

Abstract
The superoxide-producing phagocyte NADPH oxidase is activated during phagocytosis to destroy ingested microbes. The adaptor protein p40phox associates via the PB1 domain with the essential oxidase activator p67phox, and is considered to function by recruiting p67phox to phagosomes; in this process, the PX domain of p40phox binds to phosphatidylinositol 3-phosphate [PtdIns(3)P], a lipid abundant in the phagosomal membrane. Here we show that the PtdIns(3)P-binding activity of p40phox is normally inhibited by the PB1 domain both in vivo and in vitro. The crystal structure of the full-length p40phox reveals that the inhibition is mediated via intramolecular interaction between the PB1 and PX domains. The interface of the p40phox PB1 domain for the PX domain localizes on the opposite side of that for the p67phox PB1 domain, and thus the PB1-mediated PX regulation occurs without preventing the PB1–PB1 association with p67phox.
Keywords: crystal structure, NADPH oxidase, p40 phox , PtdIns(3)P, PX domain
Introduction
Upon cell stimulation, a variety of proteins are recruited to subcellular compartments, where they play their specific roles. Increasing attention has been focused on the mechanisms for stimulus-induced membrane targeting of proteins, a process that involves interactions of protein modules with membrane phospholipids (Cullen et al, 2001; Hurley and Meyer, 2001; Itoh and Takenawa, 2002; Lemmon, 2003). Among them, pleckstrin homology (PH) and phagocyte oxidase (phox) homology (PX) domains recognize specific phosphoinositides to recruit proteins to appropriate cell membranes. In general, interactions between phosphoinositides and proteins are regulated by kinases or phosphatases for inositol-containing lipids via producing specific phosphoinositides (Cullen et al, 2001; Hurley and Meyer, 2001; Itoh and Takenawa, 2002; Lemmon, 2003). For example, in response to cell stimulants, type I phosphatidylinositol 3-kinase (PI3K) is activated, leading to the synthesis of phosphatidylinositol-3,4,5-trisphosphate [PtdIns(3,4,5)P3], and thus proteins containing a PH domain that specifically binds to PtdIns(3,4,5)P3, such as the protein kinase Akt, are recruited to the membrane (Cullen et al, 2001; Hurley and Meyer, 2001; Itoh and Takenawa, 2002; Lemmon, 2003). On the other hand, there exist few examples in which changes in the protein rather than the lipids trigger the protein–lipid interaction (Karathanassis et al, 2002; Ago et al, 2003).
The phagocyte NADPH oxidase, dormant in resting cells, becomes activated during phagocytosis to produce superoxide, a precursor of microbicidal oxidants (Cross and Segal, 2004; Lambeth, 2004; Nauseef, 2004; Quinn and Gauss, 2004; Sumimoto et al, 2005). This enzyme plays a crucial role in host defense, as is exemplified by recurrent and life-threatening infections that occur in patients with chronic granulomatous disease because of the lack of the superoxide-producing system in phagocytes. The catalytic center of the phagocyte oxidase is the membrane-integrated protein gp91phox (also known as Nox2), which contains a complete electron-transporting apparatus from NADPH to molecular oxygen. Activation of gp91phox, tightly complexed with the membrane-bound protein p22phox, requires proteins present in the cytoplasm of resting cells: the small GTPase Rac and the SH3 domain-containing proteins p47phox, p67phox, and p40phox (Cross and Segal, 2004; Lambeth, 2004; Nauseef, 2004; Quinn and Gauss, 2004; Sumimoto et al, 2005). These proteins translocate upon cell stimulation to the membrane and interact with the gp91phox–p22phox complex, leading to superoxide production. In this process, p47phox localizes to the membrane via an SH3-mediated interaction with p22phox (Leto et al, 1994; Sumimoto et al, 1994; Shiose and Sumimoto, 2000) and a PX-mediated association with phosphoinositides (Ago et al, 2003), whereas the membrane recruitment of p67phox is mediated via its binding to p47phox (Heyworth et al, 1991; Kami et al, 2002). In contrast, Rac is independently targeted to the membrane and thus participates in the oxidase assembly (Heyworth et al, 1994).
The adaptor protein p40phox, starting from the N-terminus, possesses a PX domain, an SH3 domain, and a PB1 (phox and Bem 1) domain (Figure 1A): the PB1 domain mediates constitutive association with p67phox via a PB1–PB1 interaction (Ito et al, 2001; Noda et al, 2003). Although p40phox is dispensable to phagocyte oxidase activation in vitro, p40phox enhances the activation by facilitating membrane localization of the essential protein p67phox (and its associated protein p47phox) (Kuribayashi et al, 2002). The PX domain of p40phox specifically and strongly binds to phosphatidylinositol-3-phosphate [PtdIns(3)P], whereas the p47phox PX domain prefers other phosphoinositides such as phosphatidylinositol-3,4-bisphosphate [PtdIns(3,4)P2] but with a much lower affinity (Ago et al, 2001; Ellson et al, 2001a; Kanai et al, 2001). As PtdIns(3)P is enriched in the membranes of not only early endosomes but also phagosomes (Gillooly et al, 2000; Vieira et al, 2001; Ellson et al, 2001b), it is likely that p40phox participates in activation of the NADPH oxidase at the phagosomal membrane. Consistent with this, recent studies have shown that p40phox is crucial for phagocytosis-mediated activation of the oxidase (Ellson et al, 2006; Suh et al, 2006).
Figure 1.
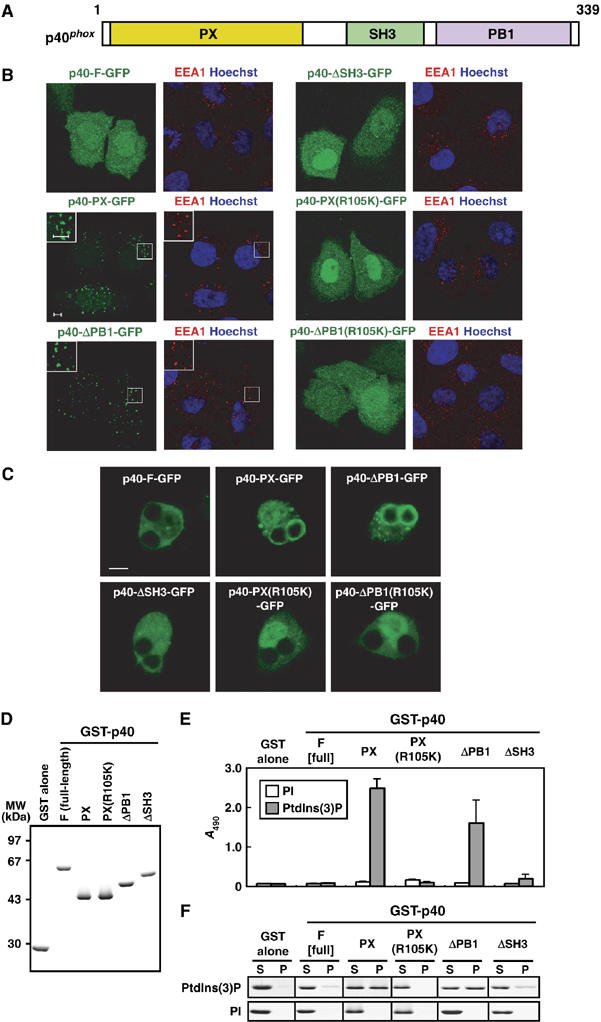
Role of the PB1 domain in p40phox binding to PtdIns(3)P. (A) A schematic diagram of p40phox. The PX, SH3, and PB1 domain are colored in yellow, green, and magenta. (B) Subcellular distribution of p40phox and its truncated proteins in transiently transfected HeLa cells. Left panel, distribution of GFP-tagged p40phox in fixed HeLa cells (green); right panel, distribution of endogenous EEA-1 (red) and Hoechst staining of the nucleus (blue) in the same field of fixed HeLa cells. The insets show the magnified views. Scale bar, 5 μm. (C) Localization of ectopically expressed p40phox and its truncated proteins in RAW264.7 cells ingesting IgG-coated beads. Scale bar, 5 μm. (D) SDS–PAGE analysis of GST-tagged p40phox proteins used in the following lipid-binding assays (E, F). Proteins were stained with Coomassie brilliant blue. (E) ELISA-format lipid-binding assay. Each well of a titer plate coated with phospholipids containing 10% of PI or PtdIns(3)P was incubated with the indicated p40phox proteins. The bound proteins were stained as described under Materials and methods. Each value represents the mean of data with bars representing s.d. in more than three independent experiments. (F) Co-sedimentation assay with liposomes. Liposomes containing 5% of PI or PtdIns(3)P were incubated with the indicated p40phox proteins. After centrifugation, proteins in the supernatant (S) and precipitate (P) fractions were analyzed by SDS–PAGE. The experiments have been repeated more than three times with similar results.
The phosphoinositide-binding activity of the p47phox PX domain is known to be regulated by protein modification although the PX domain is normally inaccessible to phosphoinositides because of its possible intramolecular interaction with the SH3 domains (Karathanassis et al, 2002; Ago et al, 2003). On the other hand, it has remained to be elucidated whether the p40phox PX domain binds to PtdIns(3)P in a regulated manner. Here we show that the PB1 domain of p40phox prevents the PX domain from interacting with PtdIns(3)P, both in vivo and in vitro: the full-length wild-type p40phox, but not the PB1-truncated one, is incapable of localizing to early endosomes or phagosomes. We have also determined the crystal structure of the full-length p40phox, which reveals that the inhibition of the PtdIns(3)P-binding activity is mediated via an intramolecular interaction between the PB1 and PX domains.
Results
Role for the PB1 domain in the recruitment of p40phox to early endosomes and phagosomes
As shown in Figure 1A, p40phox contains PX, SH3, and PB1 domains from the N-terminus. The isolated PX domain of p40phox is known to be targeted to early endosomes, abundant in PtdIns(3)P, in a manner dependent on its PtdIns(3)P-binding activity (Ago et al, 2001; Ellson et al, 2001a; Kanai et al, 2001). However, it has remained unknown whether the full-length p40phox is also recruited to early endosomes. To address this question, we expressed the full-length wild-type p40phox as a fusion-protein containing green fluorescence protein (GFP) at the C-terminus (p40-F-GFP) as well as the PX domain alone (p40-PX-GFP) in HeLa cells. As shown in Figure 1B and in agreement with previous observations (Ago et al, 2001), p40-PX-GFP co-localized with EEA1 (early endosome autoantigen 1), a marker protein of early endosomes. In contrast, p40-F-GFP was not targeted to early endosomes, suggesting that the PX-mediated PtdIns(3)P-binding activity of p40phox is suppressed by other regions of this protein. To test this possibility, we expressed a mutant protein lacking the SH3 or PB1 domain and analyzed their localization in HeLa cells. Although deletion of the SH3 domain did not lead to recruitment of the protein to early endosomes, p40-ΔPB1-GFP was targeted to early endosomes (Figure 1B). The endosomal localization is likely driven by the PtdIns(3) P-binding activity of the PX domain, as p40-ΔPB1(R105K)-GFP carrying the R105K substitution, a mutation resulting in a loss of the PtdIns(3)P-binding activity (Ago et al, 2001), failed to localize to early endosomes. The same results were obtained when N-terminally GFP-fused proteins were used instead of C-terminally GFP-fused ones (data not shown). These findings indicate that the PB1 domain, but not the SH3 domain, appears to block the endosomal localization of p40phox by preventing the PX domain from interacting with PtdIns(3)P.
It is established that PtdIns(3)P also accumulates in the phagosomal membrane in mouse macrophage-like RAW264.7 cells (Ellson et al, 2001b; Vieira et al, 2001). To investigate the role of the p40phox PB1 domain on phagosomal localization, we transfected RAW264.7 cells with cDNAs encoding the wild-type or mutant p40phox. When the transfected cells were ingesting IgG-coated Latex beads, p40-PX-GFP, but not p40-F-GFP, translocated to the phagosomal membrane (Figure 1C), suggesting that the ability to bind to PtdIns(3)P is attenuated in the full-length p40phox also in phagocytes. In addition, p40-ΔPB1-GFP was recruited to the phagosomal membrane in a manner that requires the PX-mediated binding to PtdIns(3)P, whereas p40-ΔSH3-GFP failed to localize to phagosomes (Figure 1C). Thus, localization of p40phox to phagosomes as well as to early endosomes seems to be negatively regulated by the PB1 domain. Taken together, the present findings indicate that, in cells, the PX-mediated PtdIns(3)P-binding activity of p40phox is inhibited by the PB1 domain but not by the SH3 domain.
Role for the PB1 domain in the PtdIns(3)P-binding activity of p40phox in vitro
To determine whether the p40phox PB1 domain directly inhibits the PX-mediated PtdIns(3)P-binding activity, we prepared recombinant proteins and tested their activities by two different in vitro lipid-binding assays (Figure 1D–F). First, we performed an enzyme-linked immunosorbent assay (ELISA)-format lipid-binding assay (Ghosh et al, 1996; Oikawa et al, 2004). As expected from the findings using intact cells, the full-length p40phox was incapable of binding to PtdIns(3)P coated on wells under the conditions where the isolated PX domain fully bound (Figure 1E). Although a mutant p40phox lacking the SH3 domain failed to interact with PtdIns(3)P, deletion of the PB1 domain allowed effective binding to PtdIns(3)P. This binding seems to be specific, because the PB1-truncated mutant protein did not bind to wells coated with phosphatidylinositol (PI) instead of PtdIns(3)P. Essentially the same results were obtained by a liposome co-sedimentation: the isolated PX domain and PB1-truncated p40phox co-precipitated with liposomes containing PtdIns(3)P but not with those containing PI, whereas the full-length or SH3-truncated proteins were recovered exclusively in the supernatant, indicating the lack of the PtdIns(3)P-binding activity (Figure 1F). Thus it is likely that the PB1 domain but not the SH3 domain directly interferes with the PX-mediated PtdIns(3)P-binding activity of p40phox.
Overall structure of the full-length p40phox
The present functional analyses indicate that the PX domain in intact p40phox is blocked by the PB1 domain but not by the SH3 domain both in vivo and in vitro. To directly examine this possibility, we crystallized the full-length p40phox for structure determination by X-ray diffraction studies (Honbou et al, 2006). The crystal of intact p40phox revealed that there are two p40phox molecules in the asymmetric unit (Figure 2A). One molecule has a relatively clear electron density (hereafter referred to this molecule as molecule A), but the electron density of the other molecule is sparse especially for the SH3 and PB1 domains (hereafter referred to this molecule as molecule B). For molecule A, the N-terminus 8 residues (1–8), the C-terminus 9 residues (331–339), the linker region between the SH3 and PB1 domains (154–168), and some residues in the loop regions were not observed because of the structural disorder, and thus could not be modeled in the electron density map (see Structural statistics in Table I). To investigate the relative domain orientation between molecules A and B, we superimposed the PX domain of molecule B onto that of molecule A (red and blue, respectively, in Figure 2B). The overlay of the two molecules shows that the relative position of the PX domain to the PB1 domain is fixed in p40phox, although that to the SH3 domain is not restricted. The finding suggests that the interaction between the PX and PB1 domains is not due to crystallization artifact. To our knowledge, this is the first full-length structure that has been determined among the NADPH oxidase proteins.
Figure 2.
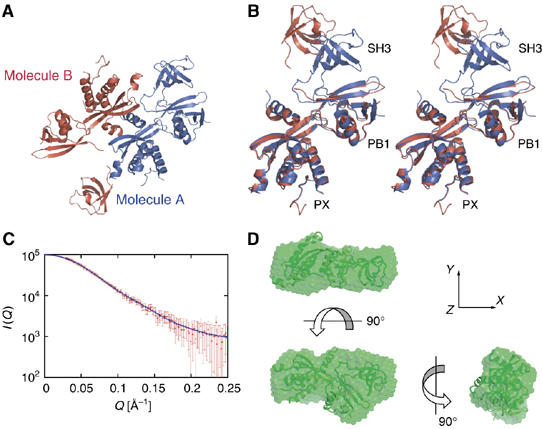
The overall structure of p40phox. (A) Structure of the two molecules of p40phox in the asymmetric unit. Molecule A, which has a clear electron density, is colored blue, and molecule B, which has a poor electron density, is colored red. (B) A stereo pair of a ribbon diagram of p40phox. Molecule B is superimposed onto Molecule A by the PX domain. Each molecule is color coded as in (A). (C) Experimental and calculated small-angle X-ray scattering curves for p40phox. The smooth curve in red corresponds to the scattering curve calculated from the dummy atom model derived using the program DAMMIN, which is superposed on the experimental curve (dots with error bars). (D) The crystal structure of p40phox (Molecule A, ribbon diagram) is superimposed onto the low-resolution model restored by the DAMMIN, shown as green surface representation. The right model is rotated by 90° around the y-axis and the upper model is rotated by 90° around the x-axis according to the arrows.
Table 1.
Data collection and refinement statistics
| p40phox | |
|---|---|
| Data collection | |
| Space group | C2221 |
| Cell dimensions | |
| a, b, c (Å) | 146.27, 189.81, 79.88 |
| Resolution (Å) | 50.0–3.00 (3.11–3.00) |
| Rmerge | 7.1 (47.9) |
| I/σI | 20.3 (3.7) |
| Completeness (%) | 99.5 (98.5) |
| Redundancy | 7.1 |
| Refinement | |
| Resolution (Å) | 40.8–3.15 |
| No. of reflections (total) | 19 197 |
| No. of reflections (test set) | 1912 |
| Rwork/Rfree | 26.3/30.2 |
| No. of atoms | |
| Protein | 4252 |
| Ligand/ion | — |
| Water | — |
| B-factors | |
| Protein | 82.3 |
| R.m.s deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (deg) | 1.6 |
| Highest-resolution shell is shown in parentheses. | |
Comparison of the molecular shapes of p40phox in solution and in crystal
To determine the global structure of p40phox in solution, we performed small-angle X-ray scattering (SAXS) experiments. A low-resolution model of p40phox was determined by ab initio molecular shape analysis using the simulated annealing program DAMMIN (Svergun, 1999). Ten independent simulated annealing calculations were carried out for small angle X-ray scattering data, and the models were superimposed to construct an averaged model. Although all the calculated models yielded nearly identical scattering curves, the best model provided a fit to the experimental data with χ2=0.41, as shown in Figure 2C. The averaged low-resolution model roughly fits the molecular shape and dimension of molecule A in the crystal structure, with respect to the molecular shape and dimension (Figure 2D). There were some slight differences between the crystal structure and the low-resolution model, probably due to the mobile nature of the SH3 domain. Thus the SAXS data support the idea that the PX–PB1 interaction is not due to the artifact of crystal packing, but rather represents an intrinsic property of intact p40phox.
Interface between the PX and PB1 domains of p40phox
The overall structure of p40phox in molecule A is shown in Figure 3A, where the PX domain is colored yellow, the PB1 domain pink, and the SH3 domain green, respectively. A close-up view of the interface between the PX and PB1 domains, which is enclosed by a rectangle in the overall structure, is displayed as a stereo in Figure 3B. The total buried surface area in the interface of about 1300 Å2 is slightly smaller than that of the average value observed for various protein–protein complexes (1600±400 Å2) (Lo Conte et al, 1999). In Figure 3B, we showed the side chains of amino-acid residues involved in the interface between the PX and PB1 domains in a stick model: F35, located on the loop between β1 and β2 of the PX domain, protrudes into the hydrophobic pocket lined by V257, P265, L273, F320, and W322 of the PB1 domain, which forms a critical interaction between the two domains. Moreover, there are ionic interactions between the basic cluster of H38, R58, and R60 in the PX domain and the acidic cluster of E259 and D269 in the PB1 domain. Intriguingly, these amino acids are evolutionarily conserved from fish to mammals (Inoue et al, 2004).
Figure 3.
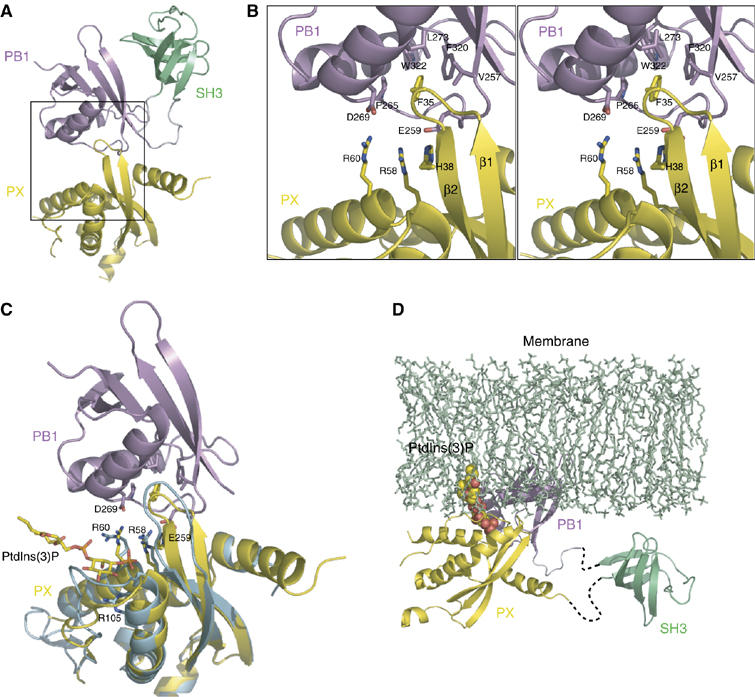
The interface between the PX domain and the PB1 domain. (A) A ribbon diagram of Molecule A. Each domain of p40phox is colored coded as in Figure 1A. (B) Stereo diagram of a close-up view (the region corresponds to the rectangle in (A)) of the interface between the PX domain and the PB1 domain. Amino-acid residues involved in the interaction are shown in a stick representation. (C) Superposition of the p40phox PX domain complexed with PtdIns(3)P (blue; PDB code 1H6H) onto that of crystal structure of p40phox (yellow). The SH3 domain is removed for clarity. PtdIns(3)P, R58, R60, and R105 of the each PX domain, and E259 and D269 of the PB1 domain, which interact with R58 and R60 respectively, are shown in a stick representation. (D) The model of the interaction between the full-length p40phox and membrane-bound PtdIns(3)P. SH3 domain is connected by dashed line, since relative position of the SH3 domain does not seem to be restricted.
To elucidate how the PB1 domain affects the binding of the PX domain to PtdIns(3)P, we superimposed the structure of the p40phox PX domain (blue in Figure 3C) complexed with a soluble PtdIns(3)P (Bravo et al, 2001) onto that of the full-length p40phox (yellow, molecule A in Figure 3C). We observed a significant structural difference between the two structures in the loop region around 31–38 of the PX domain. This structural difference enables the aromatic ring of F35 in intact p40phox to protrude into the hydrophobic pocket in the PB1 domain as indicated in Figure 3B. Although the PtdIns(3)P-binding site on the PX domain is close to the interface with the PB1 domain, the PB1 domain does not seem to restrict the access of PtdIns(3)P in intact p40phox (Figure 3C). R58 and R105 in the PX domain are reported to be specificity determinants for the recognition of PtdIns(3)P. The side chains of R58 and R105 do not change their position in either structure and thus can recognize the 3-phosphate group and the 4- and 5-OH groups of PtdIns(3)P, respectively. On the other hand, the side-chain orientation of R60 is appreciably changed to interact with the 1-phosphate group of PtdIns(3)P in the complex; however, considering that R60 plays a minor role in the binding to PtdIns(3)P (Bravo et al, 2001), the salt bridge between R60 and D269 may be maintained in the p40phox–PtdIns(3)P interaction. Thus, as shown in the model proposed here (Figure 3C), the PB1 domain does not directly interfere with binding of the polar head of PtdIns(3)P to the PX domain. Instead the PB1 domain in p40phox causes a steric clash with the membrane when the PX domain interacts with membrane-embedded PtdIns(3)P (Figure 3D). As a result, the PX domain in p40phox cannot be tethered to the membrane.
To verify the significance of specific amino-acid residues on the PX–PB1 interface, we substituted alanine for E259, D269, and F320 in the PB1 domain. As shown in Figure 4A, the three mutant proteins p40-F(E259A)-GFP, p40-F(D269A)-GFP, and p40-F(F320A)-GFP were all targeted to early endosomes. The effects of the mutations were abrogated by further introduction of the R105K substitution, indicating the requirement for the PX-mediated PtdIns(3)P-binding activity. Similarly, when expressed in RAW264.7 cells, the three mutant proteins translocated to phagosomes in a manner dependent on the binding of PtdIns(3)P (Figure 4B). In addition, mutant proteins carrying the E259A, D269A, or F320A substitution in the PB1 domain were capable of binding to PtdIns(3)P in vitro via the PX domain, as estimated by the two different lipid-binding assays: ELISA-format one (Figure 4D) and liposome co-sedimentation one (Figure 4E). Thus, E259, D269, and F320 directly interact with the PX domain, whose interaction prevents this domain from binding to PtdIns(3)P both in vivo and in vitro.
Figure 4.
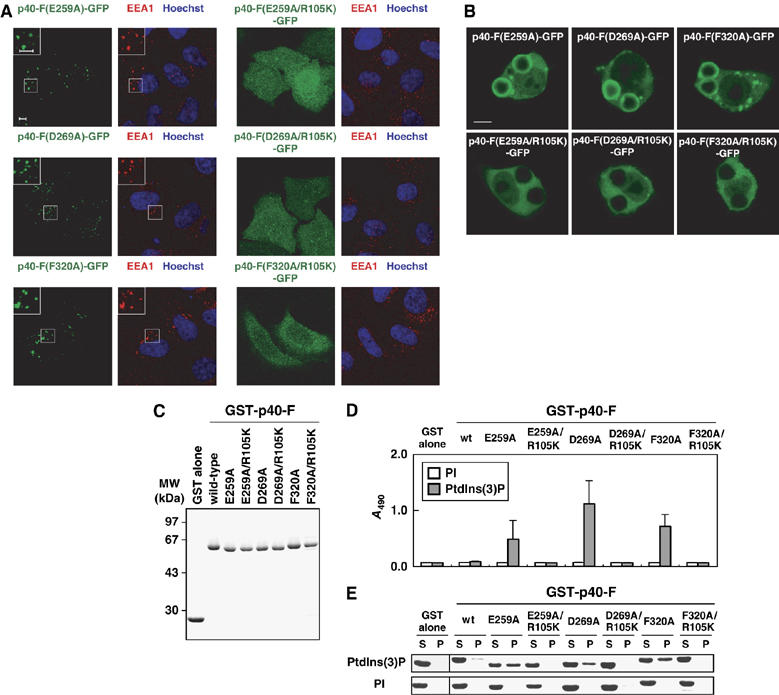
Role of E259, D269, and F320, residues in the PB1 interface for the PX domain, in the PtdIns(3)P-binding activity. (A) Subcellular distribution of p40phox and its mutant proteins carrying the indicated amino-acid substitution in transiently transfected HeLa cells. Left panel, distribution of GFP-tagged p40phox in fixed HeLa cells (green); right panel, distribution of endogenous EEA-1 (red) and Hoechst staining of the nucleus (blue) in the same field of fixed HeLa cells. The insets show the magnified views. Scale bar, 5 μm. (B) Localization of ectopically expressed p40phox and its mutant proteins in RAW264.7 cells ingesting IgG-coated beads. Scale bar, 5 μm. (C) SDS–PAGE analysis of GST-tagged p40phox and its mutant proteins carrying the indicated amino-acid substitution, which were used in the following lipid-binding assays (D, E). (D) The PtdIns(3)P-binding activity estimated by the ELISA-format phosphoinositide-binding assay as in Figure 1E. (E) The PtdIns(3)P-binding activity estimated by the liposome co-sedimentation assay as in Figure 1F.
Influence of the p40phox PX domain on the PB1–PB1 interaction between p40phox and p67phox
The PB1 domains of p40phox and p67phox are considered to constitutively form a heterodimer. The complex formation involves direct interaction of the OPCA motif presented on the p40phox PB1 domain with the conserved lysine residue on the p67phox PB1 domain (Ito et al, 2001; Noda et al, 2003). To clarify the influence of the PX domain on the heterodimerization of the PB1 domains, we superimposed the structure of the p40phox PB1 domain in the p40phox–p67phox PB1 complex onto that of intact p40phox. As shown in Figure 5A, the OPCA motif of p40phox is located on the opposite surface of the PX-binding region and therefore there is no steric clash between the p40phox PX domain and the p67phox PB1 domain. There is little structural difference except for structural disorders at the C-terminus (encircled by dotted lines in Figure 5A) and at several loop regions. Because the C-terminal region of p40phox is not included in the canonical PB1 domain but participates in the heterodimeric interaction, the structural disorder observed at the C-terminus is probably due to the absence of the p67phox PB1 domain.
Figure 5.
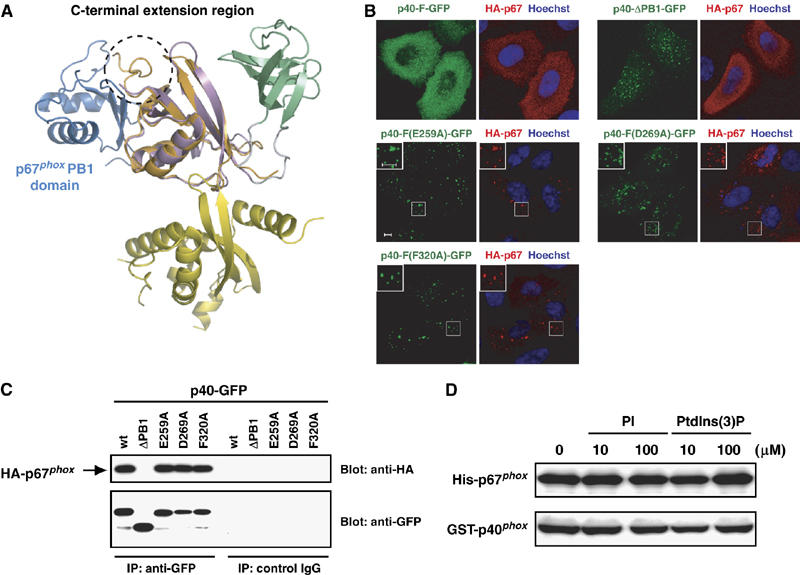
Dual roles for the PB1 domain of p40phox. (A) Ribbon diagram of the superposition of p40phox/p67phox PB1 heterodimer (blue is p67phox PB1 and orange is p40phox PB1; PDB code 1OEY) onto that of the full-length p40phox (color coded as in Figure 1A). A C-terminal extension region is shown in dashed circle. (B) Subcellular distribution of GFP-tagged p40phox and HA-tagged p67phox in transiently transfected HeLa cells. Left panel, distribution of GFP-tagged p40phox in fixed HeLa cells (green); right panel, distribution of endogenous HA-tagged p67phox (red) and Hoechst staining of the nucleus (blue) in the same field of fixed HeLa cells. The insets show the magnified views. Scale bar, 5 μm. (C) Proteins of lysates prepared from HeLa cells expressing both p40phox and p67phox were immunoprecipitated (IP) with the anti-GFP or control IgG, and then analyzed by immunoblot (Blot) with the anti-HA or anti-GFP antibodies. (D) In vitro interaction between purified GST-p40phox and His-tagged p67phox in the presence or absence of PtdIns(3)P.
To test whether the interaction of the p40phox PB1 domain with the p67phox PB1 domain occurs independently of that with the p40phox PX domain in vivo, we expressed both GFP-fused p40phox and HA-tagged p67phox proteins in HeLa cells. When the full-length p40phox was co-expressed, both p67phox and p40phox were found throughout the cytoplasm (Figure 5B). In cells co-expressing p40-ΔPB1-GFP, a mutant defective in p67phox binding, p67phox retained in the cytoplasm despite the endosomal localization of p40-ΔPB1-GFP (Figure 5B). On the other hand, p67phox co-localizes at early endosomes with mutant p40phox proteins carrying a substitution that disrupts the intramolecular PX–PB1 interaction (E259A, D269A, and F320A). Thus these mutations do not seem to affect the PB1-mediated association with p67phox, which is confirmed by the following observations: the mutant p40phox proteins (p40-F(E259A)-GFP, p40-F(D269A)-GFP, and p40-F(F320A)-GFP), but not p40-ΔPB1-GFP, were co-immunoprecipitated with HA-tagged p67phox to an extent similar to the wild-type p40phox (Figure 5C). These findings indicate that the integrity of the mutant proteins carrying an amino-acid substitution in the PB1 domain is maintained, and that the p40phox PB1 domain constitutively associates with the p67phox PB1 domain in a manner independent of the p40phox PX domain. Consistent with this, direct interaction between the full-length p40phox and p67phox was not affected by PtdIns(3)P as indicated by an in vitro pull-down assay using purified proteins (Figure 5D). In addition, the presence of the full-length p67phox did not induce the binding of the full-length p40phox to PtdIns(3)P in the liposome co-sedimentation assay (data not shown), supporting the idea that the heterodimerization of the PB1 domains does not affect the intramolecular interaction between the PB1 and PX domains in p40phox.
Discussion
The present structural and functional analyses reveal that binding of the p40phox PX domain to PtdIns(3)P is negatively regulated by its intramolecular interaction with the PB1 domain both in vivo and in vitro. It is generally considered that interactions between phosphoinositides and proteins are regulated by kinases or phosphatases for inositol-containing lipids via producing specific phosphoinositides, rather than by protein modification (Cullen et al, 2001; Hurley and Meyer, 2001; Itoh and Takenawa, 2002; Lemmon, 2003). Indeed, several proteins containing a PX domain constitutively associate with early endosomes (Xu et al, 2001a, 2001b). In contrast, the PX domain of p47phox is likely regulated by stimulus-induced phosphorylation of this protein (Karathanassis et al, 2002; Ago et al, 2003). In the resting state, the p47phox PX domain seems to be inhibited by its interaction with the SH3 domains; a phosphorylation-driven conformational change of p47phox renders the PX domain in a state accessible to phosphoinositides. This induced-binding plays a crucial role in both membrane translocation of p47phox and activation of the phagocyte NADPH oxidase (Ago et al, 2003). On the other hand, we showed here that, although p40phox also harbors an SH3 domain, the p40phox PX domain is regulated by directly interacting with the PB1 domain but not with the SH3 domain, as shown in the present structure-based analyses. It is known that two types of PI3K are activated during phagosome formation: type I PI3K is initially activated at the phagocytic cup to produce PtdIns(3,4,5)P3 and its metabolite PtdIns(3,4)P2; and after phagosome closure, when the activity of the type I enzyme is attenuated, type III PI3K becomes active to synthesize PtdIns(3)P (Ellson et al, 2001b; Vieira et al, 2001). Interestingly, the p47phox PX domain has a preference for PtdIns(3,4)P2 as its target (Ago et al, 2001, 2003; Kanai et al, 2001; Karathanassis et al, 2002), whereas the p40phox PX domain specifically binds to PtdIns(3)P. Thus the contents of these phosphoinositides and the binding activities of the PX domains are both regulated during phagocytosis. Activation of the NADPH oxidase at the phagosome may require coordinated functions of the PX domains of p47phox and p40phox: the initial oxidase complex assembles at the phagocytic cup in a manner dependent on the p47phox PX domain and PtdIns(3,4)P2, whereas the activated oxidase localizes to the closed phagosome via interaction of the p40phox PX domain with PtdIns(3)P. The strict control of the protein–lipid interaction by both phosphoinositide synthesis and protein conformational change appears to be particularly important for the phagocyte oxidase, as superoxide produced by the oxidase and its derivatives are toxic not only to invading microbes but also to host cells.
Based on the crystal structure of the full-length p40phox, we propose a model where the PX domain is inaccessible to the membrane-embedded PtdIns(3)P. The inaccessibility is due to the fact that the PB1 domain sterically hinders the access of the PtdIns(3)P-binding site towards the surface of the membrane and thus towards the membrane-embedded PtdIns(3)P, rather than due to the fact that the PB1 domain directly masks the binding site or stabilizes a conformation in which the polar head of PtdIns(3)P is unable to bind (Figure 3C and D). This model explains well the previous observation that the full-length p40phox is capable of binding to a soluble PtdIns(3)P, harboring C4 fatty acids, to the same extent as the isolated PX domain of p40phox (Bravo et al, 2001). On the other hand, the SH3 domain is mobile in p40phox, which is consistent with the present observations that this domain is not involved in inhibition of the PX domain binding to PtdIns(3)P (Figure 1). It has been reported that the full-length p40phox binds to PtdIns(3)P as well as the isolated PX domain, using surface plasmon resonance/lipid monolayer assays (Stahelin et al, 2003). Although the reason for the discrepancy is presently unknown, it might be possible that the position of PtdIns(3)P is relatively flexible in the lipid monolayer, and thus the PX domain of the full-length p40phox is accessible to the polar head of PtdIns(3)P.
The PB1 domain is a protein module that mediates a protein–protein interaction by directly binding to a PB1 domain on another protein (Ito et al, 2001; Terasawa et al, 2001; Kuribayashi et al, 2002; Noda et al, 2003; Wilson et al, 2003; Yoshinaga et al, 2003). It is well established that, in the resting state, p40phox tightly associates with p67phox via the PB1–PB1 interaction (Ito et al, 2001; Wilson et al, 2003). This implies, together with the present findings, that the intermolecular PB1–PB1 and intramolecular PX–PB1 interactions are not mutually exclusive but occur simultaneously. This is well explicable by the present crystal structure, in which the interface between the p40phox PB1 and PX domains is on the side opposite to its interface with the p67phox PB1 domain (Figure 5A). In addition, binding of p40phox to PtdIns(3)P, which is induced by disruption of the PX–PB1 interaction, does not result in an impaired association with p67phox (Figure 5B–D). Thus the PB1-mediated interaction of p40phox with p67phox is likely maintained independently of the PX-mediated binding to PtdIns(3)P. This allows p40phox to recruit the essential oxidase activator p67phox to PtdIns(3)P-rich phagosomal membranes, so that the components of the phagocyte NADPH oxidase can be assembled, producing a large amount of superoxide for microbial killing (Cross and Segal, 2004; Lambeth, 2004; Nauseef, 2004; Quinn and Gauss, 2004; Sumimoto et al, 2005). This scenario is supported by the following previous observations. Using K562 cells ectopically expressing oxidase proteins, we have previously shown that p40phox, albeit not essential for the oxidase activity, enhances oxidase activation by facilitating the membrane translocation of the indispensable proteins p67phox and p47phox, and that this enhancement requires the PB1-mediated interaction between p40phox and p67phox (Kuribayashi et al, 2002). The positive role of p40phox has recently been confirmed by experiments using neutrophils from p40phox-deficient mice (Ellson et al, 2006). Intriguingly, phagocytosis-induced oxidase activation and bacterial killing are incompletely but severely impaired in p40phox-deficient neutrophils (Ellson et al, 2006). Another recent study has shown that p40phox plays an essential role in oxidase activation during phagocytosis of IgG-coated particles in the cells ectopically expressing other oxidase proteins and the FcγIIA receptor; the effect of p40phox is totally dependent on the PX-mediated PtdIns(3)P-binding activity (Suh et al, 2006). Taken together, our and others' findings suggest that p40phox participates in oxidase activation at phagosomes by tethering p67phox (and its associated protein p47phox) to PtdIns(3)P-containing phagosomes, a process that requires the simultaneous functions of the PB1 and PX domains.
It has been shown that, during ingestion of IgG-coated particles, the full-length p40phox fused to yellow fluorescent protein, as well as to the isolated p40phox PX domain, is recruited to phagosomes of human neutrophil-like PLB-985 cells or those of the cells with exogenous gene expression for other oxidase proteins and the FcγIIA receptor (Suh et al, 2006). In contrast, in the present study, the GFP-fused full-length p40phox expressed in RAW264.7 macrophages did not translocate to phagosomes. This may suggest that a limiting quantity of an interacting partner is required to activate the ‘PtdIns(3)P-binding switch'. However, co-expression of p47phox and p67phox in RAW264.7 macrophages does not force p40phox-GFP to recognize PtdIns(3)P-rich phagosomes (R Minakami and H Sumimoto, unpublished observation). It seems thus possible that a heretofore unidentified partner of p40phox, which assists a conformational change to allow the PX domain to become accessible to PtdIns(3)P, is abundant in neutrophils but not in macrophages.
In addition, disruption of the PX–PB1 interaction in p40phox, leading to binding of the PX domain to PtdIns(3)P, might involve phosphorylation of p40phox. It has been shown that p40phox becomes phosphorylated at T154 and S315 by protein kinase C (PKC) (Bouin et al, 1998), and that PKC is activated at the phagosomal membrane (Ueyama et al, 2004); however, since T154 and S315 are not located near the PX–PB1 interface, it seems unlikely that the phosphorylation of these residues participates in the disruption of the PX–PB1 interaction. Consistent with this, the addition of phorbol myristate acetate, a potent activator of PKC, to RAW264.7 cells ingesting IgG-coated beads does not induce recruitment of the full-length p40phox to phagosomes (R Minakami and H Sumimoto, unpublished observation). Further studies are needed to determine the mechanism underlying the disruption of the PX–PB1 interaction in p40phox, which is considered to play a crucial role in assembly of the NADPH oxidase at phagosomes.
Materials and methods
Plasmid construction
The DNA fragments encoding the full-length of p40phox (p40-F; amino-acid residues 1–339), p40-PX (1–167), p40-ΔSH3 (the full-length lacking amino acids 167–233), and p40-ΔPB1 (1–233) were amplified from the human p40phox cDNA by polymerase chain reaction (PCR) with specific primers. Mutations leading to the indicated amino-acid substitutions were introduced by PCR-mediated site-directed mutagenesis. The PCR product was ligated to pGEX-6P (GE Healthcare Bio-Sciences) for bacterial expression as a protein fused to glutathione S-transferase (GST), to pEGFP-C1 (Clontech) for expression as an N-terminally GFP-tagged protein in mammalian cells, or to a modified pEGFP vector for expression as a C-terminally GFP-tagged protein in mammalian cells. For expression of HA-tagged p67phox, the cDNA was ligated to pEF-BOS as previously described (Miyano et al, 2006). All the constructs were sequenced for confirmation of their identities.
Cell culture, transfection, and confocal imaging
HeLa and mouse macrophage-like RAW264.7 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. HeLa cells were transfected using LipofectAMINE and Plus reagent (Invitrogen) and cultured for 18–24 h. Cells were fixed in 1.2% formaldehyde, permeabilized in 0.1% Triton X-100, blocked with 3% bovine serum albumin (BSA), and stained with anti-early endosome antigen 1 (EEA1) monoclonal antibody (Transduction Laboratories) or anti-HA monoclonal antibody (HA.11; Covance). Alexa Fluor 594-labeled goat anti-mouse IgG antibody (Invitrogen) was used as a secondary antibody. Cell nuclei were stained with Hoechst 33342.
Transfection of RAW264.7 cells with cDNAs for the wild-type and mutant p40phox was performed with Cell Line Nucleofector™ Kit V (amaxa) using the Nucleofector™ apparatus (amaxa), according to the manufacturer's instruction. The transfected RAW264.7 cells were plated for 3 h on 35-mm glass-bottom dishes (MatTek). For phagocytosis assay, Latex beads (Polybead Carboxylate Microspheres, 4.5 μm; Polysciences) were opsonized by sequential incubation with BSA and mouse-derived anti-BSA IgG (Sigma). After the culture medium was replaced with HEPES-buffered saline (120 mM NaCl, 5 mM KCl, 5 mM glucose, 1 mM MgCl2, 1 mM CaCl2 and 17 mM HEPES, pH 7.4), cells were incubated with IgG-opsonized beads for 10 min at 37°C, and fixed in 1.2% formaldehyde. Samples were analyzed with a laser-scanning confocal microscope LSM510 (Carl Zeiss). For live phagocytosis assays, cells were maintained at 37°C on a microscope stage incubator. Phagocytosis was initiated by adding of IgG-opsonized beads. Confocal fluorescence images of live cells were captured every 20 s with the LSM510 microscope.
In vitro lipid-binding assays
The wild-type and mutant p40phox were expressed as proteins fused to GST in the Escherichia coli strain BL21, and purified using glutathione-Sepharose-4B (GE Healthcare Bio-Sciences), as previously described (Ito et al, 2001; Ago et al, 2003). Synthetic PI or PtdIns(3)P with C16 fatty acids were purchased from Echelon Biosciences Inc.; phosphatidylethanolamine (PE) and phosphatidylcholine (PC) prepared from bovine brain were obtained from Sigma.
The ELISA-format lipid-binding assay for lipid binding was performed by the method of Ghosh et al (1996) with minor modifications (Oikawa et al, 2004). Briefly, phospholipid solutions containing PE (72%) and PC (18%) with 10% of PI, or PtdIns(3)P in ethanol, were applied to the wells of a 96-well titer plate (0.1 μg lipid for each well), and air dried for 12 h at room temperature. The remaining binding sites on the wells were blocked for 1 h with 5% BSA in PBS (137 mM NaCl, 2.68 mM KCl, 8.1 mM Na2HPO4, and 1.47 mM KH2PO4, pH 7.4). GST-fusion proteins (1.0 μg ml−1) in PBS containing 5% BSA were incubated for 45 min in the wells coated with phospholipids. The wells were washed three times with PBS containing 0.05% Tween-20, and overlaid with glutathione-conjugated peroxidase (Sigma). The bound GST-fusion proteins were measured by reaction with o-phenylenediamine dihydrochloride: after the termination of the reaction with HCl, the absorbance at 490 nm was determined with a Wallac 1420 ARVOsx ELISA plate reader (Wallac).
The co-sedimentation assay with liposomes was performed as previously described (Ago et al, 2001, 2003) with minor modifications. Briefly, liposomes were prepared by mixing PE (85%) and PC (10%) with 5% PI or PtdIns(3)P, drying the mixture under a stream of nitrogen, and resuspending it in a sample buffer (150 mM NaCl, 20 mM HEPES, pH 7.2). Liposomes (50 μM) were incubated for 10 min on ice with the indicated GST-fusion proteins (5 μg) in 50 μl of the sample buffer. The liposomes were precipitated for 20 min at 15 000 g, and bound proteins were subjected to 12% SDS–PAGE followed by staining with Coomassie brilliant blue. For estimation of the amount of proteins on the gel, densitometric analysis was performed using a LAS-1000plus image analyzer (Fuji photo film).
Structure determination of p40phox
The construct encoding the full-length wild-type p40phox (amino acids 1–339) was used for structure determination. Protein expression, purification, crystallization, data collection, and processing were performed as previously described (Honbou et al, 2006). Briefly, the full-length p40phox was expressed as a TEV-cleavable 6 × His-tagged protein in E. coli strain BL21. The protein was purified by affinity chromatography, and 6 × His tag was cleaved by digestion with TEV protease. After digestion, further purification was performed using SourceQ column (GE Healthcare) followed by Superdex 75 gel filtration column (GE Healthcare). Purified protein solution was concentrated to 10 mg ml−1 and crystallized using sitting-drop vapor diffusion against 5% PEG 20 000, 200 mM bis–tris–HCl, pH 6.5 at 293 K. The structure was solved by molecular replacement with MolRep (Vagin and Teplyakov, 1997) and the CNS program suite (Brunger et al, 1998) using the p40phox PX domain coordinates from the complex with PtdIns(3)P (PDB code 1H6H), the p40phox SH3 domain coordinates (PDB code 1W6X), and the p40phox PB1 domain coordinates from the complex with the p67phox PB1 domain (PDB code 1OEY chain J) as a search model. The correct orientations of the two PX and single PB1 domains were found by MolRep and CNS, respectively, but those of other domains were not found. Electron density map was thus calculated using phase information from two PX and one PB1 domains, and other domains were manually fitted into the density by using program O (Jones et al, 1991).
Initial refinement was performed by the torsion angle molecular dynamic-simulated annealing method and bulk-solvent correction against the maximum-likelihood amplitude target using CNS. For each cycle, the model was rebuilt manually using program O and Coot (Emsley and Cowtan, 2004) in several steps alternated with cycles of automated refinement using data to 3.15 Å resolution with R=26.3% and Rfree=30.2%, respectively. There are no residues that lie in disallowed regions in a Ramachandran analysis. The refinement statistics are summarized in Table I. All the structure figures were prepared using PyMOL (http://pymol.sourceforge.net).
SAXS data analysis and shape restoration
SAXS data were acquired at the BL-10C beamline at the Photon Factory in Tsukuba, Japan. Data on p40phox were collected at several concentrations from 2.2 to 13 mg ml−1 in the Q range 0.017 to 0.443 Å−1 to estimate the possible effects of protein concentration on the determination of structural parameters. The wavelength of the X-ray was 1.488 Å. The sample cell had a volume of 50 μl and a 1-mm path length with quartz windows. The data acquisition time was 600 s for each measurement. The identical buffer solution to the sample was recorded to measure solvent scattering. Protein scattering was obtained by subtracting the solvent scattering as the background trace. The values of Rg from the Guinier analysis were stable from the lowest (2 mg ml−1) to highest (13 mg ml−1) concentration. The distance distribution function, P(r), was calculated with the program GNOM (Svergun, 1992) with a Q-value of 0.0328<Q<0.2508 Å−1 at the protein concentration of 13 mg ml−1. The ab initio shape determination program DAMMIN (Svergun, 1999) was used to generate a low-resolution model of p40phox from the SAXS data. DAMMIN calculates a volume of a protein filled with densely packed spheres (dummy atoms) to fit the experimental scattering data by a simulated annealing minimization procedure. Ten independent fits were carried out and superimposed using the program SUPCOMB (Kozin and Svergun, 2001) and averaged by the program DAMAVER (Volkov and Svergun, 2003), highlighting common structural features.
Immunoprecipitation analysis
HeLa cells were transfected with the cDNAs encoding GFP–fused p40phox and HA-tagged p67phox, and cultured for 24 h. The cells were lysed at 4°C with a lysis buffer (150 mM NaCl, 0.4 mM EDTA, 10% glycerol, 0.5% Triton X-100, and 50 mM Tris, pH 7.5) containing 1 mM dithiothreitol and Protease inhibitor cocktail (Sigma). The lysates were centrifuged for 30 min at 20 000 g, and the resultant supernatant was precipitated with an anti-GFP monoclonal antibody (nacalai tesque) or mouse control IgG (DakoCytomation) coupled to protein G-Sepharose (GE Healthcare Bio-Sciences). The precipitates were washed three times with a buffer (150 mM NaCl, 0.05% Tween-20, and 20 mM Tris, pH 7.4) containing 1 mM dithiothreitol and Protease inhibitor cocktail, and applied to SDS–PAGE, followed by immunoblot analysis with an anti-HA monoclonal antibody (Covance) or anti-GFP polyclonal antibodies (MBL). The blots were developed using ECL-plus agents (GE Healthcare Bio-Sciences) for visualization of the antibodies.
In vitro pull-down binding assay
Purified GST-p40phox (10 μg) and His-tagged p67phox (10 μg) were incubated in 500 μl of PBS with or without liposomes containing PI or PtdIns(3)P at the indicated concentration. Proteins were pulled down with glutathione-Sepharose-4B (GE Healthcare Bio-Sciences), and precipitated proteins were eluted with 10 mM glutathione. The eluates were analyzed by immunoblot with an anti-GST monoclonal antibody (Upstate Biotechnology) or a Penta-His monoclonal antibody (QIAGEN).
Accession code
The coordinates and structure factors of the p40phox have been deposited in the Protein Data Bank with accession code 2DYB.
Acknowledgments
We are grateful to N Kubo (Kyushu University), N Yoshiura (Kyushu University), Y Kage (Kyushu University), and M Ohtsu (Kyushu University) for their technical assistance. This work was supported in part by National Project on Protein Structural and Functional Analyses Science (to FI and HS) and Grants-in-Aid for Scientific Research Science (to HS and FI) from the Ministry of Education, Culture, Sports, and Technology of Japan, as well as the CREST and BIRD projects of the Japan Science and Technology Agency (to HS).
References
- Ago T, Kuribayashi F, Hiroaki H, Takeya R, Ito T, Kohda D, Sumimoto H (2003) Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc Natl Acad Sci USA 100: 4474–4479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ago T, Takeya R, Hiroaki H, Kuribayashi F, Ito T, Kohda D, Sumimoto H (2001) The PX domain as a novel phosphoinositide- binding module. Biochem Biophys Res Commun 287: 733–738 [DOI] [PubMed] [Google Scholar]
- Bouin AP, Grandvaux N, Vignais PV, Fuchs A (1998) p40phox is phosphorylated on threonine 154 and serine 315 during activation of the phagocyte NADPH oxidase. Implication of a protein kinase C-type kinase in the phosphorylation process. J Biol Chem 273: 30097–30103 [DOI] [PubMed] [Google Scholar]
- Bravo J, Karathanassis D, Pacold CM, Pacold ME, Ellson CD, Anderson KE, Butler PJ, Lavenir I, Perisic O, Hawkins PT, Stephens L, Williams RL (2001) The crystal structure of the PX domain from p40phox bound to phosphatidylinositol 3-phosphate. Mol Cell 8: 829–839 [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Cross AR, Segal AW (2004) The NADPH oxidase of professional phagocytes—prototype of the NOX electron transport chain systems. Biochim Biophys Acta 1657: 1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen PJ, Cozier GE, Banting G, Mellor H (2001) Modular phosphoinositide-binding domains—their role in signalling and membrane trafficking. Curr Biol 11: R882–R893 [DOI] [PubMed] [Google Scholar]
- Ellson CD, Anderson KE, Morgan G, Chilvers ER, Lipp P, Stephens LR, Hawkins PT (2001b) Phosphatidylinositol 3-phosphate is generated in phagosomal membranes. Curr Biol 11: 1631–1635 [DOI] [PubMed] [Google Scholar]
- Ellson CD, Davidson K, Ferguson GJ, O'Connor R, Stephens LR, Hawkins PT (2006) Neutrophils from p40phox−/− mice exhibit severe defects in NADPH oxidase regulation and oxidant-dependent bacterial killing. J Exp Med 203: 1927–1937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellson CD, Gobert-Gosse S, Anderson KE, Davidson K, Erdjument-Bromage H, Tempst P, Thuring JW, Cooper MA, Lim ZY, Holmes AB, Gaffney PR, Coadwell J, Chilvers ER, Hawkins PT, Stephens LR (2001a) PtdIns(3)P regulates the neutrophil oxidase complex by binding to the PX domain of p40phox. Nat Cell Biol 3: 679–682 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Ghosh S, Strum JC, Sciorra VA, Daniel L, Bell RM (1996) Raf-1 kinase possesses distinct binding domains for phosphatidylserine and phosphatidic acid. Phosphatidic acid regulates the translocation of Raf-1 in 12-O-tetradecanoylphorbol-13-acetate-stimulated Madin-Darby canine kidney cells. J Biol Chem 271: 8472–8480 [DOI] [PubMed] [Google Scholar]
- Gillooly DJ, Morrow IC, Lindsay M, Gould R, Bryant NJ, Gaullier JM, Parton RG, Stenmark H (2000) Localization of phosphatidylinositol 3-phosphate in yeast and mammalian cells. EMBO J 19: 4577–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyworth PG, Bohl BP, Bokoch GM, Curnutte JT (1994) Rac translocates independently of the neutrophil NADPH oxidase components p47phox and p67phox. Evidence for its interaction with flavocytochrome b558. J Biol Chem 269: 30749–30752 [PubMed] [Google Scholar]
- Heyworth PG, Curnutte JT, Nauseef WM, Volpp BD, Pearson DW, Rosen H, Clark RA (1991) Neutrophil nicotinamide adenine dinucleotide phosphate oxidase assembly. Translocation of p47phox and p67phox requires interaction between p47phox and cytochrome b558. J Clin Invest 87: 352–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honbou K, Yuzawa S, Suzuki NN, Fujioka Y, Sumimoto H, Inagaki F (2006) Crystallization and preliminary crystallographic analysis of p40phox, a regulatory subunit of NADPH oxidase. Acta Crystallogr F 62: 1018–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurley JH, Meyer T (2001) Subcellular targeting by membrane lipids. Curr Opin Cell Biol 13: 146–152 [DOI] [PubMed] [Google Scholar]
- Inoue Y, Suenaga Y, Yoshiura Y, Moritomo T, Ototake M, Nakanishi T (2004) Molecular cloning and sequencing of Japanese pufferfish (Takifugu rubripes) NADPH oxidase cDNAs. Dev Comp Immunol 28: 911–925 [DOI] [PubMed] [Google Scholar]
- Ito T, Matsui Y, Ago T, Ota K, Sumimoto H (2001) Novel modular domain PB1 recognizes PC motif to mediate functional protein–protein interactions. EMBO J 20: 3938–3946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh T, Takenawa T (2002) Phosphoinositide-binding domains: functional units for temporal and spatial regulation of intracellular signalling. Cell Signal 14: 733–743 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kami K, Takeya R, Sumimoto H, Kohda D (2002) Diverse recognition of non-PxxP peptide ligands by the SH3 domains from p67phox, Grb2 and Pex13p. EMBO J 21: 4268–4276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai F, Liu H, Field SJ, Akbary H, Matsuo T, Brown GE, Cantley LC, Yaffe MB (2001) The PX domains of p47phox and p40phox bind to lipid products of PI(3)K. Nat Cell Biol 3: 675–678 [DOI] [PubMed] [Google Scholar]
- Karathanassis D, Stahelin RV, Bravo J, Perisic O, Pacold CM, Cho W, Williams RL (2002) Binding of the PX domain of p47 phox to phosphatidylinositol 3, 4-bisphosphate and phosphatidic acid is masked by an intramolecular interaction. EMBO J 21: 5057–5068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozin MB, Svergun DI (2001) Automated matching of high- and low-resolution structural models. J Appl Crystallogr 34: 33–41 [Google Scholar]
- Kuribayashi F, Nunoi H, Wakamatsu K, Tsunawaki S, Sato K, Ito T, Sumimoto H (2002) The adaptor protein p40phox as a positive regulator of the superoxide-producing phagocyte oxidase. EMBO J 21: 6312–6320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambeth JD (2004) NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol 4: 181–189 [DOI] [PubMed] [Google Scholar]
- Lemmon MA (2003) Phosphoinositide recognition domains. Traffic 4: 201–213 [DOI] [PubMed] [Google Scholar]
- Leto TL, Adams AG, de Mendez I (1994) Assembly of the phagocyte NADPH oxidase: binding of Src homology 3 domains to proline-rich targets. Proc Natl Acad Sci USA 91: 10650–10654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Conte L, Chothia C, Janin J (1999) The atomic structure of protein–protein recognition sites. J Mol Biol 285: 2177–2198 [DOI] [PubMed] [Google Scholar]
- Miyano K, Ueno N, Takeya R, Sumimoto H (2006) Direct involvement of the small GTPase Rac in activation of the superoxide-producing NADPH oxidase Nox1. J Biol Chem 281: 21857–21868 [DOI] [PubMed] [Google Scholar]
- Nauseef WM (2004) Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol 122: 277–291 [DOI] [PubMed] [Google Scholar]
- Noda Y, Kohjima M, Izaki T, Ota K, Yoshinaga S, Inagaki F, Ito T, Sumimoto H (2003) Molecular recognition in dimerization between PB1 domains. J Biol Chem 278: 43516–43524 [DOI] [PubMed] [Google Scholar]
- Oikawa T, Yamaguchi H, Itoh T, Kato M, Ijuin T, Yamazaki D, Suetsugu S, Takenawa T (2004) PtdIns(3,4,5)P3 binding is necessary for WAVE2-induced formation of lamellipodia. Nat Cell Biol 6: 420–426 [DOI] [PubMed] [Google Scholar]
- Quinn MT, Gauss KA (2004) Structure and regulation of the neutrophil respiratory burst oxidase: comparison with nonphagocyte oxidases. J Leukocyte Biol 76: 760–781 [DOI] [PubMed] [Google Scholar]
- Shiose A, Sumimoto H (2000) Arachidonic acid and phosphorylation synergistically induce a conformational change of p47phox to activate the phagocyte NADPH oxidase. J Biol Chem 275: 13793–13801 [DOI] [PubMed] [Google Scholar]
- Stahelin RV, Burian A, Bruzik KS, Murray D, Cho W (2003) Membrane binding mechanisms of the PX domains of NADPH oxidase p40phox and p47phox. J Biol Chem 278: 14469–14479 [DOI] [PubMed] [Google Scholar]
- Suh CI, Stull ND, Li XJ, Tian W, Price MO, Grinstein S, Yaffe MB, Atkinson S, Dinauer MC (2006) The phosphoinositide-binding protein p40phox activates the NADPH oxidase during FcγIIA receptor-induced phagocytosis. J Exp Med 203: 1915–1925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumimoto H, Kage Y, Nunoi H, Sasaki H, Nose T, Fukumaki Y, Ohno M, Minakami S, Takeshige K (1994) Role of Src homology 3 domains in assembly and activation of the phagocyte NADPH oxidase. Proc Natl Acad Sci USA 91: 5345–5349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumimoto H, Miyano K, Takeya R (2005) Molecular composition and regulation of the Nox family NAD(P)H oxidases. Biochem Biophys Res Commun 338: 677–686 [DOI] [PubMed] [Google Scholar]
- Svergun DI (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Cryst 25: 495–503 [Google Scholar]
- Svergun DI (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J 76: 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terasawa H, Noda Y, Ito T, Hatanaka H, Ichikawa S, Ogura K, Sumimoto H, Inagaki F (2001) Structure and ligand recognition of the PB1 domain: a novel protein module binding to the PC motif. EMBO J 20: 3947–3956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueyama T, Lennartz MR, Noda Y, Kobayashi T, Shirai Y, Rikitake K, Yamasaki T, Hayashi S, Sakai N, Seguchi H, Sawada M, Sumimoto H, Saito N (2004) Superoxide production at phagosomal cup/phagosome through βI protein kinase C during FcγR-mediated phagocytosis in microglia. J Immunol 173: 4582–4589 [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (1997) MOLREP: an automated program for molecular replacement. J Appl Crystallogr 30: 1022–1025 [Google Scholar]
- Vieira OV, Botelho RJ, Rameh L, Brachmann SM, Matsuo T, Davidson HW, Schreiber A, Backer JM, Cantley LC, Grinstein S (2001) Distinct roles of class I and class III phosphatidylinositol 3-kinases in phagosome formation and maturation. J Cell Biol 155: 19–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov VV, Svergun DI (2003) Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 36: 860–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MI, Gill DJ, Perisic O, Quinn MT, Williams RL (2003) PB1 domain-mediated heterodimerization in NADPH oxidase and signaling complexes of atypical protein kinase C with Par6 and p62. Mol Cell 12: 39–50 [DOI] [PubMed] [Google Scholar]
- Xu J, Liu D, Gill G, Songyang Z (2001a) Regulation of cytokine-independent survival kinase (CISK) by the Phox homology domain and phosphoinositides. J Cell Biol 154: 699–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Hortsman H, Seet L, Wong SH, Hong W (2001b) SNX3 regulates endosomal function through its PX-domain-mediated interaction with PtdIns(3)P. Nat Cell Biol 3: 658–666 [DOI] [PubMed] [Google Scholar]
- Yoshinaga S, Kohjima M, Ogura K, Yokochi M, Takeya R, Ito T, Sumimoto H, Inagaki F (2003) The PB1 domain and the PC motif-containing region are structurally similar protein binding modules. EMBO J 22: 4888–4897 [DOI] [PMC free article] [PubMed] [Google Scholar]