

Abstract
Spatial and temporal modulation of intracellular Ca2+ fluxes controls the cellular response of B lymphocytes to antigen stimulation. Herein, we identify the hematopoietic adaptor protein Dok-3 (downstream of kinase-3) as a key component of negative feedback regulation in Ca2+ signaling from the B-cell antigen receptor. Dok-3 localizes at the inner leaflet of the plasma membrane and is a major substrate for activated Src family kinase Lyn. Phosphorylated Dok-3 inhibits antigen receptor-induced Ca2+ elevation by recruiting cytosolic Grb2, which acts at this location as a negative regulator of Bruton's tyrosine kinase. This leads to diminished activation of phospholipase C-γ2 and reduced production of soluble inositol trisphosphate. Hence, the Dok-3/Grb2 module is a membrane-associated signaling organizer, which orchestrates the interaction efficiency of Ca2+-mobilizing enzymes.
Keywords: adaptor proteins, B-cell activation, Ca2+ mobilization, plasma membrane recruitment, tyrosine phosphorylation
Introduction
Development, survival and activation of B lymphocytes are tightly controlled by intracellular Ca2+ ions, which act as second messengers in a wide range of signaling pathways (Gallo et al, 2006). The regulation of Ca2+ concentrations is a key function of the B-cell antigen receptor (BCR). BCR ligation triggers elevation of intracellular Ca2+ concentrations through activation of spleen tyrosine kinase Syk and subsequent phosphorylation of the adaptor protein SLP-65 (Wienands et al, 1998) (alternatively called BLNK, (Fu et al, 1998) or BASH, (Goitsuka et al, 1998)). Phosphorylated SLP-65 recruits Bruton's tyrosine kinase (Btk) and phospholipase C-γ2 (PLC-γ2) into a trimolecular Ca2+ initiation complex (Hashimoto et al, 1999; Ishiai et al, 1999a, 1999b; Su et al, 1999; Chiu et al, 2002). This allows phosphorylation-mediated activation of PLC-γ2, which in turn hydrolyzes membrane phospholipids to yield soluble inositol trisphosphate (IP3) (Kurosaki and Tsukada, 2000). IP3 receptors are ligand-gated Ca2+ channels located in the membrane of the endoplasmic reticulum (ER), which stores intracellular Ca2+. Hence, IP3 production causes the release of Ca2+ from the ER into the cytosol. The IP3-driven intracellular Ca2+ flux is followed by entry of Ca2+ from the extracellular space through weakly characterized membrane channels (Parekh and Putney, 2005; Putney, 2005). This biphasic character of the Ca2+ response allows shaping of the Ca2+ signal in the dimensions space and time, which is thought to contribute to cell fate determination during B-cell differentiation (Dolmetsch et al, 1997, 1998). Indeed, Koncz et al (2002) and Hoek et al (2006) reported differential Ca2+ signaling in BCR-activated splenic B-cell populations, which represent distinct developmental stages and are known to respond to antigen stimulation with induction of either anergy, clonal deletion or proliferation (Niiro and Clark, 2002).
Several negative regulators of the Ca2+ activation cascade have been described. Most prominently, the SH2 domain-containing 5′-inositol phosphatase (SHIP) interferes with membrane recruitment and concomitant activation of Btk or PLC-γ2 by disrupting the lipid binding motifs for the enzyme's pleckstrin homology (PH) domains at the inner leaflet of the plasma membrane (Ono et al, 1997; Bolland et al, 1998; Okada et al, 1998; Kim et al, 1999; Brauweiler et al, 2000). Also the protein tyrosine phosphatase SHP-1 and the inhibitory C-Src kinase (Csk) are implicated in the attenuation of BCR-regulated Ca2+ elevation and inhibition of cellular activation (Ono et al, 1997; Adachi et al, 2001). Our group has recently described the downmodulation of intra- and extracellular Ca2+ fluxes by the adaptor protein Grb2 (growth factor receptor-bound protein 2) (Stork et al, 2004). Grb2 is expressed in all cell types and throughout the B-cell lineage. It is composed of a central Src homology (SH) 2 domain flanked on either side by one SH3 domain (Lowenstein et al, 1992). DT40 B-cell mutants, which were rendered deficient for Grb2 expression by gene targeting (Hashimoto et al, 1998), but not their wild-type counterparts, showed a sustained biphasic Ca2+ response following BCR engagement (Stork et al, 2004). This raised the question how Grb2-positive B cells of the peripheral lymph organs mount a full Ca2+ response, which is mandatory for their antigen-mediated activation and differentiation. It turned out that stimulation-induced recruitment of cytosolic Grb2 into the lipid raft fraction of the plasma membrane prevents Ca2+ inhibition (Stork et al, 2004). Relocalization can be achieved by transmembrane adaptor proteins such as NTAL (non-T-cell activation linker), which upon tyrosine phosphorylation bind the SH2 domain of Grb2. NTAL expression is low in developing B cells showing a weak Ca2+ response, but high in mature B cells with robust Ca2+ elevation (Stork et al, 2004; Hoek et al, 2006). A number of additional transmembrane adaptor proteins with consensus binding sites for the Grb2 SH2 domain exist (Horejsi et al, 2004) and may subrogate NTAL function, for example, in NTAL-deficient mouse mutants, which possess immunocompetent B cells (Wang et al, 2005). The effector proteins that execute Grb2-mediated Ca2+ inhibition are unknown. Here we report the identification of the critical Grb2 partner for Ca2+ inhibition as the hematopoietic adaptor protein Dok-3 (Cong et al, 1999; Lemay et al, 2000). SH2-mediated recruitment of Grb2 to tyrosine-phosphorylated Dok-3 at the plasma membrane attenuates Btk-mediated PLC-γ2 phosphorylation independently of SHIP and Csk. Unlike positive Grb2 regulators with transmembraneous and palmitoylated polypeptide anchors, Dok-3 is tethered at the inner side of the plasma membrane through its PH domain. Hence, Dok-3 appears to direct Grb2 into a distinct membrane compartment. In this location Grb2 acts as a negative regulator of Btk, resulting in diminished PLC-γ2 activity. These findings exert a molecular basis for differential Ca2+ signals in B cells and moreover, directly enforce the concept that precise membrane compartmentalization of signaling elements determines positive versus negative cellular responses.
Results
Grb2 controls inducible phosphorylation of Dok-3, the main tyrosine kinase substrate protein in DT40 B cells
To assess the signaling role of Grb2 in B cells, we analyzed BCR-induced tyrosine phosphorylation in wild-type and Grb2-deficient DT40 cells. Anti-phosphotyrosine (pTyr) immunoblotting of cleared cellular lysates revealed that the main tyrosine kinase substrate protein, migrating with an apparent molecular mass of approximately 50 kDa (p50), remains almost unphosphorylated in the absence of Grb2 (Figure 1A). The association of phosphorylated p50 with the Grb2 SH2 domain (data not shown) was employed to affinity-purify large amounts of p50 from stimulated DT40 cells in order to determine the peptide profile of tryptic digestion products by mass spectrometry (Figure 1B). The obtained peptide amino-acid sequences matched to a partial chicken EST (GenBank accession number XP_427516), which shows highest homology to the murine adaptor protein downstream of kinase-3 (Dok-3). Murine Dok-3 encompasses one PH and one PTB domain at its N-terminal end, followed by consensus tyrosine phosphorylation motifs in the C-terminal half (Lemay et al, 2000). Our cloning of the full-length avian dok-3 cDNA revealed that this overall structure is evolutionary conserved and that avian Dok-3 shares 68% and 62% amino-acid sequence homology to its murine and human orthologs, respectively (Supplementary Figure S1). The identity of p50 and Dok-3 was confirmed by anti-Dok-3 immunopurification (data not shown). Further reconstitution experiments with Grb2-deficient cells showed that efficient Dok-3 phosphorylation is independent of the N-terminal SH3 domain of Grb2, but requires the SH2 and C-terminal SH3 domains (Figure 1C, lanes 7–12). Similar to avian Dok-3, efficient tyrosine phosphorylation of murine Dok-3 is also dependent on Grb2 expression, as revealed by our analysis of Grb2-deficient mouse B-cell line Bal-17.TR and its Grb2-reconstituted transfectants (Figure 1D). As shown in Figure 1E, inducible tyrosine phosphorylation of Dok-3 is detectable in the absence of Syk (lanes 3 and 4) and Btk (lanes 5 and 6), but requires expression of the Src family kinase Lyn (lanes 7 and 8). Collectively, these data identify the intracellular adaptor protein Dok-3 as a major substrate of Src family kinases in activated B cells. The efficiency of Dok-3 tyrosine phosphorylation is, however, critically dependent on the additional presence of Grb2, which we have previously described as a negative regulator of BCR-induced Ca2+ mobilization.
Figure 1.
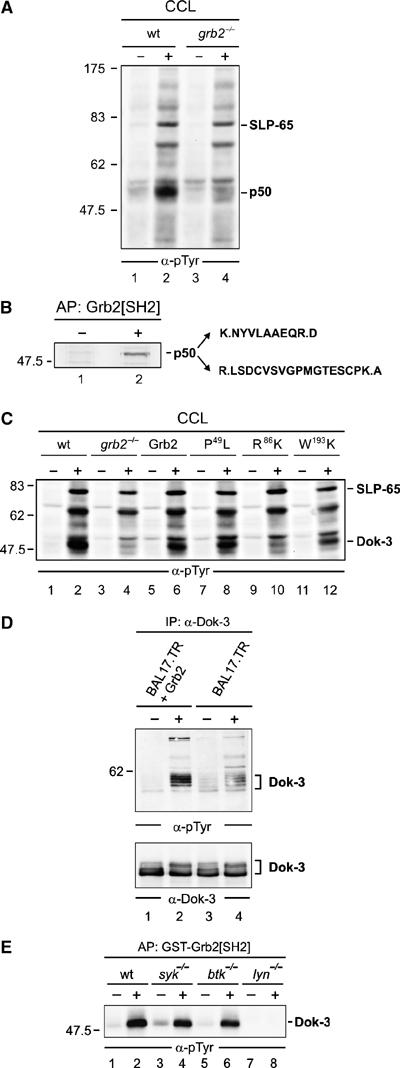
Grb2 controls Lyn-mediated phosphorylation of the adaptor protein Dok-3. (A) Wild-type (wt) and Grb2-deficient (grb2−/−) DT40 cells (lanes 1, 2 and 3, 4) were left untreated (−) or stimulated through their BCRs for 3 min (+). Equal amounts of proteins from cleared cellular lysates (CCL) were analyzed by anti-phosphotyrosine (α-pTyr) immunoblotting. (B) The major phosphotyrosine-containing protein, p50, was affinity-purified (AP) by GST-Grb2[SH2] from stimulated DT40 cells (lane 2), silver-stained, excised, digested by trypsine and peptide products were analyzed by ESI-Trap mass spectrometry. Purified proteins from unstimulated cells served as negative control (lane 1). The obtained amino-acid sequences are shown (single-letter code) with lysine (K) and arginine (R) being inferred from trypsine cleavage specificity (indicated by dots). These sequences matched a partial chicken EST (GenBank accession number XP_427516). Full-length chicken cDNA was isolated and submitted to GenBank with the accession number EF051736 (see also Supplementary Figure S1). (C) Wild-type (lanes 1 and 2) and grb2−/− DT40 cells (lanes 3 and 4) reconstituted with either wild-type Grb2 (lanes 5 and 6) or Grb2 variants, in which one of the three SH domains has been inactivated by single amino-acid substitution (N-terminal SH3 domain, P49L; SH2 domain, R86K; C-terminal SH3 domain, W193K; lanes 7–12), were left untreated (−) or stimulated through their BCRs (+). Equal amounts of proteins from CCL were subjected to anti-pTyr immunoblotting. To confirm equal loading, phospho-SLP-65 was detected separately by anti-SLP-65 immunoblotting (data not shown). (D) Murine Bal17.TR B cells, deficient for Grb2 expression, were transfected with an expression vector for Grb2 (lanes 1 and 2) or the empty vector as control (lanes 3 and 4) and left untreated (−) or stimulated through their BCRs (+). CCL were subjected to anti-Dok-3 immunoprecipitation and purified proteins were analyzed by immunoblotting with antibodies to pTyr and Dok-3 (upper and lower panels, respectively). (E) Resting (−) or BCR-activated (+) wild-type DT40 cells (lanes 1 and 2) or variants deficient for the protein tyrosine kinase Syk (lanes 3 and 4), Btk (lanes 5 and 6) or Lyn (lanes 7 and 8) were lysed and subjected to affinity purification with GST-Grb2[SH2]. Phosphorylated Dok-3 was detected by anti-pTyr immunoblotting. Relative molecular masses of marker proteins are indicated on the left in kDa.
Dok-3 is a negative regulator of BCR-induced Ca2+ mobilization
To functionally characterize Dok-3, we generated a Dok-3-deficient DT40 variant by gene targeting (see Materials and methods and Supplementary Figure S2A for details). Successful inactivation of dok-3 alleles and ablation of protein expression was confirmed by genomic PCR analysis (Supplementary Figure S2B) and anti-pTyr immunoblotting of cleared cellular lysates, Grb2[SH2]-purified proteins and anti-Dok-3-immunoprecipitates (Figure 2A and B; Supplementary Figure S2C). Note that Dok-3 tyrosine phosphorylation is considerably reduced in heterozygous dok-3+/− cells (Figure 2B, lanes 1–4).
Figure 2.
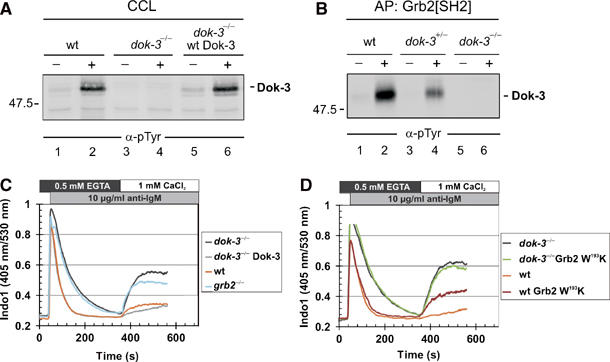
Gene targeting reveals a negative regulatory role of Dok-3. (A) Dok-3-deficient DT40 B cells were generated by targeted disruption of both dok-3 alleles (dok-3−/−, see Materials and methods for details), and absence of tyrosine-phosphorylated p50/Dok-3 in cleared cellular lysates (CCL) of resting (−) and BCR-activated (+) cells was tested by anti-pTyr immunoblotting (lanes 3 and 4). As control, wild-type DT40 and Dok-3-reconstituted dok-3−/− cells were analyzed in parallel (lanes 1 and 2 and 5 and 6, respectively). (B) Wild-type DT40 cells (lanes 1 and 2), heterozygous dok-3+/− (lanes 3 and 4) and homozygous dok-3−/− mutants (lanes 5 and 6) were left untreated (−) or stimulated through their BCRs (+). Cell lysates were subjected to affinity purification with the GST-Grb2[SH2] fusion protein and proteins so obtained were analyzed by anti-pTyr immunoblotting. Relative molecular mass of marker protein is indicated in (A) and (B) on the left in kDa. (C, D) BCR-induced intra- and extracellular Ca2+ mobilization of the indicated DT40 cells was recorded by flow cytometry as described in detail in Materials and methods. Briefly, cells were loaded with Indo-1 and release of intracellular Ca2+ was measured for 6 min in the presence of EGTA. Subsequently, extracellular Ca2+ was restored to 1mM in order to monitor Ca2+ entry across the plasma membrane. Lines represent wild-type DT40 (orange), dok-3−/− mutants (black), Dok-3-reconstituted dok-3−/− cells (gray), grb2−/− mutants (blue) and wild-type and dok-3−/− transfectants expressing the dominant-negative W193K version of Grb2 (brown and green, respectively). Data are representative of at least three independent measurements.
Given the reported role of Grb2 for BCR-induced Ca2+ signaling (Stork et al, 2004), we next tested this response in various DT40 cell lines, which are positive or negative for Dok-3 or Grb2 (Figure 2C). In marked contrast to wild-type DT40 cells, Dok-3-deficient cells show a biphasic Ca2+ profile, which is almost identical to that of Grb2-deficient cells (Figure 2C, orange, black and blue lines). The monophasic Ca2+ response of wild-type DT40 cells, which is characteristic for B cells with an immature phenotype (Koncz et al, 2002; Stork et al, 2004; Hoek et al, 2006), was restored in the Dok-3 mutant cells upon reconstitution with wild-type Dok-3 (gray line). These results show that similar to Grb2, Dok-3 is a negative regulatory element of BCR-induced Ca2+ mobilization. Moreover, both adaptor proteins appear to function in a common signaling pathway. To further confirm the latter notion, we employed a dominant-negative Grb2 mutant protein, which harbors an inactivated C-terminal SH3 domain (W193K). Expression of Grb2 W193K in DT40 cells overwrote the inhibitory function of endogenous wild-type Grb2 and allowed extracellular Ca2+ influx (Figure 2D, brown and orange lines). In marked contrast, expression of the Grb2 W193K protein in Dok-3-deficient DT40 cells had no effect on the Ca2+ profile (black and green lines), which strongly suggests that Dok-3 and Grb2 build a functional unit to attenuate BCR-induced Ca2+ mobilization.
Plasma membrane tethering and association to Grb2 are sufficient for Dok-3 to inhibit Ca2+ signaling
To elucidate the structural requirements of Dok-3 for Ca2+ inhibition, we expressed a series of HA-tagged Dok-3 mutants (see Figure 3A) in dok-3−/− cells. Inactivation of the PTB domain (R197A) had no effect on the ability of Dok-3 to prevent extracellular Ca2+ entry (Figure 3B, left panel, green and orange lines). Deletion of the PH domain (ΔPH) abolished Dok-3-mediated Ca2+ inhibition, which resulted in a biphasic response that was indistinguishable from that observed in cells with no Dok-3 expression (left panel, red and black lines). Single and dual Y-to-F amino-acid substitutions revealed that among the consensus tyrosine phosphorylation motifs of Dok-3, only that at Y331 is essential and sufficient for Ca2+ inhibition, whereas those at Y140 and Y307 are dispensable (right panel). Immunoprecipitation with anti-HA antibodies and subsequent immunoblot analysis showed that wild-type and mutant Dok-3 proteins are expressed by the transfectants at similar levels (Figure 3C, upper panel). This setting was also used to investigate the tyrosine phosphorylation status of the various Dok-3 proteins by anti-pTyr immunoblotting (Figure 3C, second panel). Inducible phosphorylation was easily and at almost identical levels detectable for wild-type Dok-3 (lanes 3 and 4) and Dok-3 mutants R197A and Y140F and Y307F (lanes 7–12), which all promoted the same biphasic Ca2+ profile (see above). In marked contrast, the Dok-3ΔPH protein did not become phosphorylated (lanes 5 and 6), and that of the Y331F mutant was strongly diminished (lanes 13 and 14). Both of these Dok-3 mutants were unable to support Ca2+ inhibition (see above). A strongly reduced tyrosine phosphorylation was also observed for the Y331 only protein (lanes 15 and 16) that, however, was fully capable of attenuating BCR-induced Ca2+ flux (see above). Collectively, we conclude that PH domain-mediated plasma membrane localization of Dok-3 is a requisite for Ca2+ inhibition, which itself is tightly associated with Dok-3 tyrosine phosphorylation. The latter event per se appears to be necessary but not sufficient for Ca2+ regulation. Rather, specific phosphorylation at Y331 is the second key element of Dok-3-mediated Ca2+ regulation.
Figure 3.
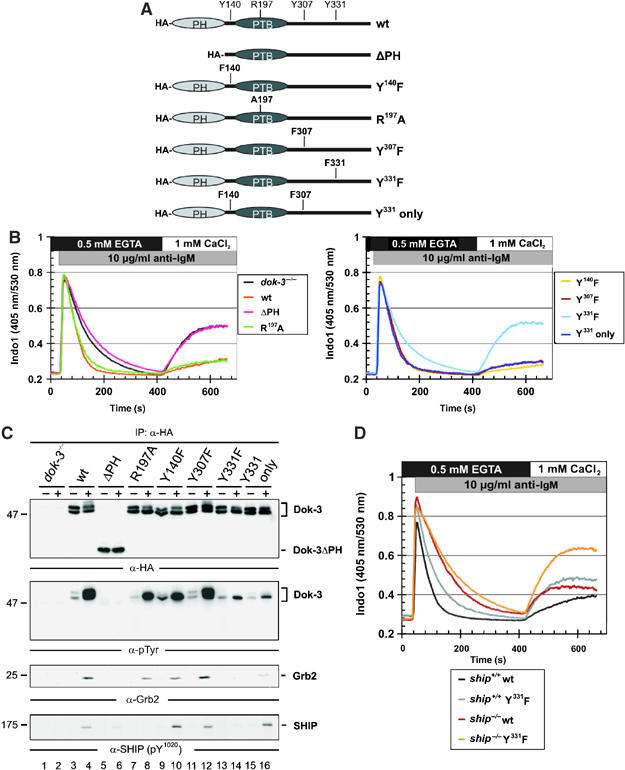
Dok-3 and Grb2 build a functional unit, that inhibits Ca2+ flux independent of SHIP. (A) Schematic representation of expression constructs encoding HA-tagged versions of wild-type Dok-3, a PH domain deletion mutant (ΔPH) or mutants encompassing amino-acid exchanges depicted in single-letter code. (B) Expression vectors were introduced by retroviral transduction in dok-3−/− mutants and BCR-induced Ca2+ mobilization of the transfectants was measured by flow cytometry, as described in the legend to Figure 2. Wild-type DT40 cells and empty vector transfectants of dok-3−/− mutants served as control (see inlay for color code). (C) Wild-type and DT40 variants described in (B) were left untreated (−) or BCR-activated (+) and lysates were subjected to anti-HA immunoprecipitation. Expression and tyrosine phosphorylation of Dok-3 proteins, as well as their association to Grb2 and SHIP, were detected by sequential immunoblotting with antibodies to HA, pTyr, Grb2 and SHIP (upper to lower panels, respectively). (D) BCR-induced Ca2+ fluxes were analyzed as described in the legend to Figure 2 in SHIP-deficient DT40 cells (ship−/−, brown) and ship−/− transfectants expressing a Dok-3 Y331F variant that counteracts Ca2+ inhibition by endogenous wild-type Dok-3 (orange). As control, parental DT40 cells, which are positive for endogenous SHIP and Dok-3 (black), and the Dok-3 Y331F transfectants (gray) were analyzed in parallel, demonstrating the dominant-negative function of Dok-3 Y331F.
Phosphorylation of Y331 creates a consensus binding site for the Grb2 SH2 domain. Indeed, the inducible association of Dok-3 with Grb2 was lost in cells expressing the Y331F mutant of Dok-3 (Figure 3C, third panel, lanes 13 and 14). Also the signaling-inactive ΔPH domain mutant did not co-immunoprecipitate with Grb2 (Figure 3C, third panel, lanes 5 and 6). For all other Dok-3 mutants, which retained their inhibitory capacity, BCR-induced complex formation with Grb2 was preserved (lanes 7–12). Hence, the biochemical property of inducible Grb2 association directly correlates with the functional ability of Dok-3 to downmodulate Ca2+ signals. This further demonstrates that Dok-3 and Grb2 together constitute a Ca2+-regulating signaling module.
Dok-3 has been previously reported to associate with SHIP and Csk via the PTB domain and phospho-Y307, respectively (Lemay et al, 2000; Robson et al, 2004). Indeed, the R197A amino-acid exchange in the PTB domain of Dok-3 abolished SHIP binding, which moreover appeared to require specific phosphorylation at Y331 (Figure 3C, lower panel). SHIP, however, is a well-known inhibitor of BCR-induced Ca2+ elevation, and it was therefore unexpected that disruption of the Dok-3/SHIP complex had no effect on the Ca2+ response. Hence, we wanted to confirm the missing role of SHIP with a second experimental setting. For this purpose, we employed the Y331F mutant of Dok-3, which counteracted Ca2+ inhibition by wild-type Dok-3, and when expressed in DT40 cells allowed for entry of extracellular Ca2+ (Figure 3D, black and gray lines). We reasoned that if Dok-3 controls Ca2+ through SHIP, expression of the Y331F dominant-negative version in SHIP-deficient cells should have no effect on the extracellular Ca2+ influx observed in these cells (Figure 3D, brown line). However, and consistent with our mutational analysis described above, expression of the Y331F mutant in ship−/− cells strongly augmented intra- and extracellular Ca2+ mobilization (orange line). This result demonstrates that inhibition of Ca2+ signals by endogenous wild-type Dok-3 is independent of SHIP expression. Final proof that SHIP is not a major downstream effector of Dok-3 came from the biochemical analysis of SHIP itself and its downstream target, the kinase Akt (alternatively called PKB). Neither phosphorylation of SHIP nor of Akt/PKB was drastically altered in the absence of Dok-3 expression (Supplementary Figure S3A). Similar to SHIP, also the catalytic activity of the Dok-3 binding partner Csk appeared unaltered in dok-3−/− cells (Supplementary Figure S3B), which further supports our mutational analysis. In summary, SHIP and Csk are both dispensable for Dok-3-mediated regulation of Ca2+, demonstrating that these proteins do not function together in a common Ca2+ signaling pathway.
PLC-γ2 is a target of Dok-3
In search for an enzymatic activity that is under the control of Dok-3, we focused on PLC-γ2. First, we tested the overall tyrosine phosphorylation status of PLC-γ2, which appeared to be very similar in the presence and absence of Dok-3 (Figure 4A). Using a site-specific antibody that detects phosphorylation of Y759 (in human PLC-γ2), we observed drastic differences between Dok-3-positive and Dok-3-negative cells (Figure 4B). The kinetic and extent of PLC-γ2 phosphorylation at this specific residue was substantially upregulated in dok-3−/− cells (lanes 5–8) compared with wild-type parental cells (lanes 1–4) or Dok-3-reconstituted transfectants (lanes 9–12). Phosphorylation of Y759 is known to be dependent on Btk and directly correlates with the enzymatic activity of PLC-γ2 (Humphries et al, 2004; Kim et al, 2004). Indeed, the hydrolysis of membrane phospholipids was more rapid and efficient in dok-3−/− cells than in reconstituted transfectants, as shown by monitoring the intracellular levels of the PLC-γ2 product IP3 (Figure 4C, left panel). The same was also true for grb2−/− cells (Figure 4C, right panel). These data identify PLC-γ2 as an effector protein of the Dok-3/Grb2 signaling module.
Figure 4.
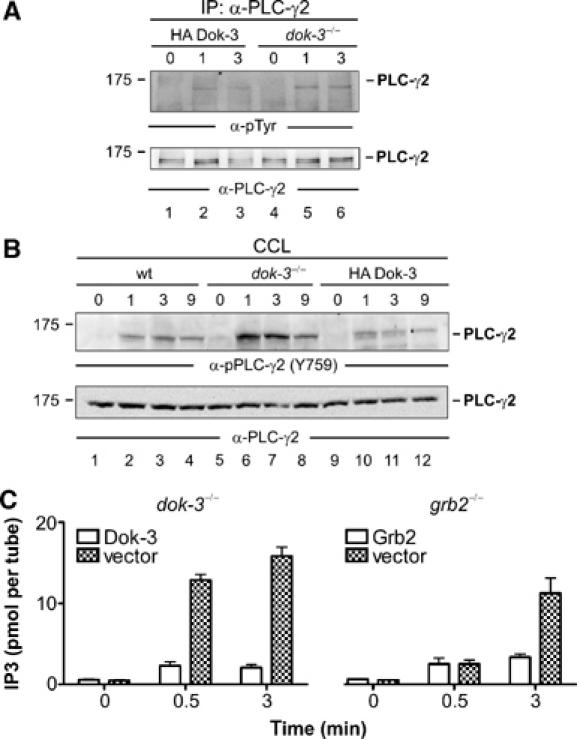
The Dok-3/Grb2 module attenuates PLC-γ2 activity. (A) Dok-3-deficient DT40 mutants (lanes 4–6) and reconstituted cells expressing HA-tagged wild-type Dok-3 (lanes 1–3) were left untreated (0) or stimulated through their BCRs for the indicated times (min). Lysates were subjected to anti-PLC-γ2 immunopurification and proteins obtained were analyzed by anti-pTyr and anti-PLC-γ2 immunoblotting (upper and lower panels, respectively). (B) Parental DT40 cells (lanes 1–4), dok-3−/− mutants (lanes 5–8) and HA-Dok-3-reconstituted transfectants (lanes 9–12) were left untreated (0) or stimulated through their BCRs for the indicated times (min). Cleared cellular lysates (CCL) were subjected to immunoblot analysis with antibodies that specifically detect PLC-γ2 phosphorylation at the Btk-dependent phospho-acceptor site corresponding to Y759 in human PLC-γ2 (upper panel). Equal protein loading was confirmed by reprobing the membrane with anti-PLC-γ2 antibodies (lower panel). Relative molecular mass of marker protein is indicated in (A) and (B) on the left in kDa. (C) DT40 mutant cells deficient for either Dok-3 (left panel) or Grb2 (right panel) and the empty vector control transfectants (open and filled bars, respectively) were left untreated (0) or BCR-activated for 0.5 or 3 min. IP3 levels in these cells were measured using a competitive binding assay with radiolabelled IP3-binding proteins. Error bars represent s.e.m. of three independent experiments with double preparation.
Membrane-bound Dok-3 controls BCR-induced relocalization of Grb2
Stimulation-dependent plasma membrane anchoring is a critical event for PLC-γ2 function (Nishida et al, 2003). This led us to investigate the in vivo subcellular localization of Dok-3 and Grb2 in resting and BCR-activated cells by confocal laser scanning microscopy (Figure 5). Dok-3-deficient DT40 cells were reconstituted with GFP-tagged versions of either wild-type or mutant Dok-3. Wild-type Dok-3 was constitutively and almost exclusively localized at the plasma membrane (Figure 5A, upper row). Nonetheless, BCR activation appeared to induce intra-membraneous relocalization of Dok-3, as indicated by the shift from uniform plasma membrane staining in resting cells to dotted fluorescence signals in stimulated cells. Membrane tethering was completely lost for the ΔPH mutant of Dok-3, which was homogeneously distributed in the cytoplasm of the cells (Figure 5A, middle row). The Y331F mutant of Dok-3 behaved like the wild-type protein (Figure 5A, lower row).
Figure 5.
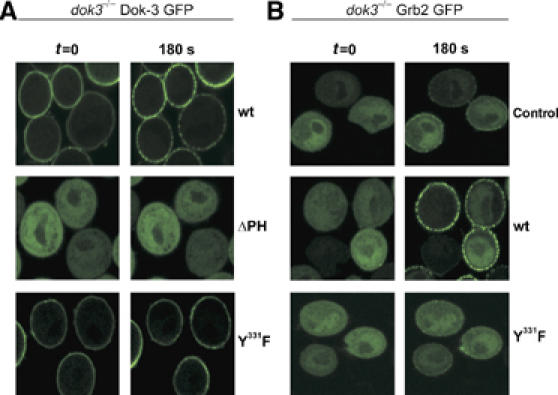
Dok-3 is permanently localized at the plasma membrane and is essential for stimulation-dependent recruitment of Grb2. (A) Dok-3-deficient DT40 mutants were transfected with expression constructs encoding fusion proteins between the green fluorescence protein (GFP) at the C terminus and either wild-type Dok-3 (upper row), Dok-3ΔPH (middle row) or Dok-3 Y331F (lower row) at the N terminus. Subcellular localization of Dok-3/GFP fusion proteins in resting (t=0) or BCR-activated (180 s) cells (left and right images) was visualized by confocal laser scanning microscopy. (B) Dok-3−/− DT40 cells expressing a Grb2/GFP fusion protein were transfected with either empty control vector (upper row) or expression vectors encoding wild-type or Y331F Dok-3 mutants (middle and lower rows). Subcellular Grb2 localization was analyzed as in (A). The Ca2+ signaling function of GFP fusion proteins was tested separately (data not shown).
Next, we assessed the role of Dok-3 for subcellular localization of Grb2. Expression of GFP-tagged Grb2 in unstimulated dok-3−/− cells resulted in fluorescence staining of the cytoplasm but not the plasma membrane (Figure 5B, upper left). Following BCR activation, minute amounts of Grb2 could be detected at the plasma membrane, but the overall staining pattern remained unchanged (Figure 5B, upper right). In marked contrast, in the presence of wild-type Dok-3, the vast majority of Grb2 translocated to the plasma membrane in a stimulation-dependent manner (Figure 5B, middle row). Expression of the Y331F mutant of Dok-3 did not support this relocalization of Grb2 (Figure 5B, lower row). Altogether, our in vivo imaging reveals that plasma membrane-bound Dok-3 recruits majority of cytosolic Grb2 in BCR-activated DT40 cells most likely by phospho-Y331/SH2-interaction. Notably, the PH domain-anchored Dok-3 itself undergoes a BCR-triggered clustering within the plasma membrane.
Dok-3 homo-oligomerizes upon BCR activation
To further dissect possible clustering processes of membrane-bound Dok-3, we coexpressed GFP-tagged Dok-3 with HA-tagged versions of wild-type or mutant Dok-3 proteins. Subsequently, their ability to form higher aggregates in resting and BCR-activated cells was biochemically investigated by co-immunoprecipitation experiments in which anti-HA-purified proteins were analyzed by Western blotting with antibodies to GFP and HA (Figure 6, upper and lower panels, respectively). In the analysis of dok-3−/− cells, parental cells served as specificity control (lanes 1 and 2). Dok-3-GFP coprecipitated with wild-type Dok-3 from both unstimulated and stimulated cells, but the efficiency strongly increased upon BCR activation (lanes 3 and 4). The stimulation-dependent, but not the constitutive association between Dok-3 proteins, was strongly diminished for those mutants that either lack the PH domain (ΔPH, lanes 5 and 6), possess an inactivated PTB domain (R197A, lanes 7 and 8) or cannot be phosphorylated at Y140 (Y140F, lanes 9 and 10). Phosphorylation of the Grb2-binding site Y331 appeared to be dispensable (Y331F, lanes 11 and 12). These data demonstrate a homotypic and inducible aggregation of Dok-3 proteins. For BCR-triggered upregulation of this interaction, Dok-3 must be localized at the plasma membrane, which allows for phosphorylation at Y140 (see also Figure 3C, lanes 5 and 6). Most likely, phospho-Y140 is then intermolecularly bound by the PTB domain of a neighboring Dok-3 molecule. The subsequently induced multimerization cascade is independent of Grb2 recruitment and vice versa, Grb2 binding and concomitant Ca2+ inhibition is independent of the Dok-3 oligomers (see also Figure 3B and C). Hence, oligomerization of and Ca2+ inhibition by Dok-3 proteins are two separate and functionally independent processes.
Figure 6.
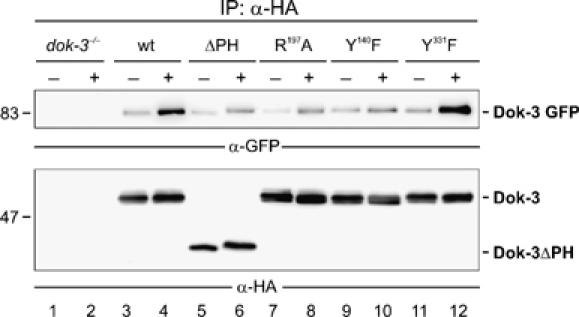
Dok-3 undergoes stimulation-dependent homo-oligomerization. Dok-3−/− mutant cells were reconstituted with Dok-3/GFP and subsequently transfected with empty control vector (lanes 1 and 2) or expression constructs for either HA-tagged wild-type Dok-3 (lanes 3 and 4) or the indicated HA-tagged Dok-3 variants (lanes 5–12; for details, see Figure 3A). Lysates were subjected to anti-HA immunoprecipitation. Proteins obtained were analyzed by immunoblotting with antibodies to GFP (upper panel) and HA peptide tag (lower panel). Relative molecular masses of marker proteins are indicated on the left in kDa.
Discussion
Intracellular elevation of Ca2+ in B cells is a tightly regulated process, which involves positive and negative control elements. Interfering with proper regulation of Ca2+ signaling is known to perturb humoral immune responses in mouse and man (Wienands, 2000). Herein, we have elucidated mechanistic details of Ca2+ inhibition through the adaptor proteins Grb2 and Dok-3. Several lines of evidence obtained by biochemical, genetic and imaging techniques show that following BCR activation, Grb2 and Dok-3 constitute a functional unit in a negative feedback control loop at the plasma membrane. First, following Lyn-mediated phosphorylation, Dok-3 associates with Grb2 by virtue of the Grb2 SH2 domain, which we have previously identified to be mandatory for Grb2-mediated Ca2+ inhibition. Interestingly, inducible tyrosine phosphorylation of Dok-3 requires the presence of the binding partner Grb2. This demonstrates the existence of a regulatory circuit, in which Grb2 ‘instructs' the kinase Lyn to create a specific docking site for the adaptor's SH2 domain. Second, grb2−/− and dok-3−/− cells exhibit almost identical Ca2+ profiles, which are characterized by robust intra- and extracellular Ca2+ fluxes. Hence, gene targeting of either grb2 or dok-3 is sufficient to convert the weak and monophasic Ca2+ response of an immature B cell such as DT40 to that of more mature B cells. Third, the dominant-negative W193K mutant of Grb2 cannot augment the Ca2+ response in dok-3−/− cells, demonstrating that Grb2 needs Dok-3 for Ca2+ signaling. Fourth and vice versa, the Y331F mutant of Dok-3, which cannot associate with Grb2 becomes hardly phosphorylated and is incapable of inhibiting BCR-induced Ca2+ mobilization. Moreover, all other tyrosine phosphorylation motifs of Dok-3, which bind other signaling proteins, are dispensable for Ca2+ inhibition. Fifth, membrane-associated Dok-3 is able to recruit and relocalize the majority of cytosolic Grb2 to the plasma membrane upon BCR activation. Loss of membrane association abrogates Dok-3 phosphorylation, association to Grb2 and concomitant Ca2+ inhibition. In summary, our data show that SH2 domain-mediated recruitment of Grb2 to Dok-3, which is tethered at the plasma membrane via its PH domain and becomes phosphorylated by Lyn, limits the quantity and quality of the BCR-induced Ca2+ signal.
Further studies presented in this paper excluded possible effector proteins of the Dok-3/Grb2 module. Importantly, we found no evidence for a role of SHIP, which was a likely candidate, because it is a reported Dok-3-binding protein (Lemay et al, 2000; Robson et al, 2004), and its lipid phosphatase activity reduces the number of membrane anchor motifs for PH domain-containing signaling proteins of the Ca2+-activating pathway (Damen et al, 1996). We confirmed the in vivo association between SHIP and the PTB domain of Dok-3, but the Dok-3/Grb2 module neither requires this interaction nor the expression of SHIP at all to inhibit Ca2+ fluxes. Also the phosphorylation-dependent association of Dok-3 with Csk is dispensable for Ca2+ signaling and inhibitory phosphorylation of Lyn. Finally, we demonstrated homo-oligomerization of Dok-3 proteins mediated by the PTB domain and phospho-Y140, but our mutational analysis of these intramolecular interaction sites excluded that this process participates in Ca2+ inhibition. Hetero-oligomerization between Dok-1 and Dok-2 had been previously reported to play a role in CD2 signaling in T cells (Boulay et al, 2005).
Two key observations provide mechanistic insight into the pathway downstream of the Dok-3/Grb2 module; that is, the substantially enhanced phosphorylation of PLC-γ2 at Y759 in dok-3−/− cells, which was accompanied by increased IP3 production and, the critical importance of the C-terminal SH3 domain of Grb2 for Ca2+ inhibition (see also Stork et al, 2004). The first results unmask Btk as the target of the Dok-3/Grb2 module because phosphorylation of Y759 in PLC-γ2 is strictly Btk-dependent and required for sustained Ca2+ elevation (Humphries et al, 2004; Kim et al, 2004). Our latter finding suggests how Dok-3-associated Grb2 can inhibit Btk function. Two conserved SH3 domain recognition motifs in the Tec homology (TH) domain of Btk are implicated in the regulation of kinase activity (Vihinen et al, 1996; Yamadori et al, 1999; Hansson et al, 2001; Okoh and Vihinen, 2002). Recruitment of Grb2 to phospho-Dok-3 may bring Grb2 into the vicinity of membrane-anchored Btk. Note that both Dok-3 and Btk are tethered at the plasma membrane through their PH domains and hence may reside in the same membrane sub-compartment. Colocalization of Dok-3/Grb2 with Btk could permit the C-terminal SH3 domain of Grb2 to bind the proline-rich regions in Btk and directly suppress its enzymatic activity. Alternatively, association between Grb2 and Btk occurs already in the cytosol, and phosphorylated Dok-3 targets the Grb2/Btk complex to a membrane compartment, where Btk cannot interact with PLC-γ2 for activation. For this purpose, PLC-γ2 needs to be located in lipid rafts, which explains why Grb2 recruitment to phosphorylated lipid raft residents, such as NTAL, facilitates sustained Ca2+ elevation (Stork et al, 2004). A sequestering function of the Dok-3/Grb2 module for Btk is supported by the ability of Dok-3 to inhibit Ras activation (Honma et al, 2006). In any case phosphorylated Dok-3 appears to provide a ‘membrane zip code' for Grb2 and the two models are not necessarily mutually exclusive (see Figure 7). A combination of biochemical and imaging methods will be required to finally elucidate the mode of action of the Dok-3/Grb2/Btk signaling module. It is tempting to speculate that the negative regulatory signal circuit described in this manuscript is involved in anergizing immature B cells upon auto-antigen encounter.
Figure 7.
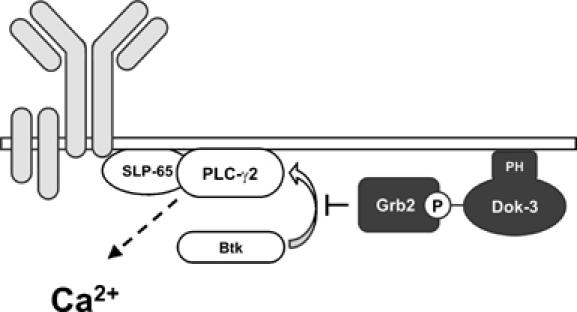
Inhibition of BCR-induced Ca2+ signaling by the Dok-3/Grb2 module. The Dok-3 adaptor protein is tethered at the inner side of the plasma membrane by virtue of its PH domain. Following tyrosine phosphorylation in activated B cells, Dok-3 recruits Grb2, which at this specific subcellular location attenuates Btk-dependent activation of PLC-γ2 by interfering with the proper formation of the SLP-65-assembled Ca2+ initiation complex and/or inhibiting the enzymatic activity of Btk. Positive and negative regulators of Ca2+ elevation are illustrated by open and filled boxes, respectively. The BCR complex is depicted in gray.
Materials and methods
Cells, Abs and reagents
Chicken DT40 cells were cultured in RPMI 1640 supplemented with 10% FCS, 1% chicken serum, 3 mM L-glutamine, 50 μM β-ME and antibiotics. Grb2- and SHIP-deficient DT40 cells are described by Ono et al (1997) and Hashimoto et al (1998). Mouse Bal17.TR B cells were kindly provided by Anthony de Franco (University of California, San Francisco, USA) and cultured in RPMI 1640 containing 10% FCS, 2 mM L-glutamine, 2 mM pyruvate, 50 μM β-ME and antibiotics (Harmer and DeFranco, 1999). Cell stimulation and lysis were performed as described by Stork et al (2004). Rabbit anti-mouse Dok-3 antibodies were kindly provided by André Veillette (IRCM, Montreal, Canada) (Lemay et al, 2000). Monoclonal antibodies to pTyr (4G10) and Grb2 (3F2) were purchased from Upstate Biotechnology (USA). Anti-Akt and phospho-specific antibodies to SHIP (Y1020), Src (Y416), Lyn (Y507) and Akt (S473) were purchased from Cell Signaling Technology (USA). Rabbit anti-PLC-γ2 (Q-20) and mAb rat anti-HA (3F10) were purchased from Santa Cruz Biotechnology (USA) and Roche (Switzerland), respectively. GST fusion proteins of Grb2[SH2] and Grb2[SH3] were prepared and used as described previously (Grabbe and Wienands, 2006).
Mass spectrometric analysis
DT40 (5 × 108) cells were stimulated with M4 and lysates were subjected to Grb2[SH2] affinity purification. After SDS–PAGE, proteins were visualized by silver staining. Proteins of interest were excised and in-gel-digested in an adapted manner according to Shevchenko et al (1996). Peptides generated were subjected to a 75-μm ID, 5-cm PepMap C18-column (Dionex, Germany). Peptide separation was performed by an acetonitrile gradient at 300 nl/min using an Ultimate/Swichos Nano-HPLC (Dionex, Germany) online coupled via a nano-spray source (Bruker, Germany) to a Esquire HCT Iontrap mass spectrometer (Bruker, Germany). Mass spectra were acquired in negative MS/MS mode, tuned for tryptic peptides. Processing of the MS/MS-spectra was performed by the use of Data Analysis and BioTools softwares from Bruker, Germany. Database search was done on the current NCBI protein database using an in-house MASCOT server.
Expression constructs and generation of Dok-3-deficient DT40 cells
The targeting vectors pDok-3-neo and pDok-3-hisD (see Supplementary Figure S2A) were constructed to insert neomycin and histidinol resistance cassettes into intron 1 of avian dok-3 alleles. The resistance cassettes were flanked by 1.8 and 2.9 kb at the 5′- and 3′-sites, respectively. For this purpose, genomic dok-3 fragments were amplified from DT40 genomic DNA using the primers 5′-TAGCACAGCTGTAGAGATGGCAGTG-3′ and 5′-AGCACATGAAGTCATCGCTTCTCC-3′ (left arm), and 5′-GCACGTTATGGGTGACATCATGGCAG-3′ and 5′-GAAGATGTTCTCATAGAGATGCTCGG-3′ (right arm). Targeting vectors were introduced by electroporation at 550 V and 25 μF. For selection, G418 was used at 2 mg/ml and histidinol at 1 mg/ml. Dok-3-deficient clones were screened by PCR and immunoblot analysis. Further details of the targeting strategy and selection of Dok-3-deficient clones are described in Supplementary Figure S2. Full-length avian dok-3 cDNA was obtained by RT–PCR with RNA from DT40 cells by using 5′-CAGTTGCTTTGGCTGAATCAGTCAC-3′ and 5′-TTTTGTTACGGCGCCCCCTGGCGG-3′ as forward and reverse primers, respectively. The GenBank accession number of avian dok-3 cDNA is EF051736. Wild-type chicken dok-3 cDNA was cloned into pCRII-TOPO vector. Coding sequences for HA tag were introduced at the 5′- and eGFP at the 3′-sites of the Dok-3 cDNA by PCR. The different Dok-3 variants were obtained using site-directed mutagenesis. All dok-3 cDNAs were directly ligated into the expression vectors pMSCV (BD Biosciences, USA) and pApuroII (Kurosaki et al, 1994). The generation of Grb2 constructs (GenBank accession number EF062570) and retroviral transduction or electroporation of expression vectors are described in Stork et al, (2004).
Calcium and IP3 measurements
For Ca2+ monitoring, 1 × 106 cells were loaded in 700 μl RPMI containing 5% FCS, 1 μM Indo1-AM (Molecular Probes) and 0.015% Pluronic F127 (Molecular Probes, USA) at 30°C for 25 min. Subsequently, the cell suspension was diluted two-fold with RPMI 10% FCS and incubated for 10 min at 37°C. Cells were washed and prepared for measurements as described earlier (Stork et al, 2004). Briefly, to discriminate between mobilization of Ca2+ from intra- and extracellular sources, BCR stimulation was performed for 6 min in the presence of 0.5 mM EGTA to remove extracellular Ca2+ and to allow for monitoring Ca2+ release from ER stores only. After restoring the extracellular Ca2+ concentration to 1 mM, entry of Ca2+ through ion channels in the plasma membrane was recorded. Changes in the ratio of fluorescence intensities at 405 and 510 nm were monitored on an LSRII cytometer (Becton Dickinson) and analyzed with FlowJo (TriStar). IP3 concentrations were determined using the IP3 Biotrak Assay (GE Healthcare) according to the manufacturer's protocol.
Confocal laser scanning microscopy
A total of 1 × 106 DT40 cells were resuspended in Krebs Ringer solution composed of 10 mM HEPES (pH 7.0), 140 mM NaCl, 4 mM KCl, 1 mM MgCl2, 1 mM CaCl2 and 10 mM glucose. After 30 min of seeding onto chambered coverglasses (Nunc, USA), cells were examined on a Leica TCS SP2 confocal laser scanning microscope. EGFP was excited at a wavelength of 488 nm and emission was recorded at 510 nm.
Supplementary Material
Supplementary Figures
Acknowledgments
We thank Drs André Veillette, Anthony de Franco and Annika Grabbe for providing Dok-3 reagents, Bal17.TR cells and recombinant Grb2 proteins, respectively. Expert technical assistance was provided by Ines Heine. This work was supported by the Deutsche Forschungsgemeinschaft through FOR 521.
References
- Adachi T, Wienands J, Wakabayashi C, Yakura H, Reth M, Tsubata T (2001) SHP-1 requires inhibitory co-receptors to down-modulate B cell antigen receptor-mediated phosphorylation of cellular substrates. J Biol Chem 276: 26648–26655 [DOI] [PubMed] [Google Scholar]
- Bolland S, Pearse RN, Kurosaki T, Ravetch JV (1998) SHIP modulates immune receptor responses by regulating membrane association of Btk. Immunity 8: 509–516 [DOI] [PubMed] [Google Scholar]
- Boulay I, Nemorin JG, Duplay P (2005) Phosphotyrosine binding-mediated oligomerization of downstream of tyrosine kinase (Dok)-1 and Dok-2 is involved in CD2-induced Dok phosphorylation. J Immunol 175: 4483–4489 [DOI] [PubMed] [Google Scholar]
- Brauweiler A, Tamir I, Dal Porto J, Benschop RJ, Helgason CD, Humphries RK, Freed JH, Cambier JC (2000) Differential regulation of B cell development, activation, and death by the Src homology 2 domain-containing 5′ inositol phosphatase (SHIP). J Exp Med 191: 1545–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CW, Dalton M, Ishiai M, Kurosaki T, Chan AC (2002) BLNK: molecular scaffolding through cis-mediated organization of signaling proteins. EMBO J 21: 6461–6472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong F, Yuan B, Goff SP (1999) Characterization of a novel member of the DOK family that binds and modulates Abl signaling. Mol Cell Biol 19: 8314–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damen JE, Liu L, Rosten P, Humphries RK, Jefferson AB, Majerus PW, Krystal G (1996) The 145-kDa protein induced to associate with Shc by multiple cytokines is an inositol tetraphosphate and phosphatidylinositol 3,4,5-trisphosphate 5-phosphatase. Proc Natl Acad Sci USA 93: 1689–1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS, Goodnow CC, Healy JI (1997) Differential activation of transcription factors induced by Ca2+ response amplitude and duration. Nature 386: 855–858 [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Xu K, Lewis RS (1998) Calcium oscillations increase the efficiency and specificity of gene expression. Nature 392: 933–936 [DOI] [PubMed] [Google Scholar]
- Fu C, Turck CW, Kurosaki T, Chan AC (1998) BLNK: a central linker protein in B cell activation. Immunity 9: 93–103 [DOI] [PubMed] [Google Scholar]
- Gallo EM, Cante-Barrett K, Crabtree GR (2006) Lymphocyte calcium signaling from membrane to nucleus. Nat Immunol 7: 25–32 [DOI] [PubMed] [Google Scholar]
- Goitsuka R, Fujimura Y, Mamada H, Umeda A, Morimura T, Uetsuka K, Doi K, Tsuji S, Kitamura D (1998) BASH, a novel signaling molecule preferentially expressed in B cells of the bursa of Fabricius. J Immunol 161: 5804–5808 [PubMed] [Google Scholar]
- Grabbe A, Wienands J (2006) Human SLP-65 isoforms contribute differently to activation and apoptosis of B lymphocytes. Blood 108: 3761–3768 [DOI] [PubMed] [Google Scholar]
- Hansson H, Smith CI, Hard T (2001) Both proline-rich sequences in the TH region of Bruton's tyrosine kinase stabilize intermolecular interactions with the SH3 domain. FEBS Lett 508: 11–15 [DOI] [PubMed] [Google Scholar]
- Harmer SL, DeFranco AL (1999) The src homology domain 2-containing inositol phosphatase SHIP forms a ternary complex with Shc and Grb2 in antigen receptor-stimulated B lymphocytes. J Biol Chem 274: 12183–12191 [DOI] [PubMed] [Google Scholar]
- Hashimoto A, Okada H, Jiang A, Kurosaki M, Greenberg S, Clark EA, Kurosaki T (1998) Involvement of guanosine triphosphatases and phospholipase C-gamma 2 in extracellular signal-regulated kinase, c-Jun NH2-terminal kinase, and p38 mitogen-activated protein kinase activation by the B cell antigen receptor. J Exp Med 188: 1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto S, Iwamatsu A, Ishiai M, Okawa K, Yamadori T, Matsushita M, Baba Y, Kishimoto T, Kurosaki T, Tsukada S (1999) Identification of the SH2 domain binding protein of Bruton's tyrosine kinase as BLNK—functional significance of Btk-SH2 domain in B-cell antigen receptor-coupled calcium signaling. Blood 94: 2357–2364 [PubMed] [Google Scholar]
- Hoek KL, Antony P, Lowe J, Shinners N, Sarmah B, Wente SR, Wang D, Gerstein RM, Khan WN (2006) Transitional B cell fate is associated with developmental stage-specific regulation of diacylglycerol and calcium signaling upon B cell receptor engagement. J Immunol 177: 5405–5413 [DOI] [PubMed] [Google Scholar]
- Honma M, Higuchi O, Shirakata M, Yasuda T, Shibuya H, Iemura S, Natsume T, Yamanashi Y (2006) Dok-3 sequesters Grb2 and inhibits the Ras–Erk pathway downstream of protein-tyrosine kinases. Genes Cells 11: 143–151 [DOI] [PubMed] [Google Scholar]
- Horejsi V, Zhang W, Schraven B (2004) Transmembrane adaptor proteins: organizers of immunoreceptor signalling. Nat Rev Immunol 4: 603–616 [DOI] [PubMed] [Google Scholar]
- Humphries LA, Dangelmaier C, Sommer K, Kipp K, Kato RM, Griffith N, Bakman I, Turk CW, Daniel JL, Rawlings DJ (2004) Tec kinases mediate sustained calcium influx via site-specific tyrosine phosphorylation of the phospholipase Cgamma Src homology 2-Src homology 3 linker. J Biol Chem 279: 37651–37661 [DOI] [PubMed] [Google Scholar]
- Ishiai M, Kurosaki M, Pappu R, Okawa K, Ronko I, Fu C, Shibata M, Iwamatsu A, Chan AC, Kurosaki T (1999a) BLNK required for coupling Syk to PLC gamma 2 and Rac1-JNK in B cells. Immunity 10: 117–125 [DOI] [PubMed] [Google Scholar]
- Ishiai M, Sugawara H, Kurosaki M, Kurosaki T (1999b) Cutting edge: association of phospholipase C-gamma 2 Src homology 2 domains with BLNK is critical for B cell antigen receptor signaling. J Immunol 163: 1746–1749 [PubMed] [Google Scholar]
- Kim CH, Hangoc G, Cooper S, Helgason CD, Yew S, Humphries RK, Krystal G, Broxmeyer HE (1999) Altered responsiveness to chemokines due to targeted disruption of SHIP. J Clin Invest 104: 1751–1759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YJ, Sekiya F, Poulin B, Bae YS, Rhee SG (2004) Mechanism of B-cell receptor-induced phosphorylation and activation of phospholipase C-gamma2. Mol Cell Biol 24: 9986–9999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koncz G, Bodor C, Kovesdi D, Gati R, Sarmay G (2002) BCR mediated signal transduction in immature and mature B cells. Immunol Lett 82: 41–49 [DOI] [PubMed] [Google Scholar]
- Kurosaki T, Takata M, Yamanashi Y, Inazu T, Taniguchi T, Yamamoto T, Yamamura H (1994) Syk activation by the Src-family tyrosine kinase in the B cell receptor signaling. J Exp Med 179: 1725–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosaki T, Tsukada S (2000) BLNK: connecting Syk and Btk to calcium signals. Immunity 12: 1–5 [DOI] [PubMed] [Google Scholar]
- Lemay S, Davidson D, Latour S, Veillette A (2000) Dok-3, a novel adapter molecule involved in the negative regulation of immunoreceptor signaling. Mol Cell Biol 20: 2743–2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowenstein EJ, Daly RJ, Batzer AG, Li W, Margolis B, Lammers R, Ullrich A, Skolnik EY, Bar-Sagi D, Schlessinger J (1992) The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70: 431–442 [DOI] [PubMed] [Google Scholar]
- Niiro H, Clark EA (2002) Regulation of B-cell fate by antigen-receptor signals. Nat Rev Immunol 2: 945–956 [DOI] [PubMed] [Google Scholar]
- Nishida M, Sugimoto K, Hara Y, Mori E, Morii T, Kurosaki T, Mori Y (2003) Amplification of receptor signalling by Ca2+ entry-mediated translocation and activation of PLCgamma2 in B lymphocytes. EMBO J 22: 4677–4688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada H, Bolland S, Hashimoto A, Kurosaki M, Kabuyama Y, Iino M, Ravetch JV, Kurosaki T (1998) Role of the inositol phosphatase SHIP in B cell receptor-induced Ca2+ oscillatory response. J Immunol 161: 5129–5132 [PubMed] [Google Scholar]
- Okoh MP, Vihinen M (2002) Interaction between Btk TH and SH3 domain. Biopolymers 63: 325–334 [DOI] [PubMed] [Google Scholar]
- Ono M, Okada H, Bolland S, Yanagi S, Kurosaki T, Ravetch JV (1997) Deletion of SHIP or SHP-1 reveals two distinct pathways for inhibitory signaling. Cell 90: 293–301 [DOI] [PubMed] [Google Scholar]
- Parekh AB, Putney JW Jr (2005) Store-operated calcium channels. Physiol Rev 85: 757–810 [DOI] [PubMed] [Google Scholar]
- Putney JW Jr (2005) Capacitative calcium entry: sensing the calcium stores. J Cell Biol 169: 381–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson JD, Davidson D, Veillette A (2004) Inhibition of the Jun N-terminal protein kinase pathway by SHIP-1, a lipid phosphatase that interacts with the adaptor molecule Dok-3. Mol Cell Biol 24: 2332–2343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68: 850–858 [DOI] [PubMed] [Google Scholar]
- Stork B, Engelke M, Frey J, Horejsi V, Hamm-Baarke A, Schraven B, Kurosaki T, Wienands J (2004) Grb2 and the non-T cell activation linker NTAL constitute a Ca(2+)-regulating signal circuit in B lymphocytes. Immunity 21: 681–691 [DOI] [PubMed] [Google Scholar]
- Su YW, Zhang Y, Schweikert J, Koretzky GA, Reth M, Wienands J (1999) Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur J Immunol 29: 3702–3711 [DOI] [PubMed] [Google Scholar]
- Vihinen M, Iwata T, Kinnon C, Kwan SP, Ochs HD, Vorechovsky I, Smith CI (1996) BTKbase, mutation database for X-linked agammaglobulinemia (XLA). Nucleic Acids Res 24: 160–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Horvath O, Hamm-Baarke A, Richelme M, Gregoire C, Guinamard R, Horejsi V, Angelisova P, Spicka J, Schraven B, Malissen B, Malissen M (2005) Single and combined deletions of the NTAL/LAB and LAT adaptors minimally affect B-cell development and function. Mol Cell Biol 25: 4455–4465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wienands J (2000) Signal transduction elements of the B cell antigen receptor and their role in immunodeficiencies. Immunobiology 202: 120–133 [DOI] [PubMed] [Google Scholar]
- Wienands J, Schweikert J, Wollscheid B, Jumaa H, Nielsen PJ, Reth M (1998) SLP-65: a new signaling component in B lymphocytes which requires expression of the antigen receptor for phosphorylation. J Exp Med 188: 791–795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamadori T, Baba Y, Matsushita M, Hashimoto S, Kurosaki M, Kurosaki T, Kishimoto T, Tsukada S (1999) Bruton's tyrosine kinase activity is negatively regulated by Sab, the Btk-SH3 domain-binding protein. Proc Natl Acad Sci USA 96: 6341–6346 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures