 produces FA-like cellular phenotypes, and we conclude that FAAP100 plays an essential role in the FA-associated DNA damage response network.
Figure 5.
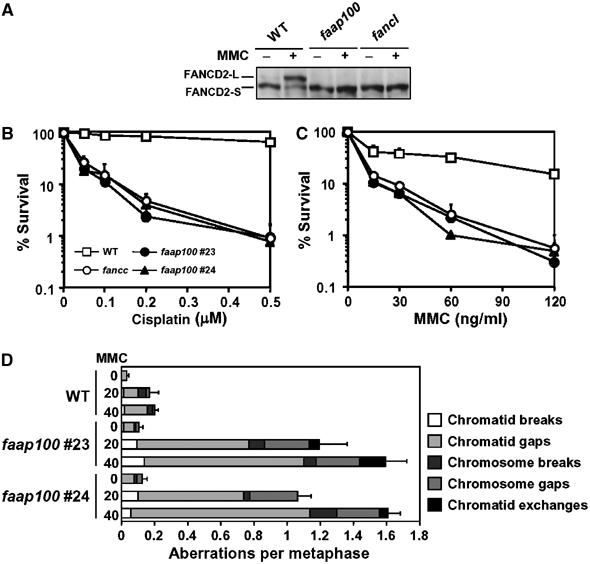
Chicken DT40 cells in which the FAAP100 gene was inactivated display hallmark features of FA cells. (A) Immunoblotting shows that DT40 cells nullizygous for FAAP100 have no detectable monoubiquitinated FANCD2. The whole-cell lysates prepared from wild-type (WT) and FANCL-deficient (fancl) cells were included as positive and negative controls, respectively. Cells were either treated with MMC (500 ng/ml) for 6 h or left untreated. (B, C) The colony survival assays show that faap100-null cells are hypersensitive to DNA crosslinking drugs cisplatin and MMC. The percentage of the surviving colonies following treatment with the drugs at the indicated concentrations are shown. Two clones of faap100 cells were included in the analysis. The data shown are mean±standard deviation of at least three separate experiments. Wild-type and FANCC-knockout (fancc) cells were used as controls. (D) The chromosomal breakage analysis revealed that faap100-null cells have increased chromosomal abberations. Wild-type and two clones of faap100 cells were treated with MMC (20 or 40 ng/ml) for 24 h or left untreated. Chromosome aberrations were counted blindly using coded slides as described (Yamamoto et al, 2003). The counting was performed only on large chromosomes (total 12). Error bars represent standard error of total aberrations per metaphase. At least 150 (for wild-type) or 50 cells (for faap100) were scored for each preparation.
Discussion
We have identified FAAP100 as a new component of the FA core complex and shown that it is required for core complex stability and a key function of the complex: FANCD2 monoubiquitination. Together with previous data showing that eight other components of this complex are all required for this ubiquitination, our results are consistent with the notion that the entire core complex works in concert as an ubiquitin ligase that monoubiquitinates FANCD2 (Meetei et al, 2005). Moreover, FAAP100 inactivation in DT40 cells results in cellular phenotypes that are hallmark features of FA cells, including hypersensitivity to DNA crosslinking drugs and MMC-induced chromosomal breakage. We therefore conclude that FAAP100 is an essential component of the FA-BRCA-associated DNA damage response network and its defects in humans could cause FA.
We have failed to identify individuals with FAAP100 mutations after extensively screening unclassified FA patients by Western blotting and sequencing (Supplementary Figure 5; Supplementary Note and data not shown). FA-I is the only complementation group whose corresponding gene has not been identified. However, the FAAP100 protein level is detectable in cell lines from FA-I patients, suggesting that the FAAP100 gene is not FANCI (data not shown). Several FA complementation groups, like L and M, have extremely few patients. A recent screening of over 1000 patients identified a total of two FA-L and one FA-M families (Meetei et al, 2003a, 2005) (data not shown). Possibly, FAAP100 patients may be as scarce as those of L and M groups, so that their number is zero in the current repositories. Knocking out the FAAP100 gene in mice may provide some clues to the function of FAAP100 in mammals.
By three-hybrid interaction study and biochemical purification, we show that FAAP100 is a crucial structural component of a subcomplex, which also contains FANCB and the ubiquitin ligase, FANCL. This subcomplex seems to be the major form in which FANCL exists in the cytosol (Figure 2C), and could also be a structural unit within the nuclear FANCL-associated complex, because it can be purified from nuclear extract in its entirety under conditions that removed the other loosely associated FA proteins (Figure 2D). Formation of the L-B-P100 subcomplex could be important in at least two aspects. First, it protects each component from degradation, because in the absence of any one subunit, the other two become unstable (Figure 3 and Supplementary Figure 4) (Meetei et al, 2004). Second, it may allow them to work as a unit, rather than as three separate proteins, during nuclear transport or other processes leading to assembly of the full complex. In support of this, each member of the L-B-P100 subcomplex has a disturbed nuclear localization in FA-A cells, although there is no detectable interaction between FAAP100 and FANCA in two- or three-hybrid interaction studies. The most plausible explanation is that FANCA interacts with FANCB and FANCL (Medhurst et al, 2006) and thereby regulates nuclear transport for the entire B-L-P100 complex, which includes FAAP100. As another line of evidence, both FAAP100 and FANCL also depend on FANCM for nuclear localization (Figure 4) (Meetei et al, 2005), which is consistent with their coregulation as one unit.
Importantly, our biochemical purification revealed a differential association between L-B-P100 subcomplex and its two regulators (FANCA and FANCM) in nuclear versus cytosolic fractions: the nuclear form of L-B-P100 associates with a near stoichiometric amount of FANCA and substoichiometric amounts of FANCM, whereas the cytoplasmic form contains a low level of FANCA and non-detectable levels of FANCM (Figure 2C and D). This differential association suggests a simple model, which could account for the positive effects of FANCA and FANCM in promoting nuclear localization of L-B-P100: the L-B-P100 complex is normally present in the cytosol, and moves to the nucleus when it is associated with FANCA and FANCM. It may be worth noting that in cells lacking FANCA, the nuclear localization of FANCM is normal (Meetei et al, 2005), whereas that of L-B-P100 is defective (Figure 4; Meetei et al, 2005), indicating that FANCM itself is insufficient to recruit L-B-P100 to nucleus. In contrast, in cells lacking FANCM, the nuclear localization of both FANCA and L-B-P100 are deficient. These data suggest a regulatory hierarchy during nuclear localization of FA core complex: FANCM recruits FANCA, which subsequently recruits L-B-P100 to nucleus. So far, FANCM is the only known FA core complex component with DNA-interacting domains and activities, which could allow it to constitutively bind DNA and recruit other components of the core complex. We recently identified a new DNA-binding component of the FA core complex, named FAAP24, which directly interacts with the C-terminal domain of FANCM and may cooperate with FANCM to target the FA core complex to damaged DNA (Ciccia et al, 2007).
We suggest that the L-B-P100 subcomplex could be an important intermediate generated during assembly of the entire FA core complex. In general, the expression levels of components in a multiprotein complex are often coregulated to ensure proper assembly and function of the entire complex (Chen and Archer, 2005). Components that fail to interact with other subunits are often degraded, possibly to avoid interference of the function of the whole complex. The FA core complex consists of at least nine proteins, and it would be difficult to envision that all of them are coregulated and assembled at the same time. The more likely scenario is that the assembly may involve several intermediate subcomplexes. On the basis of the several lines of evidence, including co-immunoprecipitation, biochemical purification and assays, two- and three-hybrid interaction studies, and interdependence of subunits on stability and nuclear localization, we speculate that the FA core complex may consist of four subcomplexes: one, FANCA and FANCG; two, FANCB, FANCL, and FAAP100; three, FANCC, FANCE, and FANCF; and four, FANCM and FAAP24 (see Figure 6 for a hypothetical model). Biochemical purification of these intermediates may allow eventual reconstitution of this 10-subunit complex and understanding of its mechanism.
Figure 6.
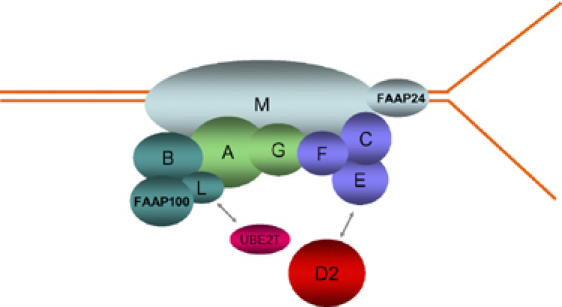
A hypothetical model for association of four subcomplexes in the FA core complex. The four predicted subcomplexes are indicated by different colors. FAAP24 is a new component of the FA core complex that interacts with the C-terminus of FANCM and may target FANCM to structured DNA that mimics intermediates formed during the replication or repair of damaged DNA (Ciccia et al, 2007). FANCD2 (red) and the E2 ubiquitin conjugating enzyme UBE2T (pink) may interact with FANCE (Pace et al, 2002) and FANCL (Machida et al, 2006), respectively.
Materials and methods
Cell culture
HeLa cells were maintained in DMEM medium with 10% fetal calf serum. Lymphoblastoid cells HSC93 (wild type), HSC72, HSC72+FANCA, EUFA867 (FA-M), EUFA868 (FA-L) and HSC230 (FA-B), and EUFA178 (FA-B) were cultured in RPMI-1640 medium with 10% fetal calf serum. Wild-type and mutant chicken DT40 cells were cultured in RPMI-1640 medium supplemented with 10% fetal calf serum, 1% chicken serum, 2 mM L-glutamine, 50 μM 2-mercaptoethanol, penicillin, and streptomycin in a 5% CO2 incubator at 39.5°C. FANCL- and FANCC-deficient DT40 cells were described previously (Hirano et al, 2005; Matsushita et al, 2005). Electroporation and subsequent selection were performed as described (Yamamoto et al, 2003).
Purification and identification of FAAP100
The FA core complex was immunopurified from HeLa nuclear extract with an antibody to FANCA as described (Meetei et al, 2003a, 2003b). The FAAP100 protein was identified by liquid chromatography and mass spectrometry analysis; the mass spectrometry data are available on request. A polyclonal rabbit antibody to FAAP100 was generated by using a chimeric protein containing a region of FAAP100 (residues 671–730) fused to the maltose-binding protein as an immunogen. This fusion protein was expressed and purified from Escherichia coli in accordance with the manufacturer's protocols (New England Biolabs) and used for both immunization and purification of the antibody. The immunoprecipitation conditions were previously described. Antibodies against FANCA, FANCB, FANCL, FANCG, FANCM, and BAF57 used for immunoprecipitation and immunoblotting have been described (Meetei et al, 2003a, 2004, 2005).
Cell extraction and fractionation
The nuclear and cytoplasmic extracts were prepared as described (Meetei et al, 2003b). For gel filtration analysis, HeLa nuclear extract (2 mg) was directly applied to a Superose 6 column (HR10/30; Amersham) equilibrated with column running buffer containing 20 mM HEPES (pH 7.9), 200 mM NaCl, 1 mM dithiothreitol, 0.1 mM phenylmethylsulfonyl fluoride, and 10% glycerol. The fractions of 0.5 ml were collected and analyzed by immunoblotting.
siRNA experiment
For experiments using regular siRNA oligos, two 19-nucleotide double-stranded siRNA oligos against FAAP100 (oligo 1: AACCAACGUUCACCUCAUCGU and oligo 2: AAGUGGCUCCUUGCUGAGAAU) and a scrambled nonspecific control oligo (GUAUAUAAGCAAGCAUUACUU) were purchased from Dharmacon Inc. FANCL and FANCM siRNA oligos have been described (Meetei et al, 2003a, 2003b, 2005). HeLa cells were transfected with the siRNA oligos by using Oligofectamine (Invitrogen) according to the manufacturer's protocol. At 48 h post-infection, cells were either untreated or treated with MMC (100 ng/ml), cisplatin (10 μM), or hydroxyurea (1 mM) for 24 h. At the 72 h time point, the cells were harvested, extracted with 8 M urea, and analyzed by immunoblotting.
For experiments using On-Targetplus SMARTpool siRNAs (Figure 3D and E and Supplementary Figure 4), the FAAP100 siRNA pool (L-017139-01, human C17orf70, NM_025161) and the non-targeting control pool (D-001810-10-20) were purchased from Dharmacon Inc. They were similarly transfected into HeLa cells by Oligofectamine. At 56 h post-infection, the cells were either untreated or treated with drugs for 16 h. At the 72 h time point, the cells were harvested, extracted with RIPA buffer (20 mM Tris–HCl (pH 7.4), 1% NP-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM EDTA, and protease inhibitor cocktail (complete)), and analyzed by immunoblotting. For immunoprecipitation-coupled Western analysis, HeLa cells were treated with siRNA or control oligos for 72 h. They were then harvested and extracted in lysis buffer (1% Triton X-100, 350 mM NaCl, and 20 mM Tris–HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, and protease inhibitor cocktail (complete)). Experimental conditions for immunoprecipitation (Meetei et al, 2003b), chromosomal breakage analysis (Meetei et al, 2003a), and MMC sensitivity assay (Meetei et al, 2005) have been previously described.
Mammalian two- and three-hybrid analyses
HEK 293 cells, either untransfected or stably transfected with wild-type FLAG-tagged FANCB were plated onto six-well plates. After 48 h, the cells were transiently transfected with FAAP100 cDNA fused to the GAL4 DNA binding domain (pM; Clontech) and the indicated FA constructs fused in-frame to the GAL-4 activation domain (pVP16; Clontech), together with a GAL4-driven reporter plasmid (G5E1bLUC, 0.2 μg). The luciferase activity was monitored after 24 h using a Dual-Luciferase Reporter Assay System (Promega) and a single-tube luminometer (DLReady, Berthold Detection Systems), according to the manufacturer's instructions. Each experimental data set was performed in triplicate.
Generation and analysis of FAAP100-deficient DT40 cells
Partial chicken FAAP100 cDNA sequences were identified by searching the NCBI database and then used for screening a chicken testis cDNA library (kindly provided by Shunichi Takeda, Kyoto University) and for RT–PCR from DT40 cDNA. The genomic fragments of chFAAP100 were isolated by screening a chicken genomic library (Stratagene) and PCR amplification from DT40 genomic DNA. The FAAP100 targeting vector was created by replacing a 3.2 kb genomic fragment containing five exons that correspond to chFaap100 amino acids 346–799, with a bsr- or his-resistant gene cassette. RT–PCR analysis, Western blotting, colony formation assay, and chromosome analysis were performed as described (Yamamoto et al, 2003; Ishiai et al, 2004).
Construction of retroviral vectors
Retroviral expression construct pMIEG3 has been described previously (Williams et al, 2000). The N-terminal His and FLAG tag bicistronic FANCL retroviral vector (pMIEG3-HF-FANCL) was constructed as follows. Two primers were designed to amplify FANCL, the forward primers contains an EcoRI and NotI site followed by a Kozak sequence, start codon, His tag and a FLAG tag (5′-ccggaattcggcggccgcGCCACCATGCATCA CATCACCATCACGACTACAAGGACGACGATGACA AG-3′) sequence and the reverse primers contains a unique sequence complimentary to FANCL followed by stop codon and an XhoI site (5′-cccaaagggctcgagTCAGTGTTTCCTTCCAG ACATTTT-3′). These primers were used to amplify FANCL from its expression vector containing a FLAG tag described previously (Meetei et al, 2003a). The amplified PCR fragment was digested with EcoRI and XhoI and cloned in EcoRI and XhoI sites of pMIEG3 vector to generate pMIEG3-HF-FANCL.
Generation of stable cell lines expressing HF-FANCL
Retroviral particles of pMIEG3-HF-FANCL were generated at the Viral Vector Core facility at Cincinnati Children's Hospital Medical Center. HeLa cells stably expressing HF-FANCL (HF-FANCL-HeLa) were obtained by infecting the HeLa cells with retroviral soup and sorting the cells according to their EGFP levels using a FACS Vantage cell sorter. Similarly, EUFA868 lymphocytes were infected with the virus using a previous procedure (Chandra et al, 2005) and the ability of HF-FANCL to complement EUFA868 was assessed by FANCD2 Western blot analysis after MMC and HU treatment.
Purification of HF-FANCL-associated proteins
Thirty 150-mm plates of HF-FANCL-HeLa cells were grown for 85–95% confluency. Cells were washed with phosphate-buffered saline and collected as a pellet. Cytoplasmic and nuclear extracts were prepared as described (Meetei et al, 2003b). Total cell lysate was made by lysing the cells in 20 ml of 250-lysis buffer (30 mM Hepes pH 7.9, 250 mM NaCl, 1 mM EDTA, 1% Triton 0.1% NP-40, and 10% glycerol) supplemented with complete protease inhibitors (Roche). Purification was performed in two steps. The first purification step is performed with anti-FLAG M2 agarose beads (Sigma). A 1.5 ml volume of the total cell lysate or the cytoplasmic or the nuclear extract was incubated with 20 μl anti-FLAG M2 agarose beads overnight, washed four times with 250-lysis buffer with 10 min rotation at 4°C, and eluted with 3 × FLAG peptide (Sigma) for 1 h. The second purification step was performed by incubating the eluate of the first step with 30 μl talon metal affinity resin (BD) for 3 h at 4°C, washing four times with 250-lysis buffer containing 10 mM imidazole, and eluted with 50 mM EDTA. The protein complex purified was resolved on an 8–16% gel and stained by either SilverQuest silver or colloidal blue staining kit according to manufacturer's instructions (InVitrogen). Immunodepletion of recombinant HF-FANCL from EUFA868 cell lysate complemented with HF-FANCL was carried out by incubating with 50 μl anti-FLAG M2 agarose beads overnight, washing four times with 250-lysis buffer and eluting with 3 × FLAG peptide (Sigma) for 1 h. The eluate was resolved on to 8–16% gel and immunoblotted with FANCL- and FAAP100-specific antibodies.
Supplementary Material
Supplementary Figures and Information
Acknowledgments
We thank all the families with Fanconi anemia for cooperating in this study and the many physicians who have referred families for participation in the research. We thank Dr S Takeda for chicken testis cDNA library, K Komatsu for anti-chicken FancD2 serum, N Sherman for mass spectrometry analysis, E Uchida for expert technical assistance, SD Batish, O Levran, and H Hanenberg for classification of FA patients. We thank National Cell Culture Center for providing HeLa cells. This work was supported in part by grants from the following agencies: the Ministry of Education, Culture, Sports, Science and Technology of Japan (MI and MT); Kawasaki Medical School (Project Research Grant 17-210T and 18-202Y to MT and MI, respectively); US National Institute of Health (NCI-R37HL32987 and NHLBI-R01CA82678 to ADA; NCI-CA112775 to MEH); Schroeder-Kurth Fund (DS); the intramural program of National Institute of Health, National institute on Aging (WW); the Dutch Cancer Society (JPdW) and Fanconi Anemia Research Fund (WW); and an American Society of Hematology Junior Faculty Award (ARM).
References
- Bridge WL, Vandenberg CJ, Franklin RJ, Hiom K (2005) The BRIP1 helicase functions independently of BRCA1 in the Fanconi anemia pathway for DNA crosslink repair. Nat Genet 37: 953–957 [DOI] [PubMed] [Google Scholar]
- Cantor S, Drapkin R, Zhang F, Lin Y, Han J, Pamidi S, Livingston DM (2004) The BRCA1-associated protein BACH1 is a DNA helicase targeted by clinically relevant inactivating mutations. Proc Natl Acad Sci USA 101: 2357–2362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Levran O, Jurickova I, Maas C, Kapur R, Schindler D, Henry R, Milton K, Batish SD, Cancelas JA, Hanenberg H, Auerbach AD, Williams DA (2005) A rapid method for retrovirus-mediated identification of complementation groups in Fanconi anemia patients. Mol Ther 12: 976–984 [DOI] [PubMed] [Google Scholar]
- Chen J, Archer TK (2005) Regulating SWI/SNF subunit levels via protein-protein interactions and proteasomal degradation: BAF155 and BAF170 limit expression of BAF57. Mol Cell Biol 25: 9016–9027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A, Ling C, Coulthard R, Yan Z, Xue Y, Meetei AR, Laghmani EH, Joenje H, McDonald N, de Winter J, Wang W, West SC (2007) Identification of FAAP24, a Fanconi anemia core complex protein that interacts with FANCM. Mol Cell 25: 331–343 [DOI] [PubMed] [Google Scholar]
- Demuth I, Wlodarski M, Tipping AJ, Morgan NV, de Winter JP, Thiel M, Grasl S, Schindler D, D'Andrea AD, Altay C, Kayserili H, Zatterale A, Kunze J, Ebell W, Mathew CG, Joenje H, Sperling K, Digweed M (2000) Spectrum of mutations in the Fanconi anaemia group G gene, FANCG/XRCC9. Eur J Hum Genet 8: 861–868 [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Kuang Y, Naf D, Wasik J, D'Andrea AD (1999) Fanconi anemia proteins FANCA, FANCC, and FANCG/XRCC9 interact in a functional nuclear complex. Mol Cell Biol 19: 4866–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7: 249–262 [DOI] [PubMed] [Google Scholar]
- Hirano S, Yamamoto K, Ishiai M, Yamazoe M, Seki M, Matsushita N, Ohzeki M, Yamashita YM, Arakawa H, Buerstedde JM, Enomoto T, Takeda S, Thompson LH, Takata M (2005) Functional relationships of FANCC to homologous recombination, translesion synthesis, and BLM. EMBO J 24: 418–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D'Andrea AD (2002) Biallelic inactivation of BRCA2 in Fanconi anemia. Science 297: 606–609 [DOI] [PubMed] [Google Scholar]
- Ishiai M, Kimura M, Namikoshi K, Yamazoe M, Yamamoto K, Arakawa H, Agematsu K, Matsushita N, Takeda S, Buerstedde JM, Takata M (2004) DNA cross-link repair protein SNM1A interacts with PIAS1 in nuclear focus formation. Mol Cell Biol 24: 10733–10741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joenje H, Patel KJ (2001) The emerging genetic and molecular basis of Fanconi anaemia. Nat Rev Genet 2: 446–459 [DOI] [PubMed] [Google Scholar]
- Kennedy RD, D'Andrea AD (2005) The Fanconi anemia/BRCA pathway: new faces in the crowd. Genes Dev 19: 2925–2940 [DOI] [PubMed] [Google Scholar]
- Levitus M, Rooimans MA, Steltenpool J, Cool NF, Oostra AB, Mathew CG, Hoatlin ME, Waisfisz Q, Arwert F, de Winter JP, Joenje H (2004) Heterogeneity in Fanconi anemia: evidence for 2 new genetic subtypes. Blood 103: 2498–2503 [DOI] [PubMed] [Google Scholar]
- Levitus M, Waisfisz Q, Godthelp BC, de Vries Y, Hussain S, Wiegant WW, Elghalbzouri-Maghrani E, Steltenpool J, Rooimans MA, Pals G, Arwert F, Mathew CG, Zdzienicka MZ, Hiom K, De Winter JP, Joenje H (2005) The DNA helicase BRIP1 is defective in Fanconi anemia complementation group. J Nat Genet 37: 934–935 [DOI] [PubMed] [Google Scholar]
- Levran O, Attwooll C, Henry RT, Milton KL, Neveling K, Rio P, Batish SD, Kalb R, Velleuer E, Barral S, Ott J, Petrini J, Schindler D, Hanenberg H, Auerbach AD (2005) The BRCA1-interacting helicase BRIP1 is deficient in Fanconi anemia. Nat Genet 37: 931–933 [DOI] [PubMed] [Google Scholar]
- Litman R, Peng M, Jin Z, Zhang F, Zhang J, Powell S, Andreassen PR, Cantor SB (2005) BACH1 is critical for homologous recombination and appears to be the Fanconi anemia gene product FANCJ. Cancer Cell 8: 255–265 [DOI] [PubMed] [Google Scholar]
- Lo Ten Foe JR, Rooimans MA, Bosnoyan-Collins L, Alon N, Wijker M, Parker L, Lightfoot J, Carreau M, Callen DF, Savoia A, Cheng NC, van Berkel CG, Strunk MH, Gille JJ, Pals G, Kruyt FA, Pronk JC, Arwert F, Buchwald M, Joenje H (1996) Expression cloning of a cDNA for the major Fanconi anaemia gene, FAA. Nat Genet 14: 320–323 [DOI] [PubMed] [Google Scholar]
- Ma Y, Creanga A, Lum L, Beachy PA (2006) Prevalence of off-target effects in Drosophila RNA interference screens. Nature 443: 359–363 [DOI] [PubMed] [Google Scholar]
- Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D'Andrea AD, Dutta A (2006) UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell 23: 589–596 [DOI] [PubMed] [Google Scholar]
- Matsushita N, Kitao H, Ishiai M, Nagashima N, Hirano S, Okawa K, Ohta T, Yu DS, McHugh PJ, Hickson ID, Venkitaraman AR, Kurumizaka H, Takata M (2005) A FancD2-monoubiquitin fusion reveals hidden functions of Fanconi anemia core complex in DNA repair. Mol Cell 19: 841–847 [DOI] [PubMed] [Google Scholar]
- Medhurst AL, Laghmani el H, Steltenpool J, Ferrer M, Fontaine C, de Groot J, Rooimans MA, Scheper RJ, Meetei AR, Wang W, Joenje H, de Winter JP (2006) Evidence for subcomplexes in the Fanconi anemia pathway. Blood 108: 2072–2080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W (2003a) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 35: 165–170 [DOI] [PubMed] [Google Scholar]
- Meetei AR, Levitus M, Xue Y, Medhurst AL, Zwaan M, Ling C, Rooimans MA, Bier P, Hoatlin M, Pals G, de Winter JP, Wang W, Joenje H (2004) X-linked inheritance of Fanconi anemia complementation group B. Nat Genet 36: 1219–1224 [DOI] [PubMed] [Google Scholar]
- Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, Hoatlin M, Joenje H, de Winter JP, Wang W (2005) A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet 37: 958–963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W (2003b) A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23: 3417–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosedale G, Niedzwiedz W, Alpi A, Perrina F, Pereira-Leal JB, Johnson M, Langevin F, Pace P, Patel KJ (2005) The vertebrate Hef ortholog is a component of the Fanconi anemia tumor-suppressor pathway. Nat Struct Mol Biol 12: 763–771 [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Lalai AS, Hoeijmakers JH (2005) Fanconi anemia (cross) linked to DNA repair. Cell 123: 1191–1198 [DOI] [PubMed] [Google Scholar]
- Niedzwiedz W, Mosedale G, Johnson M, Ong CY, Pace P, Patel KJ (2004) The Fanconi anaemia gene FANCC promotes homologous recombination and error-prone DNA repair. Mol Cell 15: 607–620 [DOI] [PubMed] [Google Scholar]
- Pace P, Johnson M, Tan WM, Mosedale G, Sng C, Hoatlin M, de Winter J, Joenje H, Gergely F, Patel KJ (2002) FANCE: the link between Fanconi anaemia complex assembley and activity. EMBOJ 21: 3414–3423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KJ (2007) Fanconi anemia and breast cancer susceptibility. Nat Genet 39: 142–143 [DOI] [PubMed] [Google Scholar]
- Reid S, Schindler D, Hanenberg H, Barker K, Hanks S, Kalb R, Neveling K, Kelly P, Seal S, Freund M, Wurm M, Batish SD, Lach FP, Yetgin S, Neitzel H, Ariffin H, Tischkowitz M, Mathew CG, Auerbach AD, Rahman N (2007) Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet 39: 162–164 [DOI] [PubMed] [Google Scholar]
- Taniguchi T, D'Andrea AD (2002) The Fanconi anemia protein, FANCE, promotes the nuclear accumulation of FANCC. Blood 100: 2457–2462 [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Tischkowitz M, Ameziane N, Hodgson SV, Mathew CG, Joenje H, Mok SC, D'Andrea AD (2003) Disruption of the Fanconi anemia-BRCA pathway in cisplatin-sensitive ovarian tumors. Nat Med 9: 568–574 [DOI] [PubMed] [Google Scholar]
- Thompson LH (2005) Unraveling the Fanconi anemi–DNA repair connection. Nat Genet 37: 921–922 [DOI] [PubMed] [Google Scholar]
- van der Heijden MS, Yeo CJ, Hruban RH, Kern SE (2003) Fanconi anemia gene mutations in young-onset pancreatic cancer. Cancer Res 63: 2585–2588 [PubMed] [Google Scholar]
- Venkitaraman AR (2004) Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat Rev Cancer 4: 266–276 [DOI] [PubMed] [Google Scholar]
- Waisfisz Q, de Winter JP, Kruyt FA, de Groot J, van der Weel L, Dijkmans LM, Zhi Y, Arwert F, Scheper RJ, Youssoufian H, Hoatlin ME, Joenje H (1999) A physical complex of the Fanconi anemia proteins FANCG/XRCC9 and FANCA. Proc Natl Acad Sci USA 96: 10320–10325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DA, Tao W, Yang F, Kim C, Gu Y, Mansfield P, Levine JE, Petryniak B, Derrow CW, Harris C, Jia B, Zheng Y, Ambruso DR, Lowe JB, Atkinson SJ, Dinauer MC, Boxer L (2000) Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood 96: 1646–1654 [PubMed] [Google Scholar]
- Xia B, Dorsman JC, Ameziane N, de Vries Y, Rooimans MA, Sheng Q, Pals G, Errami A, Gluckman E, Llera J, Wang W, Livingston DM, Joenje H, de Winter JP (2007) Fanconi anemia is associated with a defect in the BRCA2 partner PALB2. Nat Genet 39: 159–161 [DOI] [PubMed] [Google Scholar]
- Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM (2006) Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol Cell 22: 719–729 [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Ishiai M, Matsushita N, Arakawa H, Lamerdin JE, Buerstedde JM, Tanimoto M, Harada M, Thompson LH, Takata M (2003) Fanconi anemia FANCG protein in mitigating radiation- and enzyme-induced DNA double-strand breaks by homologous recombination in vertebrate cells. Mol Cell Biol 23: 5421–5430 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Information