

Abstract
High-mobility group box 1 (HMGB1) is released extracellularly upon cell necrosis acting as a mediator in tissue injury and inflammation. However, the molecular mechanisms for the proinflammatory effect of HMGB1 are poorly understood. Here, we define a novel function of HMGB1 in promoting Mac-1-dependent neutrophil recruitment. HMGB1 administration induced rapid neutrophil recruitment in vivo. HMGB1-mediated recruitment was prevented in mice deficient in the β2-integrin Mac-1 but not in those deficient in LFA-1. As observed by bone marrow chimera experiments, Mac-1-dependent neutrophil recruitment induced by HMGB1 required the presence of receptor for advanced glycation end products (RAGE) on neutrophils but not on endothelial cells. In vitro, HMGB1 enhanced the interaction between Mac-1 and RAGE. Consistently, HMGB1 activated Mac-1 as well as Mac-1-mediated adhesive and migratory functions of neutrophils in a RAGE-dependent manner. Moreover, HMGB1-induced activation of nuclear factor-κB in neutrophils required both Mac-1 and RAGE. Together, a novel HMGB1-dependent pathway for inflammatory cell recruitment and activation that requires the functional interplay between Mac-1 and RAGE is described here.
Keywords: adhesion, inflammation, integrins, neutrophils
Introduction
Leukocyte recruitment as an integral part of inflammatory processes requires multistep adhesive and signaling events including selectin-dependent rolling, chemokine-dependent leukocyte activation, and integrin-mediated firm adhesion and diapedesis (Springer, 1994). During firm endothelial adhesion of leukocytes, leukocyte β2-integrins, LFA-1 (αLβ2, CD11a/CD18), Mac-1 (αMβ2, CD11b/CD18), and p150,95 (αXβ2, CD11c/CD18), as well as β1-integrins interact with endothelial counterligands such as ICAM-1, surface-associated fibrinogen (FBG) or VCAM-1 (Gahmberg, 1997; Plow et al, 2000; Hogg et al, 2003). Among leukocyte integrins, Mac-1 plays an important role in innate immunity, as it may regulate inflammatory cell recruitment as well as pathogen recognition, phagocytosis, and neutrophil survival (Ehlers, 2000; Mayadas and Cullere, 2005). Interestingly, Mac-1 ligation on leukocytes may lead to activation of nuclear factor-κB (NF-κB) (Sitrin et al, 1998) and the activation of the consequent gene expression (Rezzonico et al, 2001), although the underlying mechanisms are poorly understood. The role of Mac-1 in innate immunity is in line with its propensity to be a highly versatile multiligand receptor (Ehlers, 2000) interacting with numerous ligands and counter-receptors. In addition, the functions of Mac-1 may be regulated by interactions in cis, that is, on the same leukocyte surface with other receptors, such as the FcγRIII or the urokinase receptor (Zhou et al, 1993; Tang et al, 1997; Petty et al, 2002; Mayadas and Cullere, 2005). Interestingly, although Mac-1 is notorious for interacting in trans with different cellular counter-receptors or matrix proteins, only a few membrane partners of Mac-1 in cis are identified that may regulate its activity (Ehlers, 2000; Petty et al, 2002).
High-mobility group box 1 (HMGB1), also named amphoterin, is a nuclear protein loosely bound to DNA that stabilizes nucleosome formation and regulates transcription (Dumitriu et al, 2005b; Lotze and Tracey, 2005). Emerging evidence has demonstrated an important role for extracellular HMGB1 as a very potent inflammatory mediator (Wang et al, 1999; Scaffidi et al, 2002; Dumitriu et al, 2005b; Lotze and Tracey, 2005). HMGB1 can be secreted into the extracellular space by activated macrophages and mature dendritic cells by an active process that may require the acetylation of the molecule in the nucleus (Bonaldi et al, 2003). Alternatively, HMGB1 is passively released by necrotic, but not apoptotic cells (Scaffidi et al, 2002), thereby representing a signal for tissue damage. Extracellular HMGB1 may interact with toll-like receptors (TLR) and/or RAGE (receptor for advanced glycation end products) (Dumitriu et al, 2005b; Lotze and Tracey, 2005). In particular, an interaction between HMGB1 and TLR-2 or TLR-4 has been demonstrated that may mediate the proinflammatory actions of HMGB1 (Park et al, 2004, 2006). On the other hand, RAGE is a multiligand receptor on vascular cells that plays a key role in inflammatory processes, especially at sites where its ligands accumulate (Schmidt et al, 2001; Chavakis et al, 2004). RAGE ligation may activate a range of signaling pathways including MAP kinases, rho GTPases, as well as activation of NF-κB (Schmidt et al, 2001; Yan et al, 2003). Recently, we established that endothelial RAGE interacts also with Mac-1 on leukocytes (Chavakis et al, 2003).
Extracellular HMGB1 evokes a strong inflammatory response; it stimulates the release of multiple proinflammatory cytokines such as tumor necrosis factor (TNF) and interleukins in macrophages and neutrophils (Andersson et al, 2000) and induces the expression of adhesion molecules on endothelial cells, such as VCAM-1 and selectins, as well as it enhances dendritic cell maturation (Dumitriu et al, 2005a, 2005b; Lotze and Tracey, 2005). Robust leukocyte recruitment is a prominent hallmark associated with HMGB1-mediated inflammation (Dumitriu et al, 2005b; Lotze and Tracey, 2005). As observed in studies that entailed HMGB1 blockade in vivo, HMGB1 is important in the pathogenesis of sepsis (Wang et al, 1999; Yang et al, 2000), as well as in arthritis (Yang et al, 2005). Recent studies also indicated that HMGB1 may mediate inflammatory cell recruitment in acute hepatic necrosis (Tsung et al, 2005) and in acute lung injury (Kim et al, 2005; Lin et al, 2005). However, the molecular mechanisms underlying this proinflammatory function of HMGB1 remain to be clarified. In particular, it is not established yet whether HMGB1 affects extravasation-related functions of leukocytes, such as adhesion and migration. Here, we identify a novel pathway for HMGB1-mediated neutrophil recruitment that requires the functional interplay between RAGE and the β2-integrin Mac-1.
Results
HMGB1-mediated neutrophil recruitment in vivo requires Mac-1
We first studied whether HMGB1 administration can elicit rapid inflammatory cell recruitment in vivo. Interestingly, intraperitoneally (i.p.) injection of HMGB1 resulted in a rapid (4 h) recruitment of leukocytes (mostly neutrophils) into the peritoneum (Figure 1A). As a comparison, we studied thioglycollate-induced peritonitis (Figure 1B) (Chavakis et al, 2002, 2003). The HMGB1-mediated effect on neutrophil recruitment was reduced in RAGE-deficient mice (Figure 1A). In addition, HMGB1-induced neutrophil emigration to the peritoneum was blocked by systemic pretreatment of wild-type mice with soluble RAGE 1 h before HMGB1 injection (data not shown). Whereas thioglycollate-induced neutrophil infiltration was blocked by blocking monoclonal antibody (mAb) against LFA-1 and, to a less extent, by blocking mAb against Mac-1, HMGB1-mediated neutrophil emigration to the peritoneum was only blocked by blocking mAb to Mac-1, but not affected by antibody against LFA-1 (Figure 1B). To define further the underlying mechanisms of HMGB1-mediated neutrophil extravasation, we engaged mice deficient in Mac-1 or LFA-1. Consistent with the antibody inhibition studies, HMGB1-induced neutrophil emigration was decreased in Mac-1-deficient mice but not in LFA-1-deficient mice (Figure 1C). In contrast, thioglycollate-induced peritonitis was prevented in LFA-1-deficient mice (Figure 1D), consistent with previous reports showing an important role of LFA-1 in thioglycollate-induced peritonitis (Coxon et al, 1996; Lu et al, 1997; Berlin-Rufenach et al, 1999; Ding et al, 1999). Together, these findings suggest that HMGB1 stimulates neutrophil recruitment in vivo and that this process requires Mac-1 as well as RAGE.
Figure 1.
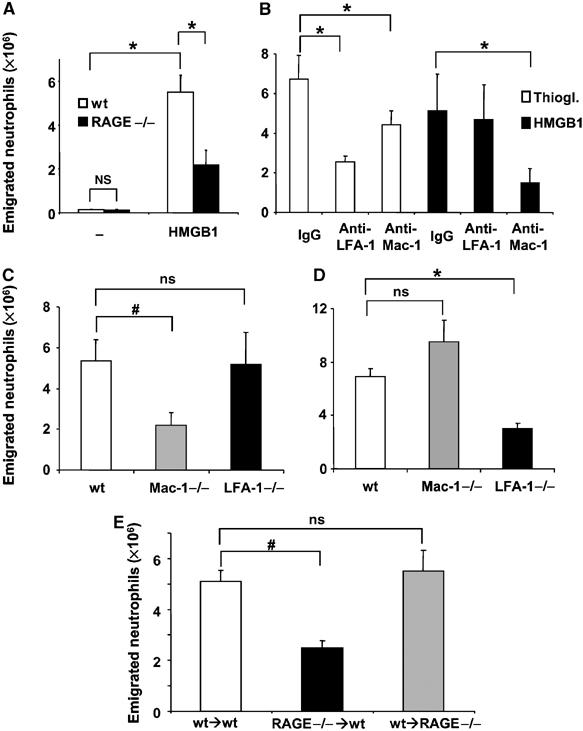
HMGB1-mediated inflammatory cell recruitment in vivo. (A) The number of neutrophils in wild-type (open bars) or RAGE−/− (filled bars) mice is shown 4 h after the i.p. injection of buffer (−) or HMGB1 (10 μg). (B) Sixty minutes before thioglycollate (open bars) or HMGB1 (filled bars) administration, wild-type mice were treated with isotype control mAb, with a blocking mAb against LFA-1 or with a blocking mAb against Mac-1 (each 100 μg). (C) HMGB1 induced peritonitis in wild-type, Mac-1−/−, and LFA-1−/− mice. (D) Thioglycollate induced peritonitis in wild-type, Mac-1−/−, and LFA-1−/− mice. (E) HMGB1 induced peritonitis in sublethally irradiated wild-type mice reconstituted with bone marrow cells from wild-type mice (wt → wt), sublethally irradiated wild-type mice reconstituted with bone marrow cells from RAGE−/− mice (RAGE−/− → wt) and sublethally irradiated RAGE−/−mice reconstituted with bone marrow cells from wild type (wt → RAGE−/−). Data are expressed as absolute numbers of emigrated neutrophils into the peritoneum. *P<0.01; #P<0.05; ns: not significant. Data are mean±s.d. (n=3–6 mice/group).
RAGE is expressed on both endothelial cells and hematopoietic cells including neutrophils (Collison et al, 2002; Yan et al, 2003). Endothelial RAGE can interact with leukocyte Mac-1 in trans (Supplementary Figure 1, and Chavakis et al, 2003). To differentiate whether endothelial- or neutrophil-associated RAGE was required for the activity of HMGB1 to stimulate inflammatory cell recruitment in vivo, we performed bone marrow transplantation experiments. In particular, wild-type mice received irradiation and were then reconstituted with bone marrow from either wild-type or RAGE-deficient mice (wt → wt and RAGE−/− → wt, respectively). In the reverse experiment, irradiated RAGE−/− mice were reconstituted with bone marrow from wild-type mice (wt → RAGE−/−). Interestingly, the decrease in HMGB1-induced neutrophil extravasation observed in RAGE−/− mice as compared to wild-type mice (Figure 1A) could be reversed in the wt → RAGE−/− group, that is, by restoring the expression of RAGE on neutrophils (Figure 1E). In contrast, HMGB1-induced neutrophil recruitment into the peritoneum was prevented in the RAGE−/− → wt group as compared to the wt → wt group (Figure 1E). The degree of reduction in HMGB1-induced neutrophil recruitment into the peritoneum owing to the hematopoietic-specific absence of RAGE was comparable to the degree of decrease of HMGB1-induced neutrophil emigration in RAGE-deficient mice (Figure 1A). Taken together, these results indicate that Mac-1 and neutrophil RAGE but not endothelial RAGE are required for the HMGB1-induced recruitment of neutrophils in vivo.
HMGB1 stimulates Mac-1-dependent leukocyte adhesion
As these observations suggested a role for HMGB1 in Mac-1-dependent leukocyte extravasation, we studied whether HMGB1 can affect neutrophil adhesion. Interestingly, Mac-1-dependent neutrophil adhesion to FBG was stimulated three-fold by HMGB1. Whereas adhesion of RAGE-deficient neutrophils to FBG was comparable to the adhesion of wild-type neutrophils, the stimulatory effect of HMGB1 on Mac-1-dependent neutrophil adhesion to FBG was abolished in the absence of RAGE (Figure 2A). Additionally, HMGB1 induced spreading of wild-type but not RAGE-deficient neutrophils on FBG (Figure 2B). In contrast, PMA-induced adhesion and spreading of neutrophils to FBG was not affected by RAGE deficiency (not shown). Moreover, Mac-1−/− neutrophils failed to adhere to FBG and HMGB1 did not stimulate the adhesion of Mac-1−/− neutrophils to FBG, whereas HMGB1 enhanced the FBG adhesion of LFA-1−/− neutrophils (Figure 2C). Thus, HMGB1 stimulates the Mac-1-dependent adhesion of neutrophils to FBG in a RAGE-dependent manner.
Figure 2.
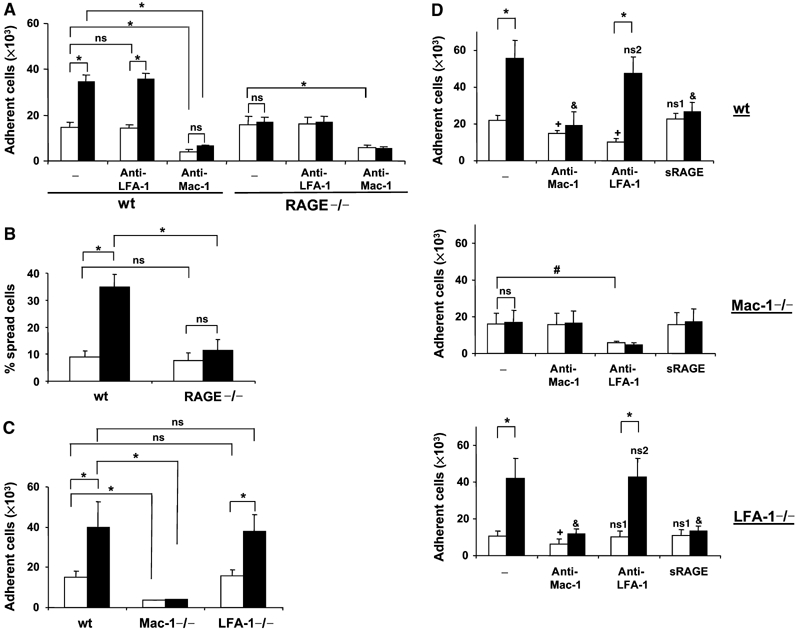
HMGB1-stimulated adhesion of mouse neutrophils. (A) Adhesion of wild-type or RAGE−/− neutrophils to immobilized FBG in the absence (open bars) or presence of HMGB1 (filled bars, 100 ng/ml) is shown without (−) or with mAb to LFA-1 or mAb to Mac-1 (each at 20 μg/ml). Cell adhesion is represented as number of adherent cells. (B) Spreading of wild-type or RAGE−/− neutrophils on immobilized FBG in the absence (open bars) or presence of HMGB1 (filled bars, 100 ng/ml). Data are represented as % spread cells. (C) Adhesion of wild-type, Mac-1−/−, or LFA-1−/− neutrophils to immobilized FBG in the absence (open bars) or presence of HMGB1 (filled bars, 100 ng/ml) is shown. Cell adhesion is represented as number of adherent cells. In (A), (B), and (C), *P<0.01; ns: not significant. (D) Adhesion of wild-type, Mac-1−/−, or LFA-1−/− neutrophils to immobilized ICAM-1 is shown in the absence (open bars) or presence of HMGB1 (100 ng/ml, filled bars) without (−) or with mAb to Mac-1, mAb to LFA-1, or soluble RAGE (each at 20 μg/ml). Cell adhesion is represented as number of adherent cells. In (D), *P<0.01; #P<0.05; ns: not significant; +P<0.05 as compared to adhesion in the absence of HMGB1 (open bars) and in the absence of competitors (−); ns1: not significant as compared to adhesion in the absence of HMGB1 (open bars) and in the absence of competitors (−); &P<0.01 as compared to adhesion in the presence of HMGB1 (filled bars) and in the absence of competitors (−); ns2: not significant as compared to adhesion in the presence of HMGB1 (filled bars) and in the absence of competitors (−). Data are mean±s.d. of three independent experiments each performed in triplicate.
Next, the effect of HMGB1 on the adhesion of neutrophils to immobilized ICAM-1, the major endothelial counter-receptor of leukocyte β2-integrins, was studied. Neutrophil adhesion to ICAM-1 is mediated by both Mac-1 and LFA-1 (Gahmberg, 1997; Hogg et al, 2003), and in the absence of HMGB1 neutrophil adhesion to ICAM-1 was blocked by inhibitory mAb to either Mac-1 or LFA-1 (Figure 2D). Both Mac-1- and LFA-1-deficient neutrophils displayed decreased adhesion to immobilized ICAM-1 (Figure 2C). HMGB1 increased adhesion of wild-type neutrophils to ICAM-1 and the HMGB1-induced effect was absent in RAGE−/− neutrophils (not shown). Moreover, the HMGB1-induced upregulation of ICAM-1 adhesion was mediated by Mac-1 but not LFA-1, as evidenced by the following observations: (i) ICAM-1 adhesion of wild-type neutrophils in the presence of HMGB1 was prevented by inhibitory mAb to Mac-1 but was not affected by inhibitory mAb to LFA-1. (ii) The HMGB1-induced stimulation of neutrophil adhesion to ICAM-1 was absent in Mac-1-deficient neutrophils. In contrast, HMGB1 increased adhesion of LFA-1-deficient neutrophils to ICAM-1. This HMGB1-induced increase in ICAM-1 adhesion of LFA-1-deficient neutrophils was prevented by inhibitory mAb to Mac-1 but not mAb to LFA-1 (Figure 2D). Moreover, although soluble RAGE did not affect the adhesion of neutrophils to ICAM-1 in the absence of HMGB1, it blocked the HMGB1-induced upregulation of wild-type or LFA-1-deficient neutrophils (Figure 2D). Furthermore, the effect of HMGB1 to stimulate Mac-1-dependent adhesion of neutrophils to FBG or ICAM-1 was dose-dependent (1–250 ng/ml; data not shown). HMGB1 was active in stimulating Mac-1-dependent adhesion independent of whether it was pre-incubated with the neutrophils and then washed away before the adhesion assay or whether it was co-incubated with the neutrophils during the course of the adhesion assay, indicating that HMGB1 primary acts on the neutrophils. Finally, adhesion of wild-type neutrophils to immobilized fibronectin (FN), which is mediated by VLA-4, was not stimulated by HMGB1 (data not shown).
Our data indicate that HMGB1 activates Mac-1 in a RAGE-dependent manner. Studies on Mac-1 activation are more feasible to perform with human Mac-1 owing to the availability of both purified human Mac-1 as well as of well-characterized antibodies against human Mac-1. First, we investigated whether HMGB1 also stimulates Mac-1-dependent adhesion of human leukocytes. Consistent with the data obtained from mouse neutrophils, HMGB1 stimulated the Mac-1-dependent adhesion of myelomonocytic THP-1 cells to immobilized FBG or ICAM-1 by three-fold (Figure 3A and B). The effect of HMGB1 was also dose-dependent (1–250 ng/ml; data not shown). HMGB1-induced Mac-1-dependent adhesion of THP-1 cells to FBG or ICAM-1 was prevented in the presence of antibody to HMGB1 or soluble RAGE. In addition, the HMGB1-induced increase of ICAM-1 adhesion of THP-1 was prevented by inhibitory mAb to Mac-1 but was not affected by inhibitory mAb to LFA-1 (Figure 3B). In contrast, PMA or monocyte chemoattractant protein-1 (MCP-1)- stimulated adhesion of THP1 cells to ICAM-1 was blocked by both antibodies to Mac-1 and LFA-1 (data not shown). Adhesion of THP-1 cells to FN was predominantly mediated by VLA-4 and was not affected by the presence of HMGB1, soluble RAGE or antibody to HMGB1 (Figure 3C). Similar results were also obtained in experiments performed with human neutrophils isolated from peripheral blood (Supplementary Figure 2). These experiments indicate that HMGB1 stimulates Mac-1-dependent adhesive events in human leukocytes in a RAGE-dependent manner.
Figure 3.
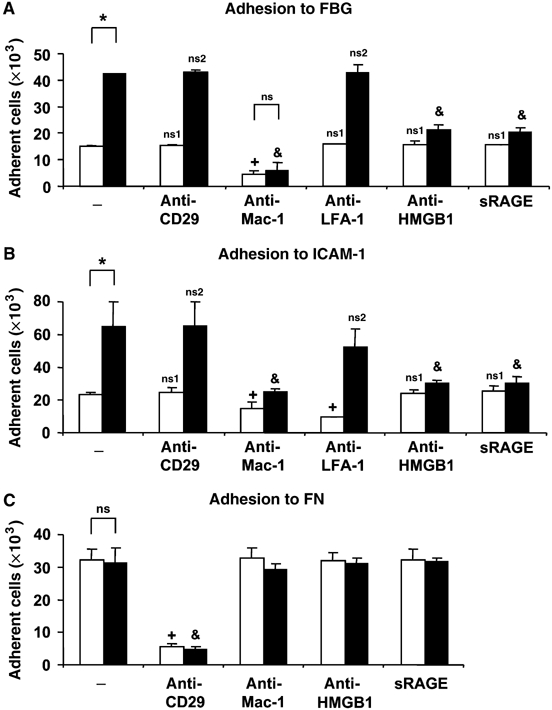
HMGB1-mediated adhesion of human leukocytes. (A, B) Adhesion of THP1 cells to immobilized FBG (A) or immobilized ICAM-1 (B) is shown in the absence (open bars) or presence of HMGB1 (100 ng/ml, filled bars), without (−) or with mAb to CD29, mAb to Mac-1, mAb to LFA-1, antibody to HMGB1 or soluble RAGE (each at 20 μg/ml). (C) Adhesion of THP-1 cells to immobilized FN is shown in the absence (open bars) or presence of HMGB1 (100 ng/ml, filled bars) without (−) or with mAb to CD29, mAb to Mac-1, antibody to HMGB1 or soluble RAGE (each at 20 μg/ml). Cell adhesion is represented as number of adherent cells. *P<0.01; ns: not significant; +P<0.05 as compared to adhesion in the absence of HMGB1 (open bars) and in the absence of competitors (−); ns1: not significant as compared to adhesion in the absence of HMGB1 (open bars) and in the absence of competitors (−); &P<0.01 as compared to adhesion in the presence of HMGB1 (filled bars) and in the absence of competitors (−); ns2: not significant as compared to adhesion in the presence of HMGB1 (filled bars) and in the absence of competitors (−). Data are mean±s.d. of three independent experiments each performed in triplicate.
Chemotactic activity of HMGB1 on neutrophils
To assess further HMGB1 as a pro-adhesive/pro-chemotactic factor, we studied whether HMGB1 stimulates lamellipodium formation. Similar to MCP-1 (not shown and Cambien et al, 2001), HMGB1 induced the polarization of THP-1 cells adhering onto FBG. This shape change corresponded with the enrichment of F-actin at the leading edge, indicating that HMGB1-induced lamellipodium formation in these cells. Moreover, consistent with previous reports (Mocsai et al, 2002; Schymeinsky et al, 2005), the non-receptor protein tyrosine kinase syk was also redistributed at the site of lamellipodium formation and colocalized with F-actin upon stimulation with HMGB1 (Figure 4A). Thus, HMGB1 resembles chemotactic factors in that it induces lamellipodium formation.
Figure 4.
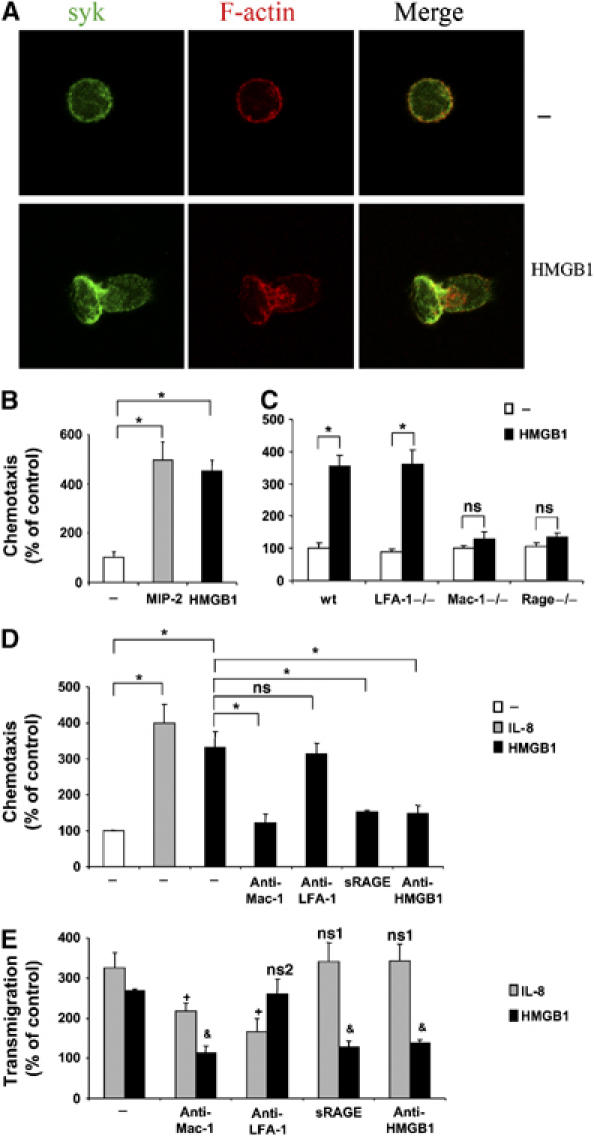
HMGB1-mediated chemotaxis and transendothelial migration of leukocytes. (A) Immunofluorescence for Syk (green) or F-actin (red) was performed followed by confocal microscopy. Representative immunofluorescence of THP-1 cells that were incubated in the absence (−) or presence of HMGB1 (50 ng/ml) for 30 min is shown. Double-stained images were merged. HMGB1 induced the enrichment of F-actin and Syk staining at the leading edge of the cell. (B) Chemotaxis of wild-type mouse neutrophils towards no chemoattractant (open bar), 50 ng/ml MIP-2 (gray bar) or 50 ng/ml HMGB1 (filled bar) is shown. (C) Chemotaxis of wild-type, LFA-1−/−, Mac-1−/−, and RAGE−/− mouse neutrophils towards no chemoattractant (open bars), or 50 ng/ml HMGB1 (filled bars) is shown. (D) Chemotaxis of human neutrophils towards no chemoattractant (open bar), 50 ng/ml IL-8 (gray bar) or 50 ng/ml HMGB1 (filled bars) is shown in the absence (−) or presence of blocking mAb to Mac-1, mAb to LFA-1, soluble RAGE, or antibody to HMGB1 (each at 20 μg/ml). In (B), (C), and (D), chemotaxis data are shown as percent of control. In (B) and (C), chemotaxis of wild-type mouse neutrophils in the absence of stimuli or competitors represents the 100% control; in (D) chemotaxis of human neutrophils in the absence of stimuli or competitors represents the 100% control. *P<0.02; ns: not significant. (E) The transmigration of human neutrophils towards 50 ng/ml IL-8 (gray bars) or 50 ng/ml HMGB1 (filled bars) across HUVEC is shown in the absence (−) or presence of blocking mAb to Mac-1, mAb to LFA-1, soluble RAGE, or antibody to HMGB1 (each at 20 μg/ml). Transmigration is represented as percent of control. Transmigration through HUVEC in the absence of stimuli or competitors represents the 100% control. +P<0.01 as compared to transmigration towards IL-8 (gray bars) and in the absence of competitors (−); ns1: not significant as compared to transmigration towards IL-8 (gray bars) and in the absence of competitors (−); &P<0.01 as compared to transmigration towards HMGB1 (filled bars) and in the absence of competitors (−); ns2: not significant as compared to transmigration towards HMGB1 (filled bars) and in the absence of competitors (−). Data are mean±s.d. of three independent experiments each performed in triplicate.
In addition, HMGB1 induced chemotaxis of mouse and human neutrophils. The chemotactic effect of HMGB1 on mouse and human neutrophils was comparable to the effects of MIP-2 and IL-8 on mouse and human neutrophils, respectively (Figure 4B and D). The chemotactic effect of HMGB1 on neutrophils required both Mac-1 and RAGE, as HMGB1 failed to induce chemotaxis of Mac-1−/− and RAGE−/− neutrophils (Figure 4C). Consistently, HMGB1-induced chemotaxis of human neutrophils was blocked by soluble RAGE and an inhibitory mAb to Mac-1 but not by inhibitory mAb to LFA-1 (Figure 4D).
As β2-integrins and Mac-1 also play an important role in neutrophil transendothelial migration, we then investigated whether HMGB1 might affect this process. In a transwell assay, HMGB1 significantly stimulated the transmigration of human neutrophils through a monolayer of human umbilical vein endothelial cells (HUVEC) and this effect was blocked by mAb against Mac-1 but not by mAb against LFA-1. In addition, HMGB1-induced transendothelial migration of neutrophils was inhibited by soluble RAGE or antibody to HMGB1. In contrast, IL-8-stimulated transmigration through cultured endothelial cells was blocked by mAb to Mac-1 as well as mAb to LFA-1, and was not affected by soluble RAGE or antibody to HMGB1 (Figure 4E). Similar results were obtained with THP-1 cells (not shown). Thus, HMGB1 exerts a chemotactic activity on neutrophils, inducing Mac-1-mediated chemotaxis and transendothelial migration in a RAGE-dependent manner.
HMGB1 increases Mac-1 activity in a RAGE-dependent manner
Our data so far suggested that HMGB1 stimulates Mac-1-mediated adhesiveness in a RAGE-dependent manner. Activation of integrin-mediated adhesiveness takes place at the level of avidity or valency (receptor density on the adhesive surface) as well as at the level of affinity for the individual ligand (Carman and Springer, 2003). We found that HMGB1-induced adhesion and spreading of THP-1 cells onto FBG-coated slides was associated with the polarization of Mac-1 to the leading edge of the THP-1 cells, as opposed to the diffuse staining on the cell surface in non-stimulated cells. In addition, we observed a strong colocalization of RAGE with Mac-1 at the leading edge of spreading THP-1 cells upon HMGB1 stimulation (Figure 5A).
Figure 5.
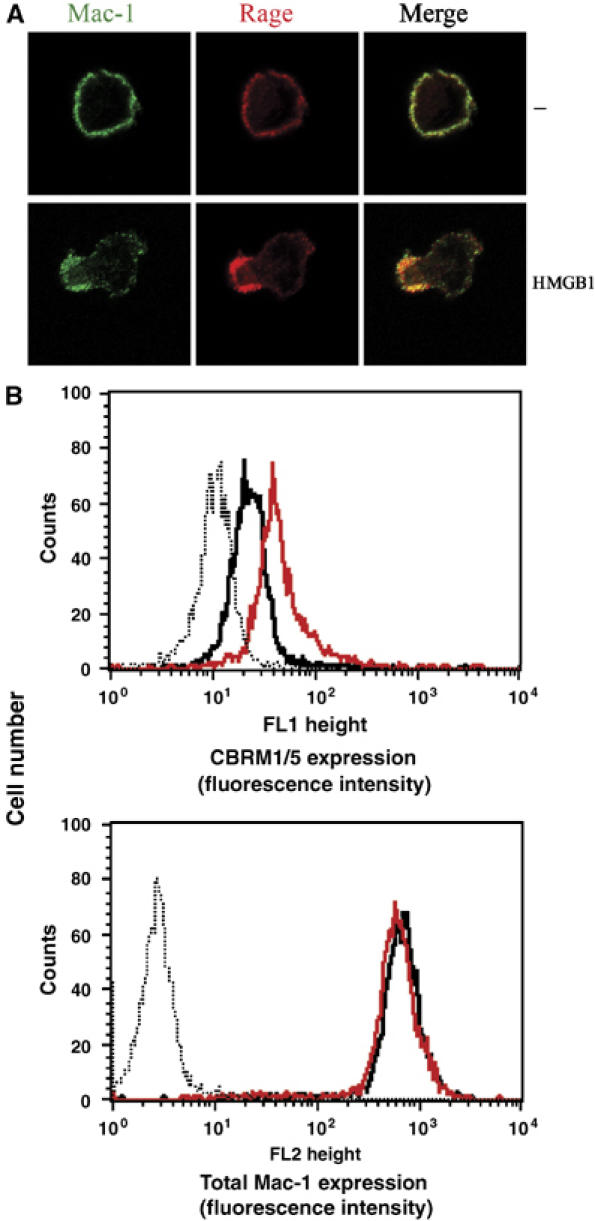
HMGB1-dependent activation of Mac-1. (A) Immunofluorescence for Mac-1 and RAGE was performed followed by confocal microscopy. Representative immunofluorescence of THP-1 cells that were incubated in the absence (−) or presence of HMGB1 (100 ng/ml) on FBG for 30 min is shown. Double-stained images were merged. (B) Human neutrophils were incubated in the absence (black curves) or presence of HMGB1 (100 ng/ml, red curves) for 20 min. Surface expression of an activation-dependent epitope (CBRM1/5) on Mac-1 was quantitated by FACS analysis. For comparison, quantitative cell surface expression of Mac-1 was analyzed using an antibody, which recognizes an epitope irrespective of the activation state of the integrin. Nonspecific fluorescence was determined using isotype-matched mouse-IgG (dotted thin curves).
Increases in the affinity of integrins are associated with conformational changes leading to increased exposure of activation-dependent epitopes on the integrin (Carman and Springer, 2003; Hogg et al, 2003). Activation of Mac-1 on the cell surface can be measured by the binding of the mAb CBRM1/5 that recognizes an activation-dependent epitope on the integrin. Activation of THP-1 cells and neutrophils with HMGB1 resulted in increased exposure of the CBRM1/5 epitope on Mac-1 (Figure 5B, data with THP-1 cells not shown). The total expression of Mac-1 remained unchanged by this short-term stimulation with HMGB1. The HMGB1-induced exposure of the CBRM1/5 epitope on Mac-1 was abolished by soluble RAGE (data not shown).
HMGB1 stimulates the interaction between Mac-1 and RAGE
The previous observations indicated that HMGB1 induces increased colocalization of Mac-1 with RAGE on the membrane of the inflammatory cell. Therefore, we continued to investigate the underlying mechanisms. In a purified system, HMGB1 did not influence the binding of FBG or ICAM-1 to immobilized Mac-1 or the binding of ICAM-1 to LFA-1 (Figure 6A and B). From these data, we can exclude that HMGB1 directly acts on the interaction of Mac-1 with its ligands FBG or ICAM-1. Interestingly, HMGB1 augmented the interaction between RAGE and immobilized Mac-1 (Figure 6C). HMGB1 interacted specifically and dose-dependently only with RAGE but not with Mac-1 (Figure 6D and Supplementary Figure 3). In addition, HMGB1 bound to wild-type neutrophils but not to RAGE−/− neutrophils (data not shown).
Figure 6.
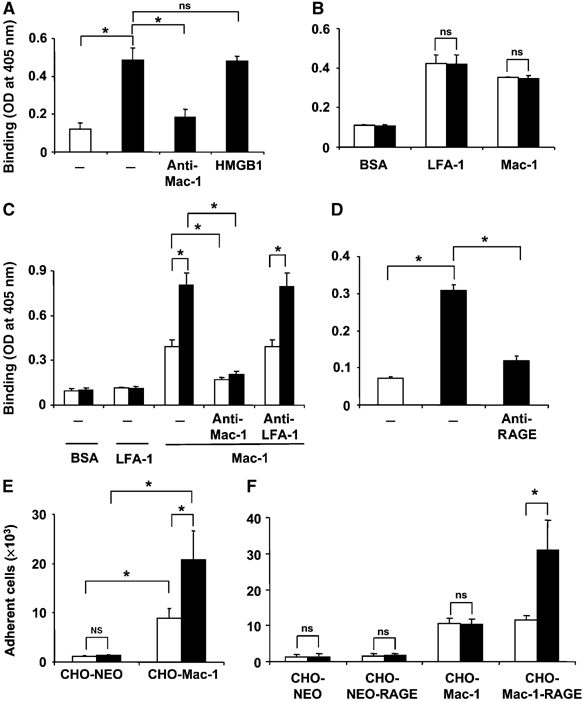
Influence of HMGB1 on the binding interactions between RAGE and Mac-1. (A) The binding of FBG to immobilized BSA (open bar) or immobilized Mac-1 (filled bars) is shown in the absence (−) or presence of mAb to Mac-1 or in the presence of HMGB1 (100 ng/ml). (B) The binding of ICAM-1 to immobilized BSA, LFA-1, or Mac-1 is shown in the absence (open bars) or presence of HMGB1 (100 ng/ml). (C) The binding of RAGE to immobilized BSA, LFA-1, or Mac-1 is shown in the absence (open bars) or presence of HMGB1 (100 ng/ml, filled bars). Binding of RAGE to Mac-1 was studied without (−) or with mAb to Mac-1 or mAb to LFA-1 (each at 20 μg/ml). (D) The binding of HMGB1 (0.1 μg/ml) to immobilized BSA (open bar) or immobilized RAGE (filled bars) is shown in the absence (−) or presence of antibody to RAGE (20 μg/ml). Binding is expressed as absorbance at 405 nm. (E) The adhesion of mock-transfected CHO cells (CHO-NEO) or CHO cells transfected with Mac-1 to immobilized RAGE is shown in the absence (open bars) or presence of HMGB1 (100 ng/ml, filled bars). (F) The adhesion of CHO-NEO cells, CHO-NEO cells transfected with RAGE, CHO-cells transfected with Mac-1 or CHO-cells transfected with both RAGE and Mac-1 to immobilized FBG is shown in the absence (open bars) or presence of HMGB1 (100 ng/ml, filled bars). Cell adhesion is represented as number of adherent cells. *P<0.01; ns: not significant. Data are mean±s.d. of three independent experiments each performed in triplicate.
The augmented Mac-1-dependent adhesion of neutrophils or THP-1 cells to FBG or ICAM-1 in the presence of HMGB1 could therefore be attributed to an HMGB1-mediated increase in the interaction between RAGE and Mac-1 in cis on the membrane of the inflammatory cell that then results in increased activity of Mac-1. To address this hypothesis, we have studied the adhesion of CHO cells transfected with Mac-1, RAGE, or both receptors. HMGB1 stimulated the adhesion of Mac-1-transfected CHO cells but not mock-transfected cells (cells transfected with the vector alone) to immobilized RAGE (Figure 6E). Mac-1-transfected cells but not mock- or RAGE-transfected cells adhered to immobilized FBG or ICAM-1 (Figure 6F, ICAM-1 adhesion data not shown). Whereas HMGB1 did not alter the adhesion of Mac-1 transfected CHO cells to immobilized FBG or ICAM-1, the adhesion of CHO cells cotransfected with Mac-1 and RAGE was stimulated by HMGB1 (Figure 6F). Thus, the presence of RAGE on the cell surface is essential for the effect of HMGB1 to enhance Mac-1-dependent adhesion to the ligands of the integrin.
HMGB1-induced activation of the transcription factor NF-κB requires both RAGE and Mac-1
Our findings up to this point suggested that HMGB1 stimulates the functional cooperation between RAGE and Mac-1. However, it is conceivable that such a cooperation between RAGE and Mac-1 on the surface of inflammatory cells may affect RAGE-dependent signaling. NF-κB activation is a well-established downstream signaling event of RAGE ligation (Schmidt et al, 2001; Chavakis et al, 2004). Notably, among β2-integrins, it is especially Mac-1 that has been linked to NF-κB activation (Schmal et al, 1998; Sitrin et al, 1998; Shi et al, 2001); however, the underlying mechanisms are poorly understood. By engaging neutrophils from Mac-1-, LFA-1-, and RAGE-deficient mice, we found that HMGB1-induced NF-κB activation was diminished by the absence of either RAGE or Mac-1 but not of LFA-1, whereas TNF-α-induced NF-κB activation was not affected by the deficiency of any of these receptors (Figure 7). Similarly, HMGB1-induced NF-κB activation in THP-1 cells could be blocked by soluble RAGE, antibody to HMGB1, and mAb to Mac-1 but not by mAb to LFA-1 (Supplementary Figure 4). Thus, HMGB1-induced NF-κB activation requires both RAGE and Mac-1.
Figure 7.
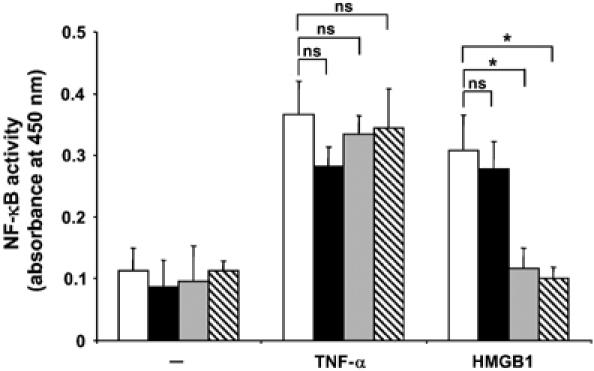
HMGB1-mediated NF-κB activation requires both RAGE and Mac-1. The activity of NF-κB was measured by ELISA in wt (open bars), LFA-1−/− (filled bars), Mac-1−/− (gray bars), or RAGE−/− (hatched bars) mouse neutrophils that were stimulated in the absence (−) or presence of TNF-α (10 ng/ml) or HMGB1 (20 ng/ml), as indicated. NF-κB activity is presented as absorbance at 450 nm. *P<0.02; ns: not significant. Data are mean±s.d. of three independent experiments each performed in triplicate.
Discussion
It has become apparent in recent years that HMGB1 is instrumental in mediating a response to tissue damage or infection. HMGB1 is released by necrotic cells or actively secreted by cells of the innate immune system upon infectious and proinflammatory stimuli and triggers a strong inflammatory response (Dumitriu et al, 2005b; Lotze and Tracey, 2005). To date, the underlying mechanisms of HMGB1-mediated inflammatory cell recruitment are poorly defined. The present report demonstrates that HMGB1 induces inflammatory cell recruitment by regulating adhesive and migratory functions of neutrophils. This novel HMGB1-dependent pathway requires the lateral interplay between RAGE and the integrin Mac-1 and may constitute a major mechanism for inflammatory cell recruitment in innate immunity and after tissue injury.
The following features are consistent with a pro-adhesive and pro-chemotactic effect of HMGB1 for inflammatory cells: (i) in vitro studies demonstrated that HMGB1 specifically promoted Mac-1-but not LFA-1-mediated adhesion of neutrophils to ICAM-1 or FBG in a RAGE-dependent manner, as the effect of HMGB1 was absent in Mac-1- and RAGE-deficient neutrophils. By using Mac-1-RAGE cotransfectants, we verified that HMGB1-induced adhesion to immobilized Mac-1 ligands requires both Mac-1 and RAGE. Additionally, HMGB1 acted chemotactically, as it induced lamellipodium formation, chemotaxis and transendothelial migration of neutrophils in a Mac-1- and RAGE-dependent manner, consistent with a previous report that antibodies to HMGB1 reduced monocyte transendothelial migration in vitro (Rouhiainen et al, 2004). The stimulation of Mac-1-dependent adhesive and migratory events was accompanied by the activation of Mac-1 by HMGB1. In particular, HMGB1 increased the direct binding between RAGE and Mac-1 in a purified system; it also induced the colocalization of RAGE with Mac-1 at the leading edge of the leukocyte. Furthermore, HMGB1 stimulated the exposure of an activation-dependent epitope on Mac-1 in a RAGE-dependent manner. (ii) In vivo, HMGB1 administration triggered acute neutrophil recruitment. This process required Mac-1, as evidenced by the diminished HMGB1-mediated neutrophil recruitment in Mac-1−/− mice, whereas the HMGB1-mediated response in LFA-1−/− mice was normal. Bone marrow chimera experiments indicated that RAGE on the surface of neutrophils, but not endothelial cell RAGE, was required for the HMGB1-induced neutrophil recruitment. Based on these findings, we postulate that HMGB1 mediates inflammatory cell recruitment by stimulating the interaction between RAGE and Mac-1 on the surface of the leukocyte, thereby activating Mac-1-dependent adhesive and migratory phenomena in a RAGE-dependent manner. However, we cannot exclude that indirect effects, such as HMGB1-induced gene expression, may also contribute to HMGB1-mediated neutrophil recruitment in vivo.
The lateral interaction of Mac-1 with RAGE triggered by HMGB1 on the leukocyte surface shares similarities with previously identified lateral interactions of Mac-1 with the urokinase receptor or FcγRIII that can regulate the activity of the integrin (Tang et al, 1997; Chavakis et al, 1999; Ross, 2002). In particular, although ligation of FcγR alone is usually sufficient to trigger activation such as phagocytosis, oxidative burst, and generation of various proinflammatory signals, this activity of FcγR is augmented further by the cooperative input from a synergistic Mac-1/FcγR complex, and vice versa Mac-1-dependent responses may be regulated by FcγR (Tang et al, 1997). In accordance, RAGE and Mac-1 are capable of triggering proinflammatory responses alone (Schmidt et al, 2001; Chavakis et al, 2004; Mayadas and Cullere, 2005); however, the functional synergism between them may result in a potentiation of the HMGB1-dependent inflammatory cell recruitment. Although the ability to react rapidly to injury or an infectious challenge is important, it is equally important that the response is in proportion to the magnitude of the threat (Ehlers, 2000). Thus, the existence of interactions between Mac-1 and FcγR or RAGE may serve to ‘fine-tune' the inflammatory response. Nevertheless, additional studies will be necessary to identify the exact structural requirements of the HMGB1-induced interaction of Mac-1 with RAGE in cis.
It was previously reported that NF-κB activation is a downstream event of the interaction of RAGE with its ligands (Schmidt et al, 2001; Chavakis et al, 2004). Interestingly, HMGB1-induced NF-κB activation in neutrophils required both RAGE and Mac-1. However, we cannot exclude that the recently described binding of HMGB1 to TLR-2 and -4 (Park et al, 2004, 2006; Lotze and Tracey, 2005; Dumitriu et al, 2005b) may also contribute to NF-κB activation or other HMGB1-related functions.
Previously, ligation of Mac-1 resulted in NF-κB activation in an IRAK-1-dependent manner; however, in contrast to Toll/IL-1 receptor signaling, Mac-1-dependent NF-κB activation is MyD88-independent (Shi et al, 2001). Although the exact pathways leading to NF-κB activation downstream of both RAGE and Mac-1 ligation still have undefined signaling intermediates, our data indicate that these two receptors may cooperate on the leukocyte surface in order to mediate NF-κB activation. Interestingly, the propensity of Mac-1 to mediate NF-κB activation in neutrophils is consistent with the role of Mac-1 to regulate apoptosis and survival of these cells as described previously (Mayadas and Cullere, 2005).
The pathway described here for HMGB1-mediated inflammatory cell recruitment is consistent with the role of both multiligand receptors, RAGE and Mac-1, as major receptors in innate immunity. Previously, RAGE-deficient mice were largely protected during cecal ligation and puncture (Liliensiek et al, 2004), whereas Mac-1 is important for leukocyte functions such as adhesion, phagocytosis, oxidative burst, and survival (Mayadas and Cullere, 2005). In addition, as opposed to LFA-1, Mac-1 has a remarkably broad capacity and diversity for ligand recognition; in this regard Mac-1 may be the most promiscuous integrin, interacting with cellular adhesion molecules, extracellular matrix proteins, complement iC3b, proteolytic factors, as well as with LPS or other microbial ligands (Ehlers, 2000; Mayadas and Cullere, 2005). Our data suggest that HMGB1 signaling converges on Mac-1 (via RAGE) for integration with signals from other ligands, as a means of fine tuning an immediate response against microbial invaders or tissue injury.
Materials and methods
Materials
Details are available in Supplementary data.
Cell culture
Details are available in Supplementary data.
In vitro ligand–receptor interactions
Binding of biotinylated-FBG or ICAM-1 to immobilized Mac-1 or LFA-1 was performed exactly as described previously (Chavakis et al, 2002, 2003; Santoso et al, 2002). Alternatively, binding of soluble RAGE (5 μg/ml) to immobilized BSA, LFA-1, or Mac-1, (each 10 μg/ml) was performed as described (Chavakis et al, 2003). Binding of HMGB1 to immobilized RAGE or Mac-1 was also studied. Details are available in Supplementary data.
Electrophoretic mobility shift assay for the detection of the activity of NF-κB
Nuclear proteins were harvested as described previously and assayed for transcription factor binding activity using the NF-κB consensus sequence 5-AGTTGAGGGGACTTTCCCAGGC-3. Specificity of binding was ascertained by competition with a 160-fold molar excess of unlabeled consensus oligonucleotides (Bierhaus et al, 2001; Sotiriou et al, 2006).
ELISA for the detection of the activity of NF-κB
Details are available in Supplementary data.
Fluorescence cell adhesion assay
Adhesion experiments were performed as described previously (Chavakis et al, 2002, 2003; Xie et al, 2006). Details are available in Supplementary data.
Chemotaxis of human and mouse neutrophils
Details are available in Supplementary data.
Transmigration
Transmigration of human neutrophils through an endothelial cell monolayer was performed as described previously (Chavakis et al, 2004a; Xie et al, 2006). Details are available in Supplementary data.
Binding of Fc-RAGE to mouse neutrophils
Details are available in Supplementary data.
Flow cytometry for the detection of activation-dependent epitopes on Mac-1
These experiments were performed as described previously (Chavakis et al, 1999). Details are available in Supplementary data.
Immunofluorescence
Details are available in Supplementary data.
Mice
The generation of RAGE−/− mice, LFA-1−/− mice, and Mac-1−/− was described previously (Lu et al, 1997; Ding et al, 1999; Liliensiek et al, 2004). All mice were backcrossed for at least seven generations to a C57BL/6 background. Wild-type C57BL/6 mice were purchased from Jackson Laboratory (Ben Harbor, Maine). Protocols were approved by the NCI Animal Care and Use Committee.
In vivo peritonitis model
Thioglycollate-induced peritonitis in wild-type, Mac-1−/−, LFA-1−/−, or RAGE−/− mice was performed as described previously (Chavakis et al, 2002, 2003, 2004a). Alternatively, mice were injected i.p. with 10 μg of HMGB1. For inhibition studies, 1 h before the injection of thioglycollate, 100 μg of mAb against mouse Mac-1 or LFA-1 were administered i.p. Control mice were treated with the same volume of PBS. To evaluate peritoneal neutrophil recruitment, mice were killed at 4 h following injection of thioglycollate or HMGB1. Thereafter, the peritoneal lavage was collected and the number of emigrated neutrophils was quantitated by FACS analysis by staining for Gr-1 (May et al, 1998). Alternatively, the number of the granulocytes was analyzed by a conventional smear with Diffquick staining (Chavakis et al, 2002, 2003, 2004a) and both methods provided almost identical results.
Radiation bone marrow chimeras were prepared exactly as described previously (Lumsden et al, 2003). Wild-type recipient mice were irradiated with 950 rad and reconstituted with 1.5 × 107 bone marrow cells from RAGE-deficient mice (RAGE−/− → wt) or from wild-type mice (wt → wt) as a control. Additionally, irradiated RAGE−/− mice received bone marrow cells from wild-type mice (wt → RAGE−/−). Reconstitution of leukocyte populations was comparable in these groups (data not shown). Peritonitis experiments were performed 7 weeks after reconstitution.
Statistical analysis
Data were analyzed by analysis of variance with post hoc analysis (Bonferroni adjustment) using the SPSS software. P-values of <0.05 were regarded as significant.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary data
Acknowledgments
We acknowledge Dr M Kruhlak for help with confocal microscopy, G Sanchez and L Stepanyan (Bioqual Inc.) for bone marrow chimera experiments, D Winkler for genotyping, Dr A Mazzoni for help with the isolation of bone marrow cells, S Sharrow, T Adams and L Granger for help with FACS analysis, and Drs JJ Oppenheim and D Segal for critically reading the manuscript. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute (TC) and the Sigrid Jusélius Foundation (CGG). The authors declare no direct financial interest in this study. However, MEB is founder and part owner of HMGBiotech, which produces HMGB1.
References
- Andersson U, Wang H, Palmblad K, Aveberger AC, Bloom O, Erlandsson-Harris H, Janson A, Kokkola R, Zhang M, Yang H, Tracey KJ (2000) High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med 192: 565–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berlin-Rufenach C, Otto F, Mathies M, Westermann J, Owen MJ, Hamann A, Hogg N (1999) Lymphocyte migration in lymphocyte function-associated antigen (LFA)-1-deficient mice. J Exp Med 189: 1467–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhaus A, Schiekofer S, Schwaninger M, Andrassy M, Humpert PM, Chen J, Hong M, Luther T, Henle T, Kloting I, Morcos M, Hofmann M, Tritschler H, Weigle B, Kasper M, Smith M, Perry G, Schmidt AM, Stern DM, Haring HU, Schleicher E, Nawroth PP (2001) Diabetes-associated sustained activation of the transcription factor nuclear factor-κB. Diabetes 50: 2792–2808 [DOI] [PubMed] [Google Scholar]
- Bonaldi T, Talamo F, Scaffidi P, Ferrera D, Porto A, Bachi A, Rubartelli A, Agresti A, Bianchi ME (2003) Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J 22: 5551–5560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambien B, Pomeranz M, Schmid-Antomarchi H, Millet MA, Breittmayer V, Rossi B, Schmid-Alliana A (2001) Signal transduction involved in MCP-1-mediated monocytic transendothelial migration. Blood 97: 359–366 [DOI] [PubMed] [Google Scholar]
- Carman CV, Springer TA (2003) Integrin avidity regulation: are changes in affinity and conformation underemphasized? Curr Opin Cell Biol 15: 547–556 [DOI] [PubMed] [Google Scholar]
- Chavakis T, Bierhaus A, Al-Fakhri N, Schneider D, Witte S, Linn T, Nagashima M, Morser J, Arnold B, Preissner KT, Nawroth PP (2003) The pattern recognition receptor (RAGE) is a counterreceptor for leukocyte integrins: a novel pathway for inflammatory cell recruitment. J Exp Med 198: 1507–1515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavakis T, Bierhaus A, Nawroth PP (2004) RAGE (receptor for advanced glycation end products): a central player in the inflammatory response. Microbes Infect 6: 1219–1225 [DOI] [PubMed] [Google Scholar]
- Chavakis T, Hussain M, Kanse SM, Peters G, Bretzel RG, Flock JI, Herrmann M, Preissner KT (2002) Staphylococcus aureus extracellular adherence protein (Eap) serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat Med 8: 687–693 [DOI] [PubMed] [Google Scholar]
- Chavakis T, Keiper T, Matz-Westphal R, Hersemeyer K, Sachs UJ, Nawroth PP, Preissner KT, Santoso S (2004a) The junctional adhesion molecule-C promotes neutrophil transendothelial migration in vitro and in vivo. J Biol Chem 279: 55602–55608 [DOI] [PubMed] [Google Scholar]
- Chavakis T, May AE, Preissner KT, Kanse SM (1999) Molecular mechanisms of zinc-dependent leukocyte adhesion involving the urokinase receptor and β2-integrins. Blood 93: 2976–2983 [PubMed] [Google Scholar]
- Collison KS, Parhar RS, Saleh SS, Meyer BF, Kwaasi AA, Hammami MM, Schmidt AM, Stern DM, Al-Mohanna FA (2002) RAGE-mediated neutrophil dysfunction is evoked by advanced glycation end products (AGEs). J Leukoc Biol 71: 433–444 [PubMed] [Google Scholar]
- Coxon A, Rieu P, Barkalow FJ, Askari S, Sharpe AH, von Andrian UH, Arnaout MA, Mayadas TN (1996) A novel role for the beta 2 integrin CD11b/CD18 in neutrophil apoptosis: a homeostatic mechanism in inflammation. Immunity 5: 653–666 [DOI] [PubMed] [Google Scholar]
- Ding ZM, Babensee JE, SimonI SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, Smith CW, Ballantyne CM (1999) Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol 163: 5029–5038 [PubMed] [Google Scholar]
- Dumitriu IE, Baruah P, Bianchi ME, Manfredi AA, Rovere-Querini P (2005a) Requirement of HMGB1 and RAGE for the maturation of human plasmacytoid dendritic cells. Eur J Immunol 35: 2184–2190 [DOI] [PubMed] [Google Scholar]
- Dumitriu IE, Baruah P, Manfredi AA, Bianchi ME, Rovere-Querini P (2005b) HMGB1: guiding immunity from within. Trends Immunol 26: 381–387 [DOI] [PubMed] [Google Scholar]
- Ehlers MR (2000) CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect 2: 289–294 [DOI] [PubMed] [Google Scholar]
- Gahmberg CG (1997) Leukocyte adhesion: CD11/CD18 integrins and intercellular adhesion molecules. Curr Opin Cell Biol 9: 643–650 [DOI] [PubMed] [Google Scholar]
- Hogg N, Laschinger M, Giles K, McDowall A (2003) T-cell integrins: more than just sticking points. J Cell Sci 116: 4695–4705 [DOI] [PubMed] [Google Scholar]
- Kim JY, Park JS, Strassheim D, Douglas I, Diaz del Valle F, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama I, Ishizaka A, Abraham E (2005) HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol 288: L958–L965 [DOI] [PubMed] [Google Scholar]
- Liliensiek B, Weigand MA, Bierhaus A, Nicklas W, Kasper M, Hofer S, Plachky J, Grone HJ, Kurschus FC, Schmidt AM, Yan SD, Martin E, Schleicher E, Stern DM, Hammerling G, Nawroth PP, Arnold B (2004) Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest 113: 1641–1650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Yang H, Sakuragi T, Hu M, Mantell LL, Hayashi S, Al-Abed Y, Tracey KJ, Ulloa L, Miller EJ (2005) Alpha-chemokine receptor blockade reduces high mobility group box 1 protein-induced lung inflammation and injury and improves survival in sepsis. Am J Physiol Lung Cell Mol Physiol 289: L583–L590 [DOI] [PubMed] [Google Scholar]
- Lotze MT, Tracey KJ (2005) High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol 5: 331–342 [DOI] [PubMed] [Google Scholar]
- Lu H, Smith CW, Perrard CW, Bullard D, Tang L, Shappell SB, Entman ML, Beaudet AL, Ballantyne CM (1997) LFA-1 is sufficient in mediating neutrophil emigration in Mac-1-deficient mice. J Clin Invest 99: 1340–1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lumsden JM, Williams JA, Hodes RJ (2003) Differential requirements for expression of CD80/86 and CD40 on B cells for T-dependent antibody responses in vivo. J Immunol 170: 781–787 [DOI] [PubMed] [Google Scholar]
- May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT (1998) Urokinase receptor (CD87) regulates leukocyte recruitment via beta 2 integrins in vivo. J Exp Med 188: 1029–1037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayadas TN, Cullere X (2005) Neutrophil beta2 integrins: moderators of life or death decisions. Trends Immunol 26: 388–395 [DOI] [PubMed] [Google Scholar]
- Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA (2002) Syk is required for integrin signaling in neutrophils. Immunity 16: 547–558 [DOI] [PubMed] [Google Scholar]
- Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E (2006) High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol 290: C917–C924 [DOI] [PubMed] [Google Scholar]
- Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E (2004) Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem 279: 7370–7377 [DOI] [PubMed] [Google Scholar]
- Petty HR, Worth RG, Todd RF III (2002) Interactions of integrins with their partner proteins in leukocyte membranes. Immunol Res 25: 75–95 [DOI] [PubMed] [Google Scholar]
- Plow EF, Haas TA, Zhang L, Loftus J, Smith JW (2000) Ligand binding to integrins. J Biol Chem 275: 21785–21788 [DOI] [PubMed] [Google Scholar]
- Rezzonico R, Imbert V, Chicheportiche R, Dayer JM (2001) Ligation of CD11b and CD11c beta(2) integrins by antibodies or soluble CD23 induces macrophage inflammatory protein 1alpha (MIP-1alpha) and MIP-1beta production in primary human monocytes through a pathway dependent on nuclear factor-kappaB. Blood 97: 2932–2940 [DOI] [PubMed] [Google Scholar]
- Ross GD (2002) Role of the lectin domain of Mac-1/CR3 (CD11b/CD18) in regulating intercellular adhesion. Immunol Res 25: 219–227 [DOI] [PubMed] [Google Scholar]
- Rouhiainen A, Kuja-Panula J, Wilkman E, Pakkanen J, Stenfors J, Tuominen RK, Lepantalo M, Carpen O, Parkkinen J, Rauvala H (2004) Regulation of monocyte migration by amphoterin (HMGB1). Blood 104: 1174–1182 [DOI] [PubMed] [Google Scholar]
- Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, Chavakis T (2002) The junctional adhesion molecule 3 (JAM-3) on human platelets is a counter-receptor for the leukocyte integrin Mac-1. J Exp Med 196: 679–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418: 191–195 [DOI] [PubMed] [Google Scholar]
- Schmal H, Czermak BJ, Lentsch AB, Bless NM, Beck-Schimmer B, Friedl HP, Ward PA (1998) Soluble ICAM-1 activates lung macrophages and enhances lung injury. J Immunol 161: 3685–3693 [PubMed] [Google Scholar]
- Schmidt AM, Yan SD, Yan SF, Stern DM (2001) The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest 108: 949–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schymeinsky J, Then C, Walzog B (2005) The non-receptor tyrosine kinase Syk regulates lamellipodium formation and site-directed migration of human leukocytes. J Cell Physiol 204: 614–622 [DOI] [PubMed] [Google Scholar]
- Shi C, Zhang X, Chen Z, Robinson MK, Simon DI (2001) Leukocyte integrin Mac-1 recruits toll/interleukin-1 receptor superfamily signaling intermediates to modulate NF-κB activity. Circ Res 89: 859–865 [DOI] [PubMed] [Google Scholar]
- Sitrin RG, Pan PM, Srikanth S, Todd RF III (1998) Fibrinogen activates NF-kappa B transcription factors in mononuclear phagocytes. J Immunol 161: 1462–1470 [PubMed] [Google Scholar]
- Sotiriou SN, Orlova VV, Al-Fakhri N, Ihanus E, Economopoulou M, Isermann B, Bdeir K, Nawroth PP, Preissner KT, Gahmberg CG, Koschinsky ML, Chavakis T (2006) Lipoprotein(a) in atherosclerotic plaques recruits inflammatory cells through interaction with Mac-1 integrin. FASEB J 20: 559–561 [DOI] [PubMed] [Google Scholar]
- Springer TA (1994) Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell 76: 301–314 [DOI] [PubMed] [Google Scholar]
- Tang T, Rosenkranz A, Assmann KJ, Goodman MJ, Gutierrez-Ramos JC, Carroll MC, Cotran RS, Mayadas TN (1997) A role for Mac-1 (CDIIb/CD18) in immune complex-stimulated neutrophil function in vivo: Mac-1 deficiency abrogates sustained Fcgamma receptor-dependent neutrophil adhesion and complement-dependent proteinuria in acute glomerulonephritis. J Exp Med 186: 1853–1863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsung A, Sahai R, Tanaka H, Nakao A, Fink MP, Lotze MT, Yang H, Li J, Tracey KJ, Geller DA, Billiar TR (2005) The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J Exp Med 201: 1135–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ (1999) HMG-1 as a late mediator of endotoxin lethality in mice. Science 285: 248–251 [DOI] [PubMed] [Google Scholar]
- Xie C, Alcaide P, Geisbrecht BV, Schneider D, Herrmann M, Preissner KT, Luscinskas FW, Chavakis T (2006) Suppression of experimental autoimmune encephalomyelitis by extracellular adherence protein of Staphylococcus aureus. J Exp Med 203: 985–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan SF, Ramasamy R, Naka Y, Schmidt AM (2003) Glycation, inflammation and RAGE. A scaffold for the macrovascular complications of diabetes and beyond. Circ Res 93: 1159–1169 [DOI] [PubMed] [Google Scholar]
- Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ (2000) Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA 101: 296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Czura CJ, Tracey KJ (2005) The cytokine activity of HMGB1. J Leukoc Biol 78: 1–8 [DOI] [PubMed] [Google Scholar]
- Zhou M, Todd RF III, van de Winkel JG, Petty HR (1993) Cocapping of the leukoadhesin molecules complement receptor type 3 and lymphocyte function-associated antigen-1 with Fc gamma receptor III on human neutrophils. Possible role of lectin-like interactions. J Immunol 150: 3030–3041 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Supplementary data