

Abstract
Activation of the apical caspase-8 is crucial to the extrinsic apoptotic pathway. Although the death effector domain (DED) of caspase-8 has been reported to be involved in death-inducing signaling complex formation, the detailed mechanism of how DED functions in regulating apoptosis remains largely unknown. Here, we demonstrate that the prodomain of the caspase-8/Mch5 can be further cleaved between two tandemly repeated DEDs (DEDa–DEDb) at the amino-acid residue Asp129 by caspase-8 itself. The DEDa fragment generated from the endogenous caspase-8 was detected in isolated nucleoli upon treatment with TRAIL (tumor necrosis factor-related apoptosis-inducing ligand). Cleaved DEDa appears to translocate into the nucleus by association with extracellular signal-regulated protein kinases-1/2 (ERK1/2). Elimination of ERK1/2 expression by RNA interference resulted in a significant attenuation of nuclear entry of DEDa and reduced caspase-8-dependent apoptosis. In the nucleus, DEDa interacts with TOPORS, a p53 and topoisomerase I binding protein, and possibly displaces p53 from TOPORS, allowing p53 to stimulate caspase-8 gene expression. In summary, we postulate a positive feedback loop involving DEDa, which enables the continual replenishment of procaspase-8 during apoptosis.
Keywords: caspase-8/Mch5, death effector domain, ERK1/2, p53, TOPORS
Introduction
Apoptosis, or programmed cell death, is orchestrated by a family of proteases known as caspases that cleave their substrates after specific aspartic acid residues (Thornberry and Lazebnik, 1998). Caspases are synthesized as catalytically inactive precursor proteins that become activated in response to specific death stimuli. The activation of initiator caspase-8, -10 and -9 usually requires the assembly of multicomponent complex such as death-inducing signaling complex (DISC) or apoptosome (Boatright et al, 2003; Pop et al, 2006). The apical caspase-8 proenzyme is transcribed into multiple mRNA transcripts, which originate from an array of complex splicing events (Boldin et al, 1996; Fernandes-Alnemri et al, 1996). FLICE, MACHα2 and MCH5 are the three procaspase-8 isoforms, all of which possess a long amino-terminal prodomain, which harbors two highly homologous death effector domains (DEDs), termed DEDa and DEDb, followed by a C-terminal protease domain that can be divided into two subunits, p18 and p11. The DED domains were originally defined as being essential for the binding of procaspase-8 to FADD, which is associated with a receptor/ligand complex. The resultant DISC will trigger the activation of procaspase-8. Generation of the mature caspase-8 protease requires two proteolytic cleavage events: cleavage and separation of the larger subunits from the smaller subunits, followed by cleavage and separation of the larger subunit from the prodomain (Srinivasula et al, 1996; Medema et al, 1997; Chang et al, 2003). The mature caspase-8 protease is then released into the cytosol, where it cleaves a number of different cellular substrates such as Bid and downstream effector caspases-3, -6 and -7, initiating a caspase cascade and the subsequent apoptotic events (Li et al, 1998; Stennicke et al, 1998; Fischer et al, 2006). However, the function of the prodomain cleaved from the proenzyme is still far less understood. Lenardo and co-workers (Siegel et al, 1998) reported that the prodomain of caspase-8 was able to form intracellular filaments termed death effector filaments (DEF) and induce apoptosis by recruiting and activating procaspase zymogens. Furthermore, studies on the roles of the DED-containing protein members offer important clues to the potential functions of this domain (for reviews, see Barnhart et al, 2003; Tibbetts et al, 2003). For example, DEDD, a DED containing DNA binding protein, is known to translocate into the nucleus upon receiving an apoptotic stimuli and induce cell death. Nuclear DEDD leads to activation of caspase-6 and inhibition of RNA polymerase I-dependent transcription (Stegh et al, 1998; Schickling et al, 2001). Other functions of DED-containing proteins are exemplified by DEDD2/Flame-3 and PEA-15, with the former directly binding to transcription factors in the nucleus and altering gene transcription (Roth et al, 2002; Zhan et al, 2002), and the latter demonstrating the ability to sequester the extracellular signal-regulated protein kinases-1/2 (ERK1/2) in the cytoplasm (Formstecher et al, 2001; Whitehurst et al, 2004). The emerging roles of DED-containing proteins in multiple signaling cascades encourage us to investigate the functions of DEDs belonging to procaspase-8. In this report, we have identified a novel procaspase-8/Mch5 self-cleavage site, which is placed right after Asp129 amino-acid residue situated between the two tandemly arranged DED domains. Upon receiving the appropriate apoptotic stimuli, such as treatment with TRAIL (tumor necrosis factor-related apoptosis-inducing ligand), this cleavage event occurs and generates the DEDa fragment. DEDa, which is normally unstable in non-apoptotic cells, can be stabilized over several hours upon TRAIL treatment. Accumulated DEDa translocates into the nucleus by binding to ERK1/2, where DEDa participates in procaspase-8 transcriptional activation by directly binding to TOPORS, a p53 and topoisomerase I binding protein (Haluska et al, 1999; Zhou et al, 1999; Lin et al, 2005). Binding of DEDa displaces p53 from TOPORS, allowing p53 to activate the expression of caspase-8. Hence, our data provide the first evidence of a novel positive feedback loop that involves activation, translocation and production of procaspase-8. Through this feedback loop involving DEDa, ERK1/2, TOPORS and p53, processed procaspase-8 can thus be continually replenished by newly synthesized procaspase-8.
Results
Processing of procaspase-8 involves a self-cleavage at Asp129 in its prodomain
Previous studies have shown that procaspase-8/Mch5 prodomain can be separated from its protease domain via auto-cleavage at D227/233, but the fate of the prodomain remains uncharacterized. To address this question, we cloned the procaspase-8/Mch5 cDNA from a HeLa cell line by reverse transcription–polymerase chain reaction (RT–PCR) and prepared a series of mutant constructs (Figure 1A). When HeLa or MCF-7 cells were transfected with pEGFP-Casp8(1–233), a cleaved band whose size corresponds roughly to GFP fused with the first DED (DEDa) was detected (Figure 1B, lanes 3 and 4). However, this band was not shown in the human dopaminergic neuroblastoma cell line SH-SY5Y (Figure 1B, lane 2), which lacks caspase-8 expression owing to its gene promoter methylation (Hopkins-Donaldson et al, 2000; Banelli et al, 2002). This observation reminds us that caspase-8 might be involved in a self-processing event occurring between the two DEDs of procaspase-8. To verify this hypothesis, various caspase-specific inhibitors were utilized. As shown in Figure 1C, only z-VAD-fmk, a broad-spectrum caspase inhibitor, and z-IETD-fmk, a selective caspase-8 inhibitor, caused an inhibition of this processing (lanes 1 and 5), whereas the caspase-3-,-7- and -9-specific inhibitors exhibited little, if any, effect on this self-cleavage activity (lanes 2–4).
Figure 1.
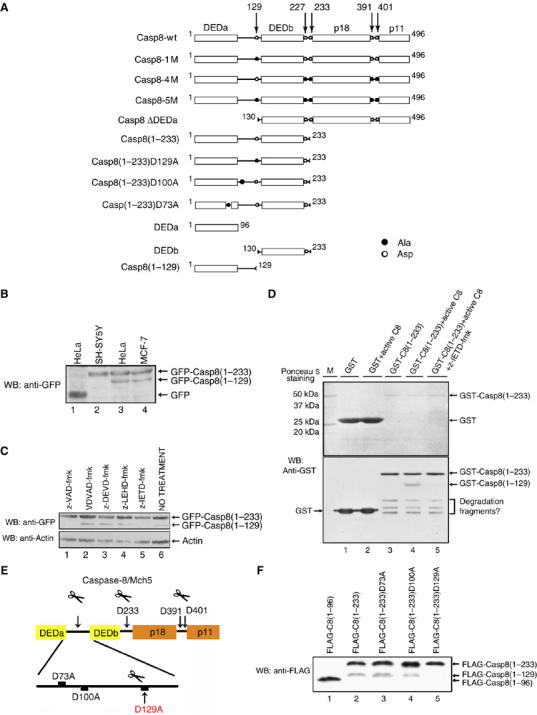
Processing of procaspase-8/Mch5 involves its self-induced cleavage at Asp129 in its prodomain. (A) Schematic representations of procaspase-8/Mch5 and its various mutants. Casp8, caspase-8; DED, death effecter domain; p18 and p11, the large and small subunits of mature caspase-8. The open circle represents the autocatalytic cleavage sites (aspartic acid) and the solid circle represents the mutations (alanine). The GFP or FLAG tag was fused to the N terminus of each indicated fragment in the plasmids pEGFP-C1 and p3XFLAG, respectively. (B) HeLa, SH-SY5Y and MCF-7 cell lines were transfected with pEGFP-Casp8(1–233), followed by immunoblotting with anti-GFP antibody. The empty vector pEGFP-C1 was used as a negative control. (C) HeLa cells were transfected with pEGFP-Casp8(1–233) in the presence of different caspase inhibitors as indicated. Cell lysates from the different treated cell groups were analyzed by Western blot using anti-GFP antibody. -Actin was used as the loading control. (D) Glutathione–agarose bead-tagged GST (lanes 1 and 2) or GST-C8(1–233) (lanes 3–5) were incubated with and without rh-caspase-8 (recombinant human caspase-8) (1 U) for 2 h at 37°C, and the resultant cleavage products were analyzed by immunoblotting with an anti-GST antibody (lower panel). The bacteria expressed GST and GST-C8(1–233) were visualized via Ponceau S staining (upper panel). The caspase-8-specific inhibitor z-IETD-fmk was preincubated with the active caspase-8 as indicated (lane 5). (E) A schematic illustration showing the auto-catalytic cleavage sites in procaspase-8/Mch5. The potential cleavage sites (D73, D100 and D129) between DEDa and DEDb were mutated as indicated. The arrowheads point to the actual cleavage sites. (F) HeLa cells were transfected with the indicated plasmids. Potential cleavage at the mutated sites was analyzed by Western blotting.
To further verify that the caspase-8 prodomain is auto-cleaved by caspase-8, an in vitro cleavage assay was performed. As shown in Figure 1D, bacterially expressed GST-Casp8(1–233) was cleaved to generate an ∼37-kDa fragment that corresponds to the GST-tagged DEDa only in the presence of active recombinant human caspase-8 (Figure 1D, lower panel, lane 3 versus 4). However, this cleavage was completely blocked when the caspase-8-specific inhibitor z-IETD-fmk was added (lane 5), indicating that the prodomain is indeed specifically cleaved by caspase-8. GST alone was used as a negative control (lanes 1 and 2).
To delineate the exact enzymatic cleavage site within the inter-region (aa 98–135) between DEDa and DEDb, three FLAG-tagged prodomain mutants, namely FLAG-Casp8(1–233)D73A, FLAG-Casp8(1–233)D100A and FLAG-Casp8(1–233)D129A (Figure 1A), were generated based on the caspase-8 consensus cleavage sites (Figure 1E). As shown in Figure 1F, only mutation at amino acid 129 (Asp → Ala) blocks the cleavage of caspase-8 prodomain (lane 5), indicating that the cleavage event indeed occurs at 129-Asp-HL, which resides between DEDa and DEDb. Caspase-8 is known to be recruited to DISC via interaction with FADD. To investigate whether DEDa or DEDb individually was able to bind to FADD, both yeast two-hybrid assay and co-immunoprecipitation (co-IP) experiments were performed (Supplementary Figure S1). Neither DEDa nor DEDb alone is able to associate with FADD, instead both DEDa and DEDb are required for the DISC assembly.
DEDa is stabilized and translocates into the nucleus upon TRAIL treatment
Ectopic expressed DEDa was very unstable, and in contrast, DEDab or DEDb was found to be expressed at much higher level than DEDa (data not shown). We therefore examined whether DEDa is subject to proteasome-mediated degradation. We first established a HeLa cell line continuously expressing GFP-DEDa. As shown in Figure 2A, DEDa was stabilized by treatment with MG132 in a time-dependent manner. Accumulation of DEDa was also observed upon stimulation of cells with TRAIL or FasL (Figure 2B), implying that DEDa was involved in the death receptor-induced apoptosis pathway. However, the detailed mechanism underlying the regulation of DEDa stability still remains unclear.
Figure 2.
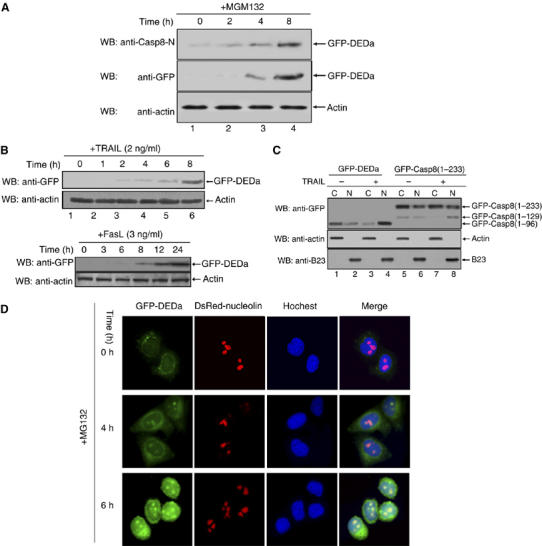
DEDa is stabilized and translocates from the cytoplasm into the nucleus upon apoptotic induction. (A) HeLa cell line stably expressing GFP-tagged DEDa was treated with MG132 (20 μM) over the indicated time periods. The expression level of GFP-DEDa was monitored by both anti-caspase-8 antibody (upper panel) and anti-GFP antibody (middle panel). β-Actin was used to verify equal loading (bottom panel). (B) HeLa cells stably expressing GFP-tagged DEDa were treated with TRAIL (upper panel) or FasL (lower panel) for the indicated time periods. Western blot was performed using anti-GFP antibody and anti-actin antibody. (C) HeLa cells were transiently transfected with pEGFP-DEDa (2 μg) or pEGFP-Casp8(1–233) (2 μg). Cells were treated with or without TRAIL (10 ng/ml) for 8 h and cytosolic and nuclear fractions from transfected cells were prepared. Subcellular localization of transfected GFP-DEDa (aa 1–96) and the cleaved DEDa (aa 1–129) was examined. The specificity of cytoplsmic or nuclear subcellular fractionation was confirmed by the detection of β-actin (a cytoplasm-specific protein) and B23 (nucleolus marker protein) respectively. (D) HeLa cells stably expressing GFP-DEDa (green fluorescence) were transfected with pDsRed1-C1/nucleolin (red fluorescence). Twenty-four hours post-transfection, cells were treated with MG132 (25 μM) for 0, 4 and 6 h, as indicated. In addition, the transfected cells were stained with Hoechst 33342 (10 μg/ml) for 15 min to mark the nuclei (blue fluorescence) before being subjected to fluorescence microscopy.
To examine the subcellular localization of DEDa, an N-terminal GFP-tagged DEDa or GFP-tagged caspase-8 prodomain construct was transiently transfected into HeLa cells and both nuclear and cytoplasmic fractions were analyzed by Western blot with the GFP antibody. The data reveal that in non-stimulated cells, GFP-DEDa was detectable in both nuclear and cytoplasmic fractions (Figure 2C, lanes 1 and 2). However, upon treatment of cells with TRAIL, GFP-DEDa was found predominantly in the nuclear fraction (lanes 3 and 4). Similarly, in cells transfected with GFP-Casp8(1–233), the cleaved DEDa was mainly found in the nuclear fraction after TRAIL treatment (Figure 2C, lane 8), suggesting that DEDa accumulates in the nucleus upon stimulation with TRAIL. To further demonstrate the subcellular localization of DEDa more directly, the HeLa cell line stably expressing GFP-DEDa was transfected with pDsRed1-C1/nucleolin (red fluorescent fusion protein). Twenty-four hours post-transfection, cells were incubated with MG132 for the indicated times and the localization of GFP-DEDa and nucleolin was visualized by immunofluorescence microscopy. Four hours after treatment with MG132, the levels of DEDa gradually increased and began to accumulate in the nucleus (Figure 2D). Of note, DEDa was not uniformly distributed within the nucleoplasm, but rather accumulated in distinct speckles, which appeared to be the nucleoli, where nucleolin is exclusively located. Superimposition of the image of DsRed1-C1/nucleolin and GFP-DEDa revealed that DEDa colocalized precisely with nucleolin.
ERK1/2 is involved in the DEDa nuclear translocation
It has been reported that the single DED-containing proteins PEA-15 and vanishin bind ERK1/2. Amino-acid sequence comparisons exhibit moderate similarity among DEDa, PEA-15 and vanishin (Figure 3A). We therefore asked whether DEDa of caspase-8 is also able to interact with ERK1/2. To address this issue, a co-IP experiment was performed. The analysis confirmed that FLAG-caspase-8 prodomain (aa 1–233) and its cleaved product DEDa (aa 1–129) (lane 1) and FLAG-DEDa (aa 1–96) (lane 2) associate with endogenous ERK-p42/p44 proteins (Figure 3B). Interaction of endogenous DEDa with endogenous ERK1/2 was further detected in the nucleolar fractions of TRAIL-treated HeLa cells using anti-caspase-8 antibody recognizing the amino terminus (aa 2–20) of procaspase-8 (anti-casp8-N, BD Pharmingen Ab 551234) (Figure 3C, lane 2) (details of nucleolar fractionation are shown in Supplementary Figure S2B). As a negative control, no endogenous ERK1/2 was detected in the anti-casp8-N immunoprecipitates in the presence of the caspase inhibitor z-VAD-fmk (lane 1). The association between ERK1/2 and DEDa was also validated by an immunostaining experiment (Supplementary Figure S2A).
Figure 3.
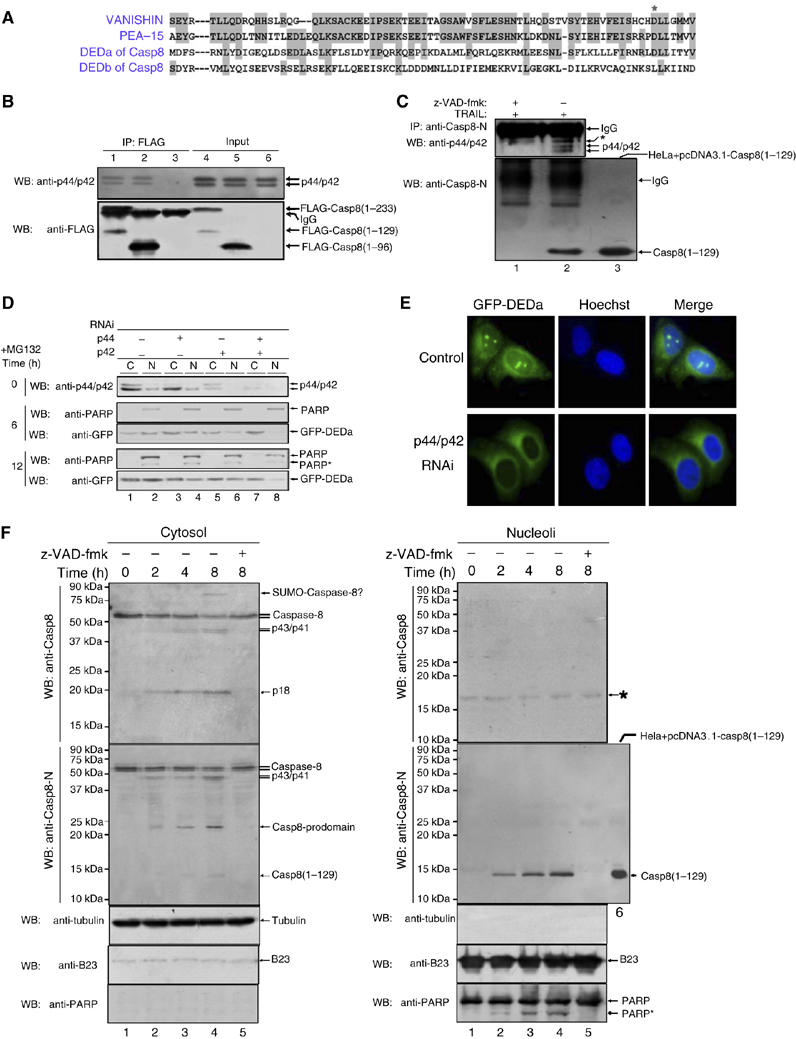
ERK1/2 associates with DEDa and is involved in DEDa nuclear translocation. (A) Alignment of amino-acid sequences among DEDa, DEDb and the DED regions of vanishin and PEA-15. Gray shading indicates identical residues. ‘*' indicates the conserved amino acid critical for ERK binding in PEA-15 and vanishin. (B) HeLa cells were transfected with p3XFLAG-Casp8(1–233) (lower panel, lane 4), p3XFLAG-DEDa (lower panel, lane 5) or the empty vector (lower panel, lane 6). Equal amounts of the cell extracts were then immunoprecipitated with an anti-FLAG antibody and analyzed by immunoblotting with an anti-p44/42 antibody to detect co-immunoprecipitated endogenous p42/p44 (top panel, lanes 1–3). The precipitates were also probed with an anti-FLAG antibody (bottom panel). (C) HeLa cells were treated with TRAIL for 12 h in the presence (lane 1) or absence of z-VAD-fmk (lane 2). The nucleoli were purified and the interaction of endogenous DEDa and p44/p42 was analyzed by co-IP using anti-caspase-8 (rabbit polyclonal antibody, BD Pharmingen), followed by Western blotting using rabbit anti-p44/42 antibody. The nuclear fraction of HeLa cells transfected with pcDNA3.1-Casp8(1–129) (untagged C8 1–129) was loaded as a migration control (lane 3). (D) HeLa cells stably expressing GFP-DEDa were transfected with or without siRNAs specific to p44 and p42, or both, as indicated. Seventy-two hours after two consecutive siRNA transfections with a 24-h interval, MG132 was added to allow the accumulation of DEDa for indicated periods of time (0, 6 and 12 h). Cytoplasmic and nuclear fractionations were prepared and further analyzed by immunoblotting using the indicated antibodies. Nuclear protein PARP was used as the nuclear marker and as the loading control as well. ‘*' denotes the cleaved fragments of PARP. (E) HeLa cells stably expressing GFP-DEDa were treated with or without p44/p42 siRNA as described above. MG132 (25 μM) was added for 8 h. Cells were stained with Hoechst 33342 to visualize the nuclei. Knockdown of endogenous ERK greatly diminished the nuclear localization of GFP-DEDa (visualized by green fluorescence). (F) HeLa cells (1 × 108) were treated with TRAIL (2 ng/ml, R&D) in the absence or presence of z-VAD-fmk (lane 5) for the indicated time points and then fractionated into cytosolic and nucleolar fractions. Each fraction was probed with antibodies against p18 (anti-Casp8) or N-terminal (anti-Casp8-N) of caspase-8. A specific protein of about 15 kDa was detected by the anti-N-terminal caspase-8 antibody in the nucleolar fraction of HeLa cells treated with TRAIL. Untagged C8 1–129-transfected cell lysate was loaded as a migration control. B23, tubulin and PARP were used as markers.
ERK1/2 is a nucleocytoplasmic shuttling protein, which enters the nucleus through a nuclear localization sequence (NLS)-independent active transport pathway. Failing to ascertain any classical NLS in DEDa, we thereby asked whether ERK1/2 is involved in the nuclear entry of DEDa. To test our hypothesis, we eliminated ERK1/2 expression by an RNA interference (RNAi) approach. HeLa cells stably expressing DEDa were transfected with small interfering RNAs (siRNAs) directed specifically against ERK1 and ERK2 either singly or in combination. Seventy-two hours post-transfection, MG132 was added for further incubation (6 and 12 h) to allow the accumulation of GFP-DEDa. The treated cells were then subjected to subcellular fractionation and both ERK1/2 and GFP-DEDa protein levels were analyzed at three different time points (0, 6 and 12 h). As shown in Figure 3D, in mock-treated control cells, nuclear DEDa is increased to a greater extent when we prolonged the MG132 incubation period from 6 to 12 h (Figure 3D, middle panel, lane 2 versus bottom panel, lane 2). Importantly, compared with siRNA single inhibition (p42−/p44+ or p42+/p44−), which showed partial inhibition of nuclear entry of DEDa (lanes 4 and 6 versus lane 2), double siRNA (p42−/p44−) treatment caused a nearly complete inhibition of nuclear translocation of DEDa at 6 h (lane 8 versus 2). At 12 h, DEDa could be scarcely detected in the nuclear fraction, which can be explained by the hypothesis that accumulation of cytoplasmic DEDa may help the residual unsuppressed ERK1/2 to translocate DEDa into the nucleus. Consistently, fluorescence imaging study (Figure 3E) showed that in mock-treated control, GFP-DEDa exhibited a nuclear accumulation, whereas in ERK1/2-specific siRNA-treated cells, none of the GFP-DEDa can be detected in the nucleus, indicating that ERK1/2 is involved in the translocation of DEDa from the cytosol to the nucleus.
To investigate the intracellular localization of endogenous DEDa, HeLa cells were treated with TRAIL to induce apoptosis and were then fractionated into cytosolic and nucleolar fractions. To monitor the processing and cellular localization of endogenous DEDa, each fraction was double checked using two antibodies directed against p18 fragment (anti-Casp8) and the N-terminal 2–20 aa (anti-Casp8-N) of human caspase-8, respectively. As shown in Figure 3F, left part, the cleavage product p18 was detected in the cytosol by the anti-Casp8 antibody as early as 2 h (upper panel), and the cleaved prodomain (1–233) and DEDa (1–129) were detected by the anti-Casp8-N antibody (second panel from the top). The figure on the right shows the gradually increasing amount of DEDa in the nucleoli detected by the anti-Casp8-N antibody (second panel from the top). In contrast, processed and unprocessed caspase-8 fragments containing p18 were not detected by the anti-Casp8 antibody in the nucleolar fraction (top panel). As expected, caspase-8-processed fragments were not detected in the presence of z-VAD-fmk (lane 5).
DEDa upregulates the expression of procaspase-8
Next, a time-course RT–PCR and Western blotting were performed to study the inter-relation between DEDa and caspase-8 expression (Figure 4A). HeLa cells stably expressing DEDa were treated with MG132, allowing for the accumulation of DEDa over the indicated periods of time, followed by RT–PCR amplification of caspase-8. An increase in both gene transcription and protein expression of caspase-8 was detected (right panel). This upregulation appears to be caspase-8-specific, as neither the RNA nor the protein levels of caspase-9 were affected by the increase of DEDa (bottom panels). The modulation of caspase-8 expression by MG132 did not occur in HeLa cells not expressing DEDa (Figure 4A, left panel). Together, these data confirmed the dependence of caspase-8 gene expression on DEDa.
Figure 4.
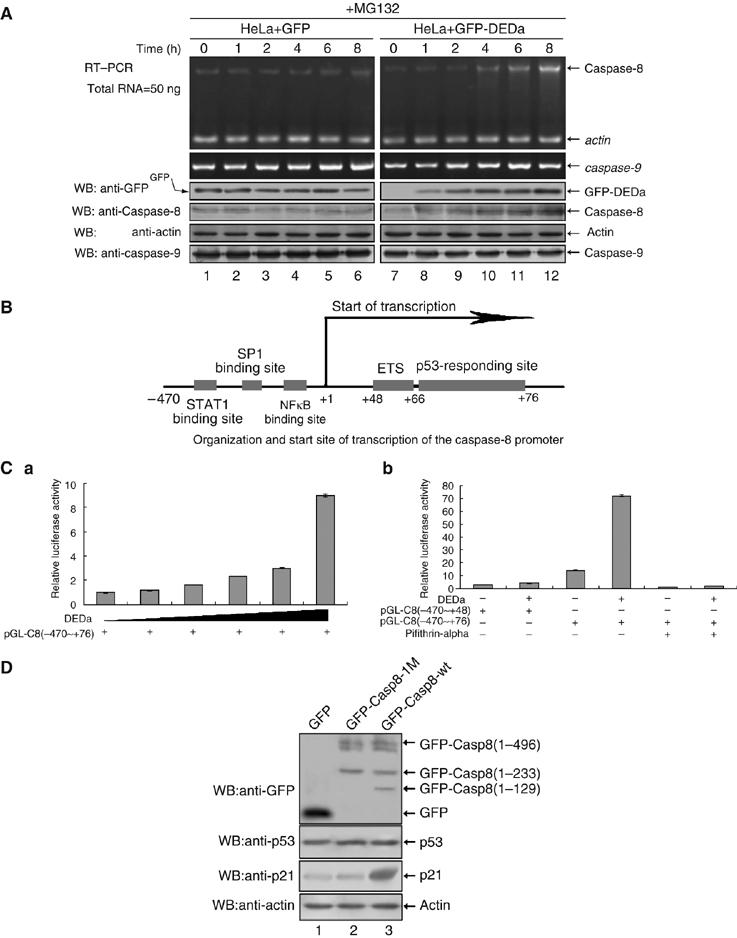
DEDa upregulates caspase-8 in a p53-dependent manner. (A) HeLa cells stably transfected with GFP-DEDa were treated with MG132 to allow the accumulation of DEDa during the indicated time periods (0–8 h). Total RNA was isolated at each time point and analyzed by semiquantitative RT–PCR using primers specific for caspase-8 and caspase-9. The protein levels of GFP-DEDa, endogenous caspase-8 and caspase-9 were also determined by immunoblotting using the respective antibodies, as indicated. HeLa cells stably expressing GFP alone were examined as a negative control. (B) Schematic representation of the caspase-8 promoter. The transcriptional start site is indicated as +1. The sequence is numbered with respect to the start site. DNA binding sites of distinct transcription factors and the p53-responding element are denoted by gray boxes. (C) (a) A549 cells were cotransfected with a fixed amount of the luciferase reporter construct pGL3-C8(−470∼+76) (1 μg) and increasing amounts of p3XFLAG-DEDa (0, 25, 50, 100, 200 and 500 ng). The total DNA concentration in each transfection was kept constant by adjusting it with an empty vector. (b) A549 cells were cotransfected with either pGL3-C8(−470∼+48) (1 μg) or pGL3-C8(−470∼+76) (1 μg) in combination with p3XFLAG-DEDa or p3XFLAG vector (1 μg). Pifithrin-alpha (20 μM) was added (lanes 5 and 6) to inhibit p53-dependent gene transcription. For both (a) and (b), Renilla luciferase.plasmid pRL-CMV (3 ng) was introduced into all the transfected cells as an internal control. Luciferase activity was measured and plotted after normalizing with respect to Renilla luciferase activity. Data shown in both (a) and (b) are representative of three independent experiments. Vertical error bars are the average s.d.s of three independent values. (D) SH-SY5Y cells were transfected with an equal amount (1 μg) of pEGFP, pEGFP-Casp8-wt or pEGFP-Casp8-1M separately. The protein levels of endogenous p53 and its transcription target p21 were analyzed using Western blot.
DEDa activates caspase-8 gene expression via p53
Trautwein and co-workers (Liedtke et al, 2003) reported that the human caspase-8 promoter can be upregulated by a p53-dependent mechanism (Figure 4B). However, the detailed mechanism by which p53 induces caspase-8 transcription was unclear. To verify that p53 is responsible for caspase-8 gene activation, a genomic fragment containing the predicted p53-responsive sequence (−470 to +76) from the caspase-8 promoter region was isolated and subcloned into a pGL3 luciferase reporter plasmid. A549 cells were cotransfected with pGL3-C8(−470∼+76) and increasing amounts of FLAG-tagged DEDa (from 0 to 500 ng). As shown in Figure 4C-a, an increasing amount of DEDa led to an augmented activation of pGL3-C8(−470∼+76), suggesting that DEDa is involved in caspase-8 activation. To further define whether the p53-responsive element located in the caspase-8 promoter region (+66 to +76) is involved in this DEDa-induced caspase-8 activation, A549 cells were transfected with another luciferase reporter construct pGL3-C8(−470∼+48), which lacks the p53-responsive element. As shown in Figure 4C-b (lane 2 versus 4), DEDa was unable to induce the activity of the reporter pGL3-C8(−470∼+48) efficiently, indicating that DEDa-dependent enhanced activation of caspase-8 requires the p53-responsive element. Consistently, in the presence of pifithrin-alpha, which inhibits p53-dependent gene transcription, the effect of DEDa on the activation of pGL3-C8(−470∼+76) was completely abolished (lane 4 versus 6). To further support the assumption that DEDa enhances p53-mediated transcriptional activity, SH-SY5Y cells were transfected individually with pEGFP, pEGFP-wt-Casp8 or pEGFP-Casp8-1M (D129A). The Casp8-1M mutant is unable to generate the DEDa fragment, yet can still produce the mature protease domain. As SH-SY5Y cells do not express endogenous caspase-8, only the introduced caspase-8 was examined. As shown in Figure 4D, GFP-wt-Casp8, but not GFP-C8-1M, underwent proteolytic cleavage at Asp129 (lanes 2 and 3), and as a result, the protein levels of p21, a target of p53, were markedly increased (second panel from the bottom, lane 3), demonstrating that DEDa somehow enhances the transcriptional activity of p53.
DEDa displaces p53 from the TOPORS/p53 complex
To investigate the molecular mechanisms underlying the upregulation of procaspase-8 by DEDa, we made use of a yeast two-hybrid system to screen a pretransformed Human Fetal Brain cDNA library using DEDa as bait. TOPORS, a DNA topoisomerase I and p53 binding protein, was detected as its novel binding partner. The interaction of endogenous DEDa with endogenous TOPORS was further verified by co-IP experiments. HeLa cells were fractionated into cytosolic, nuclearplasmic and nucleolar fractions, and endogenous TOPORS was mainly present in the nuclear and nucleolar fractions, as detected by immunoblotting with a polyclonal antibody (kindly provided by Dr Yuki Takada, Riken Yokohama Institute, Japan) (Figure 5A-a). TOPORS was able to be co-immunoprecipitated with DEDa in TRAIL-treated cells (Figure 5A-b, lane 4) but not in untreated cells (lane 3).
Figure 5.
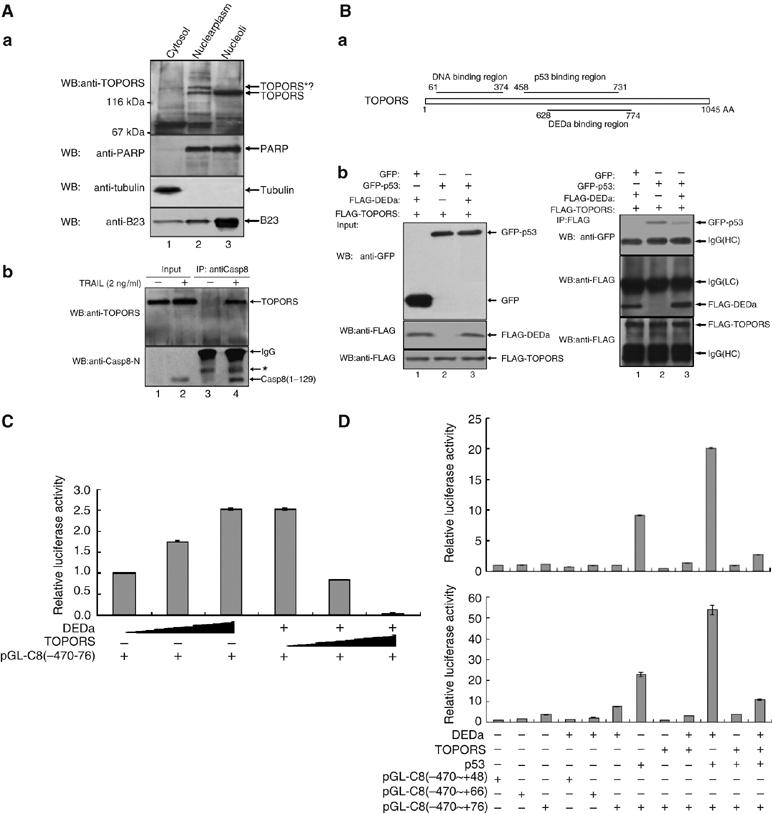
DEDa interacts with TOPORS and displays p53 from the TOPORS/p53 complex. (A) (a) Cellular distribution of endogenous TOPORS was examined in cytosolic, nuclearplasmic and nucleolar fractions by immunoblotting (upper panel). PARP, tubulin and B23 were used to verify the purity of the fractions. (b) The nucleolar fractions isolated from HeLa cells treated with or without TRAIL were immunoprecipitated with anti-Casp8-N antibody. Co-precipitated TOPORS and DEDa were analyzed by immunoblotting using the indicated antibodies. (B) (a) A schematic illustration of the regions of TOPORS required for association with p53 and DEDa. Numbers indicate amino acid residues. (b) H1299 (p53−/−) cells were cotransfected with pCATCH-TOPORS (2 μg) and either pEGFP, pEGFP-p53 or pEGFP-p53 plus p3XFLAG-DEDa. Ten percent of the total cell lysate was subjected to immunoblotting to confirm an equal expression of transfected proteins (left panel). The remaining cell extracts from each transfectant were immunoprecipitated with an anti-FLAG antibody and subsequently immunoblotted with anti-GFP and anti-FLAG antibodies (right panel). (C) A549 cells were cotransfected with pGL3-C8(−470∼+76) (1 μg) and either with an increasing amount of p3XFLAG-DEDa (0–0.5 μg) (lanes 1–3) or a fixed amount of p3XFLAG-DEDa (0.5 μg) coupled with an increasing amount of pCATCH-TOPORS (0–0.5 μg) (lanes 4–6). Luciferase assay was performed identically as described above. (D) p53-null H1299 (upper panel) and p53 wild-type A549 cells (lower panel) were respectively transfected with either pGL3-C8(−470∼+48), pGL3-C8(−470∼+66) or pGL3-C8(−470∼+76) in various combinations with p3XFLAG-DEDa, pCATCH-TOPORS or p3XFLAG-p53 as indicated. Luciferase assay was performed as described above.
We noticed that the region of TOPORS required for interaction with DEDa overlaps the reported binding site for p53 (Weger et al, 2002), implying a competitive interaction with TOPORS between DEDa and p53 (Figure 5B-a). As shown in Figure 5B-b, in the presence of DEDa, the amount of p53 that bound to TOPORS was markedly decreased (right panel, lane 2 versus 3), suggesting that DEDa may compete with p53 to bind to TOPORS. To test whether TOPORS has any effect on the DEDa-mediated activation of caspase-8, pGL3-C8(−470∼+76) was introduced into A549 cells coexpressing DEDa and an increasing amount of FLAG-tagged TOPORS (0–0.5 μg) (Figure 5C). Activation of the procaspase-8 promoter by DEDa was significantly repressed (lanes 4–6), confirming that TOPORS has an inhibitory effect on DEDa.
To delineate the interactive roles that DEDa, p53 and TOPORS play in the transactivation of caspase-8, p53 null H1299 (p53−/−) and p53 wild-type A549 (p53+/+) cells were separately cotransfected with expression plasmids encoding DEDa, p53 and TOPORS together with one of the three luciferase reporter plasmids in various combinations as indicated in Figure 5D. The results showed that DEDa greatly enhanced p53-mediated caspase-8 transactivation (Figure 5D, lane 7 versus 10). On the other hand, p53 was essential for the transactivation of the caspase-8 promoter in the presence of DEDa (lane 6, upper panel versus lane 6, bottom panel). The effect of p53 on caspase-8 promoter could be abrogated by coexpression of TOPORS (lane 7 versus 11); however, the repression of the caspase-8 promoter by TOPORS was attenuated by an increased expression of DEDa (lane 11 versus 12).
DEDa sensitizes HeLa cells to TRAIL-induced apoptosis
To confirm that DEDa is essential for enhancing the effect of caspase-8 promoter activity, various caspase-8 mutants were constructed in which the auto-cleavage sites (aspartic acid residue) were substituted with alanine residues as indicated in Figure 1A. Each mutant was transfected into SH-SY5Y or A549 cells together with pGL3-C8(−470∼+76). Compared with wild-type caspase-8 (Casp8-wt), Casp8-1M significantly impaired the activation of the caspase-8 promoter in both SH-SY5Y and A549 cells, as no DEDa can be produced owing to D129A mutation (Figure 6A, lane 1 versus 4). In A549 cells, Casp8-4M was less capable than Casp8-wt to enhance the reporter gene expression. This was expected because the amount of DEDa produced from Casp8-4M is less than that from Casp8-wt. Additionally, Casp8-5M, in which all cleavage sites were mutated, displayed a minimal level of transactivation of the caspase-8 promoter in both cell lines. It is interesting to note that both Casp8-1M and Casp8-4M were still able to activate the reporter gene to a small extent in A549 cells. This can be explained by the assumption that overexpression of Casp8-1M led to the activation of endogenous caspase-8, which activates the reporter gene. In the case of Casp8-4M, which fails to yield functional mature caspase-8, yet can produce DEDa by endogenous active caspase-8, transactivation of the reporter gene still occurs. In contrast, in caspse-8-deficient SH-SY5Y cells, compared with Casp8-wt, all the caspase-8 self-cleavage mutants displayed little, if any, enhancing effects on the caspase-8 promoter activity owing to the absence of endogenous caspase-8 to complement the mutant function derived from transfected mutants. Taken together, these data strongly suggest a role for DEDa in the upregulation of caspase-8 expression.
Figure 6.
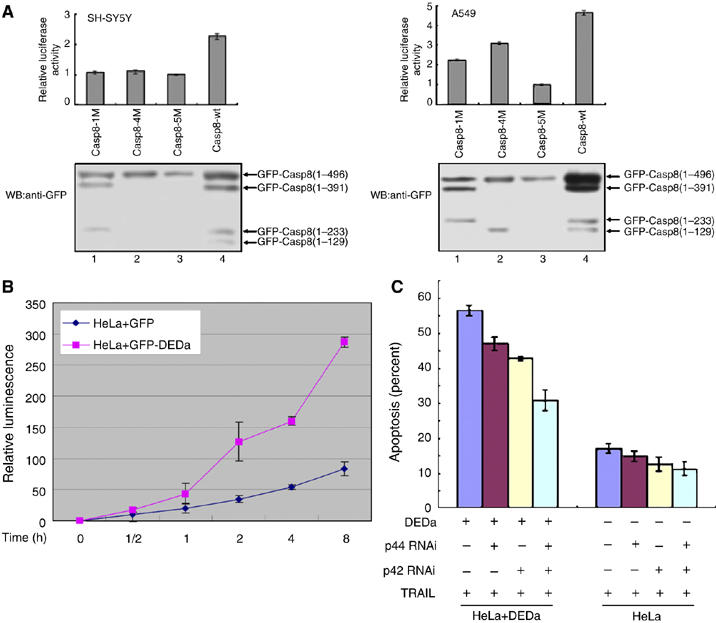
DEDa leads to increased amount of active caspase-8 and sensitizes HeLa cells to TRAIL-induced apoptosis. (A) SH-SY5Y and A549 cells were separately cotransfected with pGL3-C8(−470∼+76) (1 μg) and an equal amount (0.5 μg) of either pEGFPC1-Casp8-1M, pEGFPC1-Casp8-4M, pEGFPC1-Casp8-5M or pEGFPC1-Casp8-wt. Transfection efficiency was standardized against Renilla luciferase activity. Data are shown as fold induction of the luciferase activity versus control (luciferase activity obtained in cells cotransfected with pGL3-C8(−470∼+76) and pEGFPC1-Casp8-5M is defined as one-fold shown in lane 3). Immunoblotting was performed to ensure the expression and processing of these ectopic proteins using an anti-GFP antibody. (B) HeLa cells stably transfected with pEGFP-DEDa or pEGFP were treated with TRAIL (10 ng/ml, Sigma) for the indicated time periods. Activity of caspase-8 was measured using the Caspase-Glo™ 8 Assay kit (Promega) following the manufacturer's protocol. The data represent mean±s.d. of triplicate samples. (C) HeLa cell lines stably expressing GFP and GFP-DEDa were separately transfected with or without siRNAs specific for p44, p42 or both, as described in Figure 3D. Forty-eight hours after the second transfection with siRNA, cells were treated with TRAIL (10 ng/ml, Sigma). Apoptotic values were calculated as the percentage of apoptotic cells (condensed nuclei stained by Hoechst 33342) relative to the total number of cells in each random field (>200 cells) and represent the average of three independent experiments ±s.d.
If the conclusion that DEDa upregulates procaspase-8 is true, we would expect that increase of DEDa would lead to increased caspase-8 activity. To test this hypothesis, HeLa cells stably expressing either GFP or GFP-DEDa were treated with TRAIL for indicated times and a luminescent assay was performed to measure caspase-8 activity at each time point. Treatment of GFP-DEDa stable transfected cells with TRAIL led to a significantly increased activity of capase-8 as compared with GFP-only-transfected cells (Figure 6B).
As described above, when p44/p42 was siRNA-silenced, nuclear translocation of DEDa was disrupted and the upregulation of caspase-8 was weakened. To further determine whether ERK-mediated DEDa nuclear entry affects the cellular response to TRAIL-induced apoptosis, siRNA was used to knock down the expression of p44/p42 in both non-transfected HeLa and GFP-DEDa stably expressing HeLa cells. Seventy-two hours after siRNA-mediated p44/p42 inhibition, cells were treated with TRAIL for another 24 h. As shown in Figure 6C and Supplementary Figure S3, knockdown of either p44 or p42 suppressed the effect of DEDa on cell sensitivity to TRAIL treatment in the GFP-DEDa stably expressing HeLa cell line. However, combined siRNA inhibition on p44/p42 showed a more significant suppression of apoptosis than single inhibition. A similar effect was also observed in control HeLa cells, but to a much lesser extent (Figure 6C, columns 5–8). These results further demonstrate that nuclear translocation of DEDa promotes caspase-8-dependent apoptosis.
Discussion
We report here the identification of a novel cleavage site (Asp129) between two tandem DEDs within the caspase-8/Mch5 prodomain, which requires active caspase-8 for its proteolytical cleavage. It is well known that the prodomain of caspase-8 is involved in DISC formation (Medema et al, 1997). Our data, consistent with the result reported by Tsukumo and Yonehara (1999), show that the intact prodomain, but neither DEDa nor DEDb alone, is required for the association of procaspase-8 with FADD. It has been reported that GFP-DEDb, but not GFP-DEDa, is able to form DEF to induce apoptosis (Siegel et al, 1998). Naturally, one will ask what is the fate for DEDa? Quite unexpectedly, we found that transiently transfected DEDa is hardly detectable in non-stressed cells owing to its quick degradation through the proteasome pathway. However, DEDa can be stabilized in response to death receptor-related apoptotic signal triggered by TRAIL or FasL. This implies that DEDa of procaspase-8 may be involved in the extrinsic apoptotic cellular mechanisms. We showed that, upon TRAIL induction, DEDa of caspase-8 was stabilized and translocated into the nucleus. The nuclear compartmentalization of DEDa suggested that it might exert some effect on the gene transcription machinery. Indeed, we found that nuclear accumulation of DEDa led to an augmented expression of procaspase-8 at both mRNA and protein levels. The same effect does not occur for caspase-9 or -3, suggesting that DEDa initiates a specific gene transactivation pathway. The observation that caspase-8 mRNA level is elevated during apoptosis suggests that the regulation of procaspase-8 at the transcriptional level might be an important mechanism, which has been overlooked previously. It is of interesting to note that Asp129 is not conserved between human and mouse. Based on current knowledge, we reason that it is difficult to compare the conservation among different species. First of all, the number of caspase-8 isoforms varies greatly with species, for example, human has eight isoforms, whereas both mouse and chimpanzee have only one caspase-8 isoform. Second, Asp129 is found to be conserved only in human and chimpanzee, which indicates that Asp129 cleavage site may be a relatively recent evolutionary event and is conserved only in primates. However, this speculation needs to be further investigated.
DEDa shares moderate sequence similarity with PEA-15 and vanishin. Moreover, the amino acid Asp74, which is critical for ERK binding in PEA-15 or vanishin (Hill et al, 2002; Sur and Ramos, 2005), is well conserved in DEDa but not in DEDb. ERK is known to enter the nucleus by a carrier-independent import mechanism that involves a direct interaction with nuclear pore complex proteins (Whitehurst et al, 2002); it is reasonable to assume that the nuclear entry of DEDa is in association with ERK. Results from our RNAi and immunofluorescence studies strongly support this conclusion (Figure 4). Subcellular localization is an important determining factor for ERK-mediated signaling (Volmat and Pouyssegur, 2001). Our preliminary data showed that nuclear entry of DEDa resulted in a significant reduction in the overall phosphorylation state of ERK1/2 (Z Yao and M Wu, unpublished observation), and the detailed mechanism is currently being investigated. Although ERK signaling is generally considered to be pro-survival, we found that knockdown of ERK by the siRNA approach weakened the sensitivity to TRAIL-induced apoptosis, indicating that ERK may participate in certain proapoptotic pathways. Hence, the exact role ERK plays with respect to pro-apoptotic versus antiapoptotic regulation is an intriguing question deserving further investigation.
To elucidate the role of DEDa in the nucleus, we screened a Matchmaker library and identified TOPORS as a DEDa binding partner. Interestingly enough, the region within TOPORS, which is required for interaction with DEDa, overlaps with that for p53, implying that DEDa may compete with p53 for binding to TOPORS. In this study, we demonstrated that p53 is able to activate effectively the caspase-8 promoter, which is consistent with a previous report (Liedtke et al, 2003). More importantly, this effect is further enhanced when DEDa is coexpressed with p53. However, ectopic expression of TOPORS greatly suppresses transactivation of the caspase-8 promoter caused by DEDa and p53. It is worthwhile to mention that DEDa may not interact with p53 directly, as we were unable to detect the physical binding of DEDa to p53 in vivo (data not shown). Nevertheless, we were able to demonstrate that the association of p53 with TOPORS was much diminished by the addition of DEDa, which can be best explained by the assumption that DEDa displaces p53 from the p53/TOPORS complex, thereby allowing p53 to transactivate the caspase-8 promoter. However, neither p53 nor TOPORS was found to directly bind to the caspase-8 promoter by the chromatin immunoprecipitation experiment (data not shown). Hence, further studies are needed to characterize the downstream factor(s) that directly involve(s) the transcription of caspase-8.
A working model depicting the role of DEDa during apoptosis is proposed in Figure 7. Upon an apoptotic stimulus, such as upon TRAIL induction, the prodomain of procaspase-8 is autocatalytically cleaved at Asp129. The first DED domain, DEDa, will be stabilized by a yet unknown mechanism and associates with ERK1/2 in the cytoplasm. ERK1/2 may function as chaperone for DEDa to translocate across the nuclear membrane. In the nucleus, DEDa displaces p53 from the TOPORS/p53 complex, allowing p53 to promote caspase-8 gene transactivation. The processing of procaspase-8 is an irreversible event; therefore, DEDa initiates an amplification loop, which results in an upregulated caspase-8 expression and ensures continual replenishment for the processed caspase-8 during TRAIL-induced apoptosis. It is worthwhile to note that TRAIL treatment has a more profound effect on cell apoptosis at early time than expected in this study. This may be explained by a number of factors. Binding of DEDa to ERK1/2 may decrease the pro-survival effect of ERK. Another possibility is that p53 displaced from p53/TOPORS complex by DEDa might increase expression of some other apoptotic genes. Overall, our data represent a novel mechanism that links signaling from death receptors to nuclear events such as gene transactivation. Components identified in this signaling pathway will undoubtedly provide an opportunity to maximize the caspase-8 response and sensitize tumor cells to drug treatments.
Figure 7.
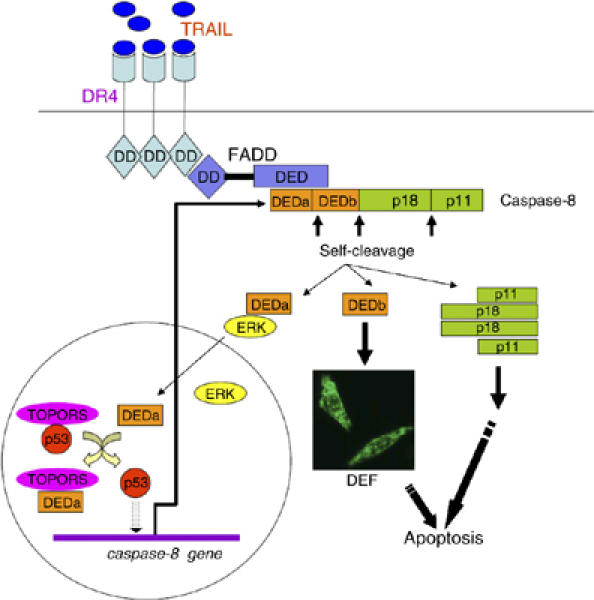
A proposed model depicting a novel positive feedback circuit as an integral part of caspase-8-mediated apoptosis (see Discussion).
Materials and methods
Cell culture and transfection
HeLaA549 and H1299 cell lines were cultured in DMEM containing 10% heat-inactivated fetal bovine serum (FBS). The human neuroblastoma SH-SY5Y cells were maintained in a 1:1 mixture of DMEM and Ham's F12 medium supplemented with 10% FBS. HeLa cells stably transfected with GFP-DEDa were initially selected in medium containing 700 μg/ml G418 (GIBCO, Grand Island, NY, USA) and maintained in medium containing 100 μg/ml G418. Transfection of cells with various mammalian expression constructs by lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was according to the methods provided by the manufacturer's specification.
Plasmid construction
The cDNA encoding Mch5 was amplified by RT–PCR from total RNA from HeLa cell line. The generated fragment was sequenced and found to be identical to the published caspase-8/Mch5 sequence (NM_001228). The caspase-8/Mch5 gene and its various mutants were subcloned into pEGFP-C1 (Clontech, Palo Alto, CA, USA) or the p3XFLAG-myc-CMV™-24 expression vector (Sigma). The primers used are listed in Supplementary data.
Subcellular fractionation, IP and Western blotting
Subcellular fractionation and isolation of nucleoli was performed as described previously (Andersen et al, 2002). Western blot analysis and IP were performed as described elsewhere (Zheng et al, 2001).
RNA interference
p44/42ERK siRNA were purchased from Cell Signaling Technology and transfected into cells using oligofectamine (Invitrogene) according to the manufacturer's recommendations (Cell Signaling Technology Inc., Beverly, MA). Briefly, HeLa cells stably expressing GFP-DEDa were cultured in 12-well plates to approximately 30% confluence and transfected twice over a 24-h interval with 100 nM p44 siRNA and 20 nM p42 siRNA either alone or together.
Luciferase reporter gene assay
Different caspase-8 promoter fragments were generated by PCR amplification using genomic DNA isolated from HeLa cells as template and further cloned into the pGL3-Basic luciferase reporter vector (Promega). The luciferase reporter assay was performed using the Dual-Luciferase Reporter assay system according to the manufacturer's instructions (Promega). The quantification of luciferase activities and calculation of the relative ratios in intensity were carried out with the Lumat luminomerter (LB9509, Berthold Technologies, Pittsburgh, PA). Transfection efficiency was normalized with respect to Renila luciferase activity.
In vitro cleavage assay
GST-C8(1–233) fusion protein or GST protein attached to glutathione–agarose beads (5 μl) was separately incubated with 1 U of active human recombinant caspase-8 (Chemicon, Temecula, CA, USA) in a reaction solution containing 50 mM Hepes, 50 mM NaCl, 5% glycerol, 0.1% CHAPS, 10 mM EDTA and 10 mM DTT at 37°C for 2 h. To inhibit caspase-8 activity, the caspase-8-specific inhibitor Z-IETD-FMK was preincubated with the active caspase-8 for 30 min at room temperature before adding the substrates. The beads were collected by centrifugation, washed and boiled in 2 × SDS loading buffer. The samples were resolved on an SDS–PAGE gel and analyzed by Western blotting.
Supplementary Material
Supplementary data
Supplementary Figures
Acknowledgments
We are grateful to Dr Koseki and Dr Takada (RIKEN Yokohama Institute, Japan) for kindly providing the pcDNA3-myc-TOPORS plasmid and anti-TOPORS antibody and Dr Weger for the pCATCH-TOPORS plasmid (Free University of Berlin, Germany). We are grateful to Ms S Ayyadhury for editorial assistance and Dr Yun Wah Lam (University of Dundee) for helpful discussion on the nucleoli isolation method. This research was supported by grants from the National Natural Science Foundation of China (30530200 and 30121001), grants (2002CB713702 and 2006CB910300) from the Ministry of Science and Technology of China, a grant from Chinese Academy of Sciences to WM (KSCX1-YW-R-57), a grant (KD2004034) from University of Science and Technology of China to Zhan Y and an ARC grant (ARC-3/05-M45080006) to KH from the Ministry of Education, Singapore.
References
- Andersen JS, Lyon CE, Fox AH, Leung AK, Lam YW, Steen H, Mann M, Lamond AI (2002) Directed proteomic analysis of the human nucleolus. Curr Biol 12: 1–11 [DOI] [PubMed] [Google Scholar]
- Banelli B, Casciano I, Croce M, Di Vinci A, Gelvi I, Pagnan G, Brignole C, Allemanni G, Ferrini S, Ponzoni M, Romani M (2002) Expression and methylation of CASP8 in neuroblastoma: identification of a promoter region. Nat Med 8: 1333–1335 [DOI] [PubMed] [Google Scholar]
- Barnhart BC, Lee JC, Alappat EC, Peter ME (2003) The death effector domain protein family. Oncogene 22: 8634–8644 [DOI] [PubMed] [Google Scholar]
- Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H, Pedersen IM, Ricci JE, Edris WA, Sutherlin DP, Green DR, Salvesen GS (2003) A unified model for apical caspase activation. Mol Cell 11: 529–541 [DOI] [PubMed] [Google Scholar]
- Boldin MP, Goncharov TM, Goltsev YV, Wallach D (1996) Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 85: 803–815 [DOI] [PubMed] [Google Scholar]
- Chang DW, Xing Z, Capacio VL, Peter ME, Yang X (2003) Interdimer processing mechanism of procaspase-8 activation. EMBO J 22: 4132–4142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Armstrong RC, Krebs J, Srinivasula SM, Wang L, Bullrich F, Fritz LC, Trapani JA, Tomaselli KJ, Litwack G, Alnemri ES (1996) In vitro activation of CPP32 and Mch3 by Mch4, a novel human apoptotic cysteine protease containing two FADD-like domains. Proc Natl Acad Sci USA 93: 7464–7469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer U, Stroh C, Schulze-Osthoff K (2006) Unique and overlapping substrate specificities of caspase-8 and caspase-10. Oncogene 25: 152–159 [DOI] [PubMed] [Google Scholar]
- Formstecher E, Ramos JW, Fauquet M, Calderwood DA, Hsieh JC, Canton B, Nguyen XT, Barnier JV, Camonis J, Ginsberg MH, Chneiweiss H (2001) PEA-15 mediates cytoplasmic sequestration of ERK MAP kinase. Dev Cell 1: 239–250 [DOI] [PubMed] [Google Scholar]
- Haluska P Jr, Saleem A, Rasheed Z, Ahmed F, Su EW, Liu LF, Rubin EH (1999) Interaction between human topoisomerase I and a novel RING finger/arginine–serine protein. Nucleic Acids Res 27: 2538–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JM, Vaidyanathan H, Ramos JW, Ginsberg MH, Werner MH (2002) Recognition of ERK MAP kinase by PEA-15 reveals a common docking site within the death domain and death effector domain. EMBO J 21: 6494–6504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N (2000) Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res 60: 4315–4319 [PubMed] [Google Scholar]
- Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94: 491–501 [DOI] [PubMed] [Google Scholar]
- Liedtke C, Groger N, Manns MP, Trautwein C (2003) The human caspase-8 promoter sustains basal activity through SP1 and ETS-like transcription factors and can be up-regulated by a p53-dependent mechanism. J Biol Chem 278: 27593–27604 [DOI] [PubMed] [Google Scholar]
- Lin L, Ozaki T, Takada Y, Kageyama H, Nakamura Y, Hata A, Zhang JH, Simonds WF, Nakagawara A, Koseki H (2005) topors, a p53 and topoisomerase I-binding RING finger protein, is a coactivator of p53 in growth suppression induced by DNA damage. Oncogene 24: 3385–3396 [DOI] [PubMed] [Google Scholar]
- Medema JP, Scaffidi C, Kischkel FC, Shevchenko A, Mann M, Krammer PH, Peter ME (1997) FLICE is activated by association with the CD95 death-inducing signaling complex (DISC). EMBO J 16: 2794–2804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pop C, Timmer J, Sperandio S, Salvesen GS (2006) The apoptosome activates caspase-9 by dimerization. Mol Cell 22: 269–275 [DOI] [PubMed] [Google Scholar]
- Roth W, Stenner-Liewen F, Pawlowski K, Godzik A, Reed JC (2002) Identification and characterization of DEDD2, a death effector domain-containing protein. J Biol Chem 277: 7501–7508 [DOI] [PubMed] [Google Scholar]
- Schickling O, Stegh AH, Byrd J, Peter ME (2001) Nuclear localization of DEDD leads to caspase-6 activation through its death effector domain and inhibition of RNA polymerase I dependent transcription. Cell Death Differ 8: 1157–1168 [DOI] [PubMed] [Google Scholar]
- Siegel RM, Martin DA, Zheng L, Ng SY, Bertin J, Cohen J, Lenardo MJ (1998) Death-effector filaments: novel cytoplasmic structures that recruit caspases and trigger apoptosis. J Cell Biol 141: 1243–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasula SM, Ahmad M, Fernandes-Alnemri T, Litwack G, Alnemri ES (1996) Molecular ordering of the Fas-apoptotic pathway: the Fas/APO-1 protease Mch5 is a CrmA-inhibitable protease that activates multiple Ced-3/ICE-like cysteine proteases. Proc Natl Acad Sci USA 93: 14486–14491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stegh AH, Schickling O, Ehret A, Scaffidi C, Peterhansel C, Hofmann TG, Grummt I, Krammer PH, Peter ME (1998) DEDD, a novel death effector domain-containing protein, targeted to the nucleolus. EMBO J 17: 5974–5986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stennicke HR, Jurgensmeier JM, Shin H, Deveraux Q, Wolf BB, Yang X, Zhou Q, Ellerby HM, Ellerby LM, Bredesen D, Green DR, Reed JC, Froelich CJ, Salvesen GS (1998) Pro-caspase-3 is a major physiologic target of caspase-8. J Biol Chem 273: 27084–27090 [DOI] [PubMed] [Google Scholar]
- Sur R, Ramos JW (2005) Vanishin is a novel ubiquitinylated death-effector domain protein that blocks ERK activation. Biochem J 387: 315–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA, Lazebnik Y (1998) Caspases: enemies within. Science 281: 1312–1316 [DOI] [PubMed] [Google Scholar]
- Tibbetts MD, Zheng L, Lenardo MJ (2003) The death effector domain protein family: regulators of cellular homeostasis. Nat Immunol 4: 404–409 [DOI] [PubMed] [Google Scholar]
- Tsukumo SI, Yonehara S (1999) Requirement of cooperative functions of two repeated death effector domains in caspase-8 and in MC159 for induction and inhibition of apoptosis, respectively. Genes Cells 4: 541–549 [DOI] [PubMed] [Google Scholar]
- Volmat V, Pouyssegur J (2001) Spatiotemporal regulation of the p42/p44 MAPK pathway. Biol Cell 93: 71–79 [DOI] [PubMed] [Google Scholar]
- Weger S, Hammer E, Heilbronn R (2002) Topors, a p53 and topoisomerase I binding protein, interacts with the adeno-associated virus (AAV-2) Rep78/68 proteins and enhances AAV-2 gene expression. J Gen Virol 83: 511–516 [DOI] [PubMed] [Google Scholar]
- Whitehurst AW, Robinson FL, Moore MS, Cobb MH (2004) The death effector domain protein PEA-15 prevents nuclear entry of ERK2 by inhibiting required interactions. J Biol Chem 279: 12840–12847 [DOI] [PubMed] [Google Scholar]
- Whitehurst AW, Wilsbacher JL, You Y, Luby-Phelps K, Moore MS, Cobb MH (2002) ERK2 enters the nucleus by a carrier-independent mechanism. Proc Natl Acad Sci USA 99: 7496–7501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan Y, Hegde R, Srinivasula SM, Fernandes-Alnemri T, Alnemri ES (2002) Death effector domain-containing proteins DEDD and FLAME-3 form nuclear complexes with the TFIIIC102 subunit of human transcription factor IIIC. Cell Death Differ 9: 439–447 [DOI] [PubMed] [Google Scholar]
- Zheng L, Schickling O, Peter ME, Lenardo MJ (2001) The death effector domain-associated factor plays distinct regulatory roles in the nucleus and cytoplasm. J Biol Chem 276: 31945–31952 [DOI] [PubMed] [Google Scholar]
- Zhou R, Wen H, Ao SZ (1999) Identification of a novel gene encoding a p53-associated protein. Gene 235: 93–101 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary Figures