

Abstract
Monoallelic RUNX1 mutations cause familial platelet disorder with predisposition for acute myelogenous leukemia (FPD/AML). Sporadic mono- and biallelic mutations are found at high frequencies in AML M0, in radiation-associated and therapy-related myelodysplastic syndrome and AML, and in isolated cases of AML M2, M5a, M3 relapse, and chronic myelogenous leukemia in blast phase. Mutations in RUNX2 cause the inherited skeletal disorder cleidocranial dysplasia (CCD). Most hematopoietic missense mutations in Runx1 involve DNA-contacting residues in the Runt domain, whereas the majority of CCD mutations in Runx2 are predicted to impair CBFβ binding or the Runt domain structure. We introduced different classes of missense mutations into Runx1 and characterized their effects on DNA and CBFβ binding by the Runt domain, and on Runx1 function in vivo. Mutations involving DNA-contacting residues severely inactivate Runx1 function, whereas mutations that affect CBFβ binding but not DNA binding result in hypomorphic alleles. We conclude that hypomorphic RUNX2 alleles can cause CCD, whereas hematopoietic disease requires more severely inactivating RUNX1 mutations.
Keywords: cleidocranial dysplasia, core binding factors, leukemia, Runx1 , Runx2
Introduction
Monoallelic mutations in RUNX1 and RUNX2 include deletions and missense, nonsense, and frameshift mutations (Lee et al, 1997; Mundlos et al, 1997; Song et al, 1999; Michaud et al, 2002; Walker et al, 2002). Large intragenic deletions in RUNX1 and RUNX2 were identified in both familial platelet disorder with predisposition for acute myelogenous leukemia (FPD/AML) and cleidocranial dysplasia (CCD) patients, respectively, indicating that haploinsufficiency is one mechanism for these diseases (Lee et al, 1997; Mundlos et al, 1997; Song et al, 1999). The majority of missense mutations in Runx1 and Runx2 are clustered within their DNA-binding Runt domains (RDs). The Runx1 and Runx2 RDs are 92% identical and recognize the same DNA sequence, and therefore should have the same structures and biophysical properties. The RD of Runx1 is an S-type immunoglobulin fold that is structurally related to the DNA-binding domains of p53, NF-κB, NFAT, Brachyury, and the STAT proteins (Berardi et al, 1999; Nagata et al, 1999). Although some missense mutations in Runx2 in CCD involve DNA-contacting residues in the DNA-binding loops, most CCD mutations are located in the β-barrel of the RD (Figure 1A) and are predicted to globally or locally disrupt the RD fold or impair its heterodimerization with CBFβ. On the other hand, Runx1 mutations in hematopoietic disorders are located almost exclusively at the DNA interface, primarily in the DNA-binding loops, and were predicted to disrupt DNA binding without affecting the RD fold (Nagata and Werner, 2001; Tahirov et al, 2001). These observations suggested that the CCD and hematopoietic disease mutations in Runx2 and Runx1, respectively, are functionally distinct. It has been proposed that several of the Runx1 mutations create dominant-negative alleles, particularly those involving amino acids at the DNA interface that result in an RD unable to bind DNA, but that is folded correctly and able to bind CBFβ (Imai et al, 2000; Nagata and Werner, 2001; Michaud et al, 2002; Osato, 2004). Here, we determined whether mutations found in hematopoietic diseases and CCD are indeed functionally distinct by introducing them into Runx1 and analyzing their effects on the thermal stability, DNA binding, and CBFβ binding of the RD, and on hematopoiesis in vivo.
Figure 1.
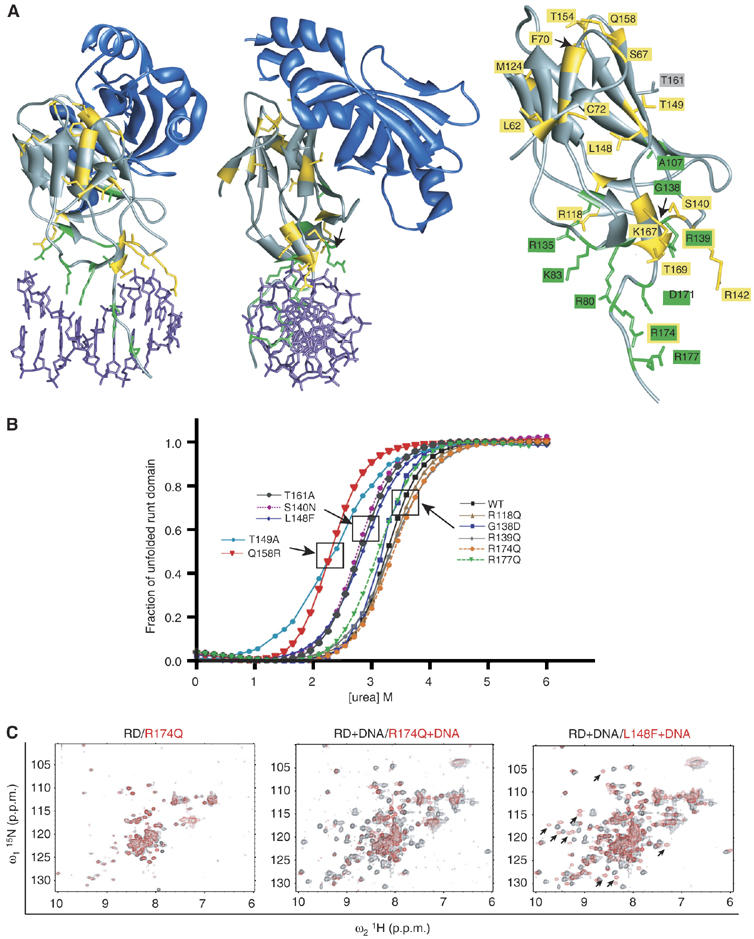
Effects of missense mutations on RD stability. (A) Ribbon diagram of the RD:CBFβ:DNA ternary complex (Bravo et al, 2001; Tahirov et al, 2001) and mutated residues. The RD and CBFβ are shown in gray and blue, respectively, and DNA is purple. Amino acids mutated in Runx2 in CCD (Lee et al, 1997; Quack et al, 1999; Zhou et al, 1999; Yoshida et al, 2002; Zhang et al, 2003) are yellow, whereas green indicates amino acids mutated in Runx1 in FPD/AML, AML M0 subtype, radiation-associated and therapy-related myelodysplastic syndrome and AML, AML M2, M5a, M3 relapse, and chronic myelogenous leukemia in blast phase (Osato et al, 1999; Song et al, 1999; Preudhomme et al, 2000; Buijs et al, 2001; Michaud et al, 2002; Walker et al, 2002; Harada et al, 2003; Roumier et al, 2003; Harada et al, 2004). R139 and R174, which are mutated in both CCD and in hematopoietic diseases, are indicated with yellow and green labels. Shown in gray is T161, an energetic hot spot at the CBFβ interface (Zhang et al, 2003). (B) The fraction of unfolded RD in the presence of increasing concentrations of urea. The boxes indicate proteins that (from right to left) had unaltered, moderately compromised, and more severely compromised stability. (C) 15N-1H-HSQC spectra of WT and mutated RDs. Each panel contains the spectrum of the WT RD (black) overlaid on the RD mutant spectrum (red). The R174Q+WT RD spectra were recorded in the absence (left panel) and presence (middle panel) of DNA. L148F was recorded in the presence of DNA. Arrows on the L148F spectra indicate several examples of peaks specific to the DNA-bound form of the RD that are shifted in the L148F spectrum relative to the WT spectrum, indicative of a conformational change in the DNA-bound L148F protein.
Results
Effects of mutations on RD stability
We analyzed the impact of missense mutations found in Runx1 and Runx2 on the thermal stability of the Runx1 RD by urea denaturation measurements (Zhang et al, 2003). Mutations located within the β barrel identified in CCD patients, including T149A, Q158R, and L148F, destabilized the RD, observed as a decrease in D50, the concentration of urea at which half of the molecules are unfolded (Figure 1B and Table I). The S140N mutation found in CCD (located in the βE′–βF loop) also compromised the RD stability, as did T161A. The T161A mutation has not been found in patients but was of interest to us because the T161 side-chain contributes more energy (2.0 kcal/mol) than that of any other residue to heterodimerization with CBFβ, and is an energetic hot spot at the CBFβ interface (Zhang et al, 2003).
Table 1.
Biochemical properties of Runx1 RD mutants
| Structure | DNA binding | CBFβ binding | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mutation | Classa | Disease (references)b | Contactsc | D50 (M)d | 15N–1H HSQCe | K2 (M) f EMSA | K2 g one-hybrid | K4 g modified one-hybrid | K1 (M) h FRET | K3 (M) f EMSA |
| WT | 3.3 | Normali,j | 5.0 (±0.3) × 10−11 | + | + | 1.1 (±0.4) × 10−7 | 1.4 (±0.2) × 10−8 | |||
| L62R | DB | CCD (1) | +/− | + | 2.1 (±1.4) × 10−6 | |||||
| S67R | B | CCD (1) | + | + | 3.0 (±2.3) × 10−6 | |||||
| F70C | DB | CCD (1) | +/− | + | 4.2 × 10−5 | |||||
| C72R | DB | CCD (2) | − | + | 3.1 × 10−6 | |||||
| R118Q | DB | CCD (2) | 3.4 | 6.7 (±11) × 10−10 | − | + | 6.7 (±3.2) × 10−7 | 7.0 (±1.0) × 10−8 | ||
| M124R | DB | CCD (2,3) | − | − | 3.7 × 10−6 | |||||
| S140N | DB | CCD (3) | 2.8 | 1.3 (±1.4) × 10−8 | − | − | 9.7 (±0.1) × 10−7 | |||
| R142C | DB | CCD (2) | DNA | − | + | 3.6 (±3.8) × 10−6 | ||||
| L148F | DB | CCD (2) | 2.8 | Perturbedj | 5.0 (±4.0) × 10−10 | − | + | 6.2 (±5.7) × 10−6 | 2.0 (±1.3) × 10−6 | |
| T149A | B | CCD (2) | CBFβ | 2.3 | Perturbedj,k | 9.2 (±1.4) × 10−11 | + | + | 2.0 (±1.0) × 10−7k | |
| T154G | DB | CCD (1) | +/− | + | 1.0 (±0.2) × 10−6 | |||||
| Q158R | B | CCD (2) | CBFβ | 2.3 | + | + | 1.2 (±0.6) × 10−6 | |||
| T161A | B | None (4) | CBFβ | 2.8 | Normalj,k | 2.3 (±1.4) × 10−11 | + | + | 2.9 (±2.0) × 10−6 | 5.6 (±2.0) × 10−7k |
| K167N | DB | CCD (5) | DNA | 3.0 | − | − | 1.6 (±0.2) × 10−6 | |||
| T169I | DB | CCD (5) | DNA | 1.7 | − | − | 1.3 (±0.4) × 10−6 | |||
| R80C | DB | CML BP l,m (6) | DNA | − | − | 1.0 × 10−6 | ||||
| K83N | D | AML M3 relapsem (6) | DNA | 1.0 (±7.4) × 10−8 | − | +/− | 1.1 (±0.1) × 10−7 | |||
| K83E | D | FPD/AML (7) | DNA | − | +/− | 1.0 (±0.2) × 10−7 | ||||
| A107P | DB | FPD/AML (8) | − | − | 3.7 × 10−6 | |||||
| R135G | DB | AML M0 n (9,10) | DNA | − | +/− | 2.6 (±1.9) × 10−6 | ||||
| G138D | D | AML M0 n (9) | 3.4 | − | +/− | 2.3 × 10−7 | ||||
| R139Q | D | FPD/AML, CCD (2,11) | DNA | 3.3 | Normalj | 1.4 (±1.1) × 10−6 | − | + | 1.1 × 10−7 | |
| D171Y | DB | FPD/AML (12) | DNA | − | + | 6.9 × 10−7 | ||||
| R174Q | D | FPD/AML, AML M0 m, CCD (2,9,10,11) | DNA | 3.4 | Normali,j | 2.3 (±2.0) × 10−6 | − | − | 9.9 × 10−8 | |
| R177Q | D | AML M2l and M5am (6,9) | DNA | 3.2 | − | + | 9.7 (±0.4) × 10−8 | |||
| R177X | FPD/AML, AML M0 o (6,10,11) | DNA | − | + | 1.3 × 10−7 | |||||
| Class D mutations are defined as those that decrease DNA binding ⩾10-fold (K2 in yeast one-hybrid or EMSA assays) and decrease CBFβ binding by ⩽5-fold (K1 by FRET or K3 by EMSA). Class DB mutations decrease DNA binding moderately or severely (+/− or—in the K2 columns) and CBFβ binding by ⩾5-fold. Class B mutations disrupt CBFβ binding, but not DNA binding. | ||||||||||
| (1) Quack et al (1999); (2) Zhou et al (1999); (3) Lee et al (1997); (4) Zhang et al (2003); (5) Yoshida et al (2002); (6) Osato et al (1999); (7) Michaud et al (2002); (8) Walker et al (2002); (9) Preudhomme et al (2000); (10) Roumier et al (2003); (11) Song et al (1999); (12) Buijs et al (2001). | ||||||||||
| Amino acids that directly contact DNA or CBFβ, from crystal structures (Bravo et al, 2001; Tahirov et al, 2001). | ||||||||||
| D50=urea concentration at which 50% of the RD molecules were denatured. Values represent the average of two experiments. | ||||||||||
| Mutants with spectra that had a WT appearance with a limited number of peaks showing significant chemical shift changes were classified as normal, whereas mutants displaying spectra with large numbers of shifted peaks or many missing peaks were characterized as perturbed. | ||||||||||
| EMSA values represent average of three experiments±s.d. | ||||||||||
| One- and modified one-hybrid filter assays were scored as positive (+) if the β-galactosidase signal was equivalent to that of the WT RD, moderate (+/−) if the level of β-galactosidase activity was between that of the RD and a negative control consisting of the GAL4 AD alone (<10-fold perturbation of DNA binding), and negative (−) if β-galactosidase activity was undetectable (⩾10-fold decrease). | ||||||||||
| FRET values represent the average of two experiments with the s.d. in parentheses. Values without s.d. represent experiments performed once. | ||||||||||
| Recorded in the absence of DNA. | ||||||||||
| Recorded on a RD–DNA complex. | ||||||||||
| From Zhang et al (2003). | ||||||||||
| CML BP, chronic myelogenous leukemia, blast phase. | ||||||||||
| Monoallelic mutation. | ||||||||||
| Monoallelic with deletion of the other RUNX1 allele. | ||||||||||
| Biallelic. | ||||||||||
Mutations in five of seven residues at the DNA interface, including R118Q, G138D, R139Q, R174Q, and R177Q, did not alter the RD's stability (Figure 1B and Table I). The T169I mutation, on the other hand, severely destabilized the RD, and the K167N mutation more moderately destabilized the protein. Thus, although in general, mutations in DNA-contacting residues tended not to affect the RD's stability, there were at least two exceptions to this trend.
We also assessed the degree to which the fold of the RD was perturbed for several mutant RDs by15N-1H heteronuclear single quantum correlation (HSQC) spectroscopy. Figure 1C shows an overlay of spectra for the wild-type (WT) and two mutant RDs. NMR spectra of the WT RD in the absence of DNA are relatively poor due to signal broadening caused by conformational exchange (Berardi et al, 1999; Nagata et al, 1999; Pérez-Alvarado et al, 2000). Despite this technical limitation, it is clear that peaks from the WT RD and the R174Q RD in the absence of DNA almost completely overlap, consistent with the fold of the R174Q mutant being unperturbed (Figure 1C, left panel). In the presence of DNA, the peaks from the WT RD are more broadly dispersed (Figure 1C, middle panel), whereas the R174Q+DNA spectrum is essentially identical to that of R174Q in the absence of DNA. The R174Q mutation decreased DNA binding by >10 000-fold (Table I); therefore, even at the relatively high protein and DNA concentrations (∼0.5 mM) used to collect the R174Q+DNA spectrum, R174Q is not bound to DNA. In summary, the R174Q mutation results in a correctly folded, but non-DNA-bound RD.
The L148F mutation decreases DNA binding by a relatively modest 10-fold (Table I), and at the concentrations used to record the spectrum the L148F RD should be DNA bound. Indeed, the L148F+DNA 15N-1H HSQC spectrum is more well dispersed due to its DNA binding. However, the L148F+DNA spectrum is significantly altered relative to that of the WT RD+DNA with numerous residues showing significant chemical shift changes. When an amino-acid substitution does not perturb a protein's fold, only a very limited subset of residues, those spatially close to the site of the mutation, will show chemical shift changes. In contrast, an amino-acid substitution that perturbs the fold will result in large numbers of chemical shift changes as a result of the conformational alteration (Corazza et al, 2004; Gorbatyuk et al, 2004; Ren et al, 2005; Sanderson et al, 2005). The large number of chemical shift changes in the L148F spectrum is consistent with a perturbation of the RD fold. This is supported by the decreased thermodynamic stability of the L148F RD observed in the urea denaturation analysis (Table I).
Effects of mutations on DNA binding
We examined DNA binding by all of the mutated RDs alone, and as RD-CBFβ heterodimers using yeast one- and modified one-hybrid assays, and independently validated several of the yeast one-hybrid results with quantitative electrophoretic mobility shift assays (EMSAs) (Figure 2 and Table I). The yeast one-hybrid assay measures the ability of an RD-Gal4 activation domain fusion protein (RD-Gal4 AD) to activate transcription of a lacZ reporter gene driven by three RD-binding (core) sites (Figure 2E). The modified yeast one-hybrid assay measures the activity of an RD-Gal4 AD/CBFβ heterodimer in the same yeast reporter strain (Figure 2F). All of the mutations identified in Runx1 in the hematopoietic disorders decreased DNA binding by ⩾10-fold (indicated by ‘−' in the one-hybrid column of Table I). EMSAs confirmed that several of the mutations affected DNA binding quite severely; K83N, R139Q, and R174Q decreased DNA binding (increased K2) by 200-, 28 000-, and 46 000-fold, respectively (Table I). We previously showed that alanine substitutions at the other DNA-contacting residues increased K2 by 27-fold (R177A), 80-fold (R135A), 115-fold (R80A), and 185-fold (D171A) (Li et al, 2003). As an alanine substitution generally removes only side-chain contacts and does not introduce a bulky or inappropriately charged side chain that could directly interfere with DNA binding, the K2 values associated with the disease mutations are likely, if anything, to be even higher than those found with alanine mutations.
Figure 2.
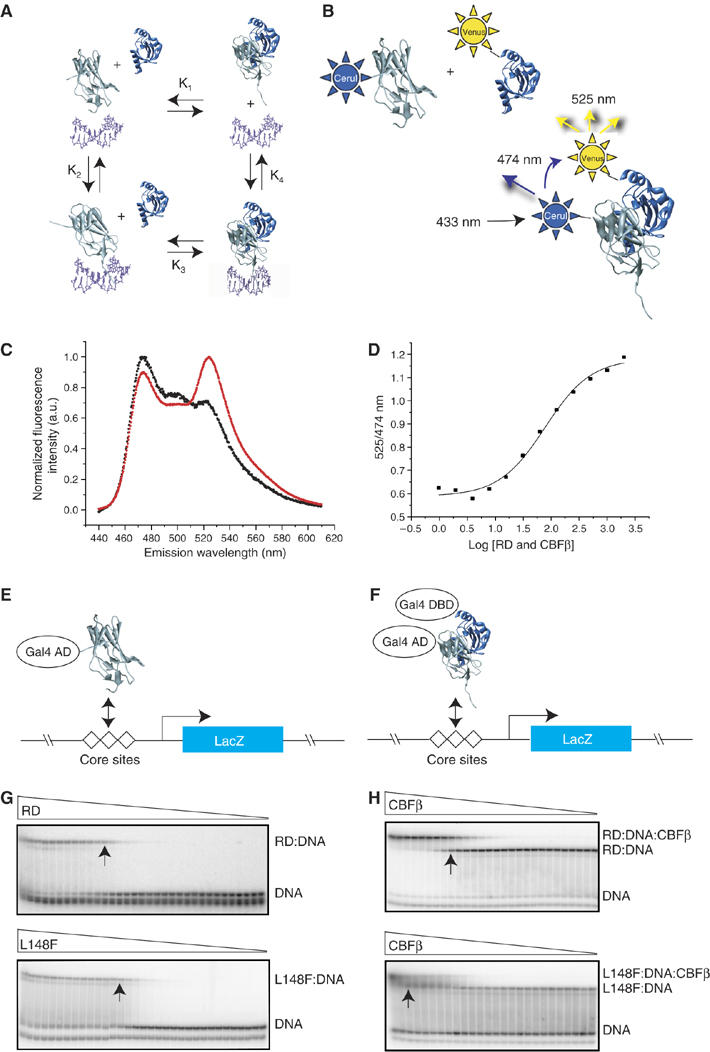
DNA and CBFβ binding by mutant RDs. (A) Schematic diagram of the potential interactions between the RD (gray), CBFβ (blue), and DNA (purple). These interactions can be described by four equilibrium constants: K1, the dissociation constant for the RD-CBFβ heterodimer in the absence of DNA; K2, the dissociation constant for the RD–DNA complex; K3, the dissociation constant for CBFβ binding to the RD–DNA complex; and K4 which describes binding of the RD–CBFβ heterodimer to DNA. (B) FRET assay to determine K1. Cerulean (Cer), an optimized version of the cyan fluorescent protein (Rizzo et al, 2004), was fused to the N-terminus of the RD, and the YFP derivative Venus (Rizzo et al, 2004) was fused to the N-terminus of CBFβ. Cerulean was excited at 433 nm and emission from Cerulean and Venus detected at 474 and 525 nm, respectively. (C) Fluorescence spectra of Cerulean-RD and Venus-CBFβ showing the FRET effect. The black curve is the spectrum of Cerulean-RD+Venus-CBFβ at a concentration of 25 nM (4.5-fold below K1). The red curve is the spectrum of Cerulean-RD+Venus-CBFβ at a concentration of 400 nM (3.6-fold above K1). (D) FRET assay of the WT RD binding to CBFβ. The samples were excited at 433 nm, and the ratio of Venus-CBFβ/Cerulean-RD emission peaks (525/474 nm) was plotted at different protein concentrations to generate a binding curve. (E) Yeast one-hybrid assay for RD binding to three core sites driving lacZ expression. Mutations that increase K2 by ⩾10-fold decrease β-galactosidase activity in a yeast one-hybrid filter assay to undetectable levels (Li et al, 2003). Visible but decreased β-galactosidase activity in the filter assay reflects K2 increases in the 3–10-fold range. (F) Modified yeast one-hybrid assay to measure binding of the RD:CBFβ heterodimer to DNA. Although the Gal4 DNA-binding domain is fused to CBFβ in the modified yeast one-hybrid assay, there are no Gal4-binding sites on the promoter driving lacZ and therefore CBFβ's activity is mediated only through the core sites. CBFβ increases the affinity of the RD for DNA by approximately 10-fold. RD mutants that can bind CBFβ and have K2 increases in the 10–90-fold range and correspondingly no β-galactosidase activity in the one-hybrid assay, yield β-galactosidase signals in the modified one-hybrid assay (Li et al, 2003). On the other hand, RD mutants with K2 values that are >100-fold higher than the WT RD produce very weak or no β-galactosidase activity in the modified yeast one-hybrid assay (Li et al, 2003). (G) EMSA measuring the affinity of the WT RD (top) and the L148F RD (bottom) for DNA (K2). Triangles indicate decreasing concentrations of RDs (WT RD, 2 × 10−6 to 4 × 10−15 M; L148F, 1 × 10−5 to 3 × 10−14 M). Arrows indicate lanes in which the RD concentration approximates K2. (H) EMSA measuring the affinity of the WT RD:DNA complex (top) and L148F RD:DNA complex (bottom) for CBFβ (K3). Triangles indicate decreasing concentrations of CBFβ (for the WT RD, 6 × 10−6–4 × 10−14 M; L148F, 2 × 10−5–4 × 10−12 M). Arrows indicate lanes in which the CBFβ concentration approximates K3.
The majority (11/16) of CCD mutations, on the other hand, did not impair DNA binding as severely as the hematopoietic mutations. For example, the S67R, T149A, and Q158R RDs had WT (+) DNA-binding activity, and L62R, F70C, and T154G decreased DNA binding in the one-hybrid assay in the relatively moderate 3- to 10-fold range (+/−; Table I). CBFβ increases the affinity of the RD for DNA by 10-fold; thus a detectable galactosidase signal in the modified yeast one-hybrid assay represents a <100-fold decrease in DNA-binding affinity. DNA binding was indistinguishable from the WT RD in the modified one-hybrid assay for 11 of the 16 RD mutations found in CCD that we analyzed (in contrast, only three of 10 RD mutations found in hematopoietic diseases had activity indistinguishable from the WT RD in the modified yeast one-hybrid assay). The CCD mutations that more severely decreased DNA binding were M124R, S140N, K167N, and T169I that are found only in CCD, and the R174Q mutation found in both CCD and in hematopoietic diseases. The M124R, S140N, K167N, and T169I (CCD-only) mutations that severely decreased DNA binding also impaired CBFβ binding, and thus the RD structure was clearly affected as none of these residues are at the CBFβ interface (Figure 1A). The R174Q RD, on the other hand, is a stably folded protein. R174 is located in the C-terminal tail of the RD, far from the CBFβ interface, and does not interact with the remainder of the protein.
Table I also shows that the thermodynamic stability as assessed by urea denaturation did not always correlate with the affinity of the mutant RDs for DNA. For example, the T149A, T161A, and Q158R mutations, all of which involve CBFβ-contacting residues, decreased the thermodynamic stability of the RD but did not affect DNA binding, which is mediated by residues in loops at the opposite side of the RD from the CBFβ interface.
In summary, the general trend we observed was that the majority of mutations identified only in CCD tended to affect DNA binding less severely than those found in hematopoietic diseases. However, there were exceptions to this trend, in that mutations that severely impair DNA binding are also found in CCD.
Effects on CBFβ binding
We used two assays to examine CBFβ binding by the RD mutants, EMSA and fluorescence resonance energy transfer (FRET). EMSA was used to measure K3 values (Figure 2A and H). However, K3 values were technically difficult to measure for RDs with severely impaired DNA binding; therefore, we used FRET to measure the RD-CBFβ interaction in the absence of DNA (K1) (Figure 2A–D).
All of the RD mutations found exclusively in CCD decreased the affinity of the RD for CBFβ (Table I). In most cases, this correlated with a decrease in the RD's thermodynamic stability, although not necessarily with the magnitude of this change. Seven of the eight CCD-only mutations that we analyzed by urea denaturation had impaired CBFβ binding, and the thermodynamic stability was also compromised. The remaining CCD-only mutation, R118Q, is a non-DNA-contacting residue at the DNA interface. R118Q had a stable fold, and only a modestly (five-fold) decreased affinity for CBFβ.
In contrast, six out of the 10 missense mutations found in hematopoietic diseases did not decrease the RD's affinity for CBFβ. Mutations in hematopoietic diseases that impaired CBFβ binding (increased K3, which include R80C, A107P, R135G, and D171Y) also impaired DNA binding, indicating that the RD's structure was affected. In no case was a mutation found in hematopoietic disease that impaired CBFβ binding alone.
In summary, the general trend we observed was that the majority of CCD mutations (11/16) moderately decreased DNA binding, impaired CBFβ binding, and therefore presumably had a global or local effect on the RD structure. In seven out of eight cases, we confirmed that the presumed structural perturbation correlated with a decrease in thermodynamic stability. However, this was only a trend, and there were several exceptions. Some mutations in CCD (3/16) affected only CBFβ binding and not DNA binding. All three of these mutations (T149A, Q158R, and T161A) involved CBFβ-contacting residues. The other two exceptions were the R139Q and R174Q mutations in DNA-contacting residues that decreased DNA binding but not CBFβ binding.
Approximately two-thirds of the hematopoietic mutations (6/10), on the other hand, decreased DNA binding, did not affect CBFβ binding, and therefore did not perturb the RD structure. The remaining hematopoietic mutations (R80C, A107P, R135G, and D171Y) impaired both DNA and CBFβ binding and thus globally or locally affected the RD structure. All three of these hematopoietic mutations (R80C, R135G, and D171Y) involved DNA-contacting residues. Thus, the prevailing model that mutations in DNA-contacting residues only impair DNA but not CBFβ binding or the RD structure (Nagata and Werner, 2001; Tahirov et al, 2001) turns out to be an oversimplification of the actual situation.
Mutations that disrupt CBFβ binding result in hypomorphic Runx1 alleles
We introduced five missense mutations that conferred different biochemical properties into the endogenous murine Runx1 locus to determine if they resulted in hypomorphic, nonfunctional, or dominant-negative alleles. All five mutations were introduced into exon 4 so that differences in targeting strategies could not contribute to any variations observed between the alleles (Figure 3A). We left the neomycin resistance gene (Neo) in intron 4, and controlled for its potential effect at that position with a similarly targeted floxed Runx1 locus (Runx1f) (Growney et al, 2005; Figure 3C). Runx1 protein levels were not affected by having Neo in the locus (Supplementary Figure 1).
Figure 3.
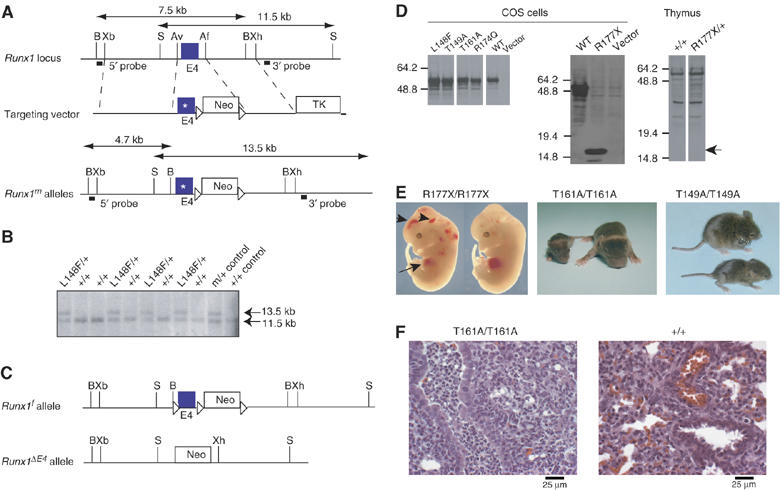
Generation and analysis of mutant Runx1 alleles. (A) Targeting vector. Point mutations were engineered into exon 4 of Runx1. A neomycin resistance gene flanked by loxP sites is in intron 4. The location of probes and restriction length fragments from the WT and targeted Runx1 alleles are indicated. B, BamHI; Xb, XbaI; S, SalI; Av, AvrII; Af, AflII; Xh, XhoI. (B) Southern blot screening of L148F/+ ES cell clones with the 3′ probe. (C) Other Runx1 alleles used in these experiments, which include a floxed locus to control for the presence of Neo in intron 4 (Runx1f) (Growney et al, 2005), and an exon 4-deleted allele (Runx1ΔE4) (Wang et al, 1996a). (D) Western blot of nuclear extracts from COS cells transfected with cDNAs encoding mutant Runx1 proteins (left and middle panels), and from thymus extracts prepared from 6-week-old R177X/+ mice and +/+ littermates (right panel). No truncated R177X protein (expected position of the 17.9 kDa protein indicated by arrow) was detected in thymus extracts. (E) R177X/R177X 12.5 d.p.c. fetuses, post-natal day 7 (P7) T161A/T161A mice, and P14 T149A/T149A mice. WT littermates are shown to the right (R177X and T161A) or on top (T149A). Note the pale liver (arrow) and hemorrhages (arrowheads) in the R177X/R177X fetuses, which is characteristic of Runx1 deficiency (Okuda et al, 1996; Wang et al, 1996a). (F) Histologic sections of lung from 2-day-old T161A/T161A and +/+ littermates (× 400). Arrows indicate an airway with infiltration of neutrophils in the T161A/T161A animal and an unaffected airway in the +/+ animal. Bronchopneumonia was diagnosed in three of the four T161A/T161A neonates analyzed. The accumulation of red blood cells in the +/+ animal is an artifact caused by decapitation.
T149A was found in a CCD pedigree in which some family members had mild and others classical CCD (Zhou et al, 1999). The clinical hallmarks of CCD are short stature, delayed closure of cranial fontanels and sutures, Wormian bones, frontal bossing, supernumerary and late erupting teeth, rudimentary or absent clavicles, wide pubic symphysis, and other skeletal anomalies (Mundlos, 1999). Milder forms of CCD are characterized by dental anomalies with minimal or absent clavicular features. As some family members with the T149A mutation had mild CCD, it was hypothesized that it creates a hypomorphic RUNX2 allele (Zhou et al, 1999). The T149A mutation decreased CBFβ binding by approximately 14-fold, destabilized the RD fold, but did not affect DNA binding. We also introduced a similar but more deleterious T161A mutation in the Runx1 locus that decreased CBFβ binding by 40-fold, but did not affect DNA binding (Zhang et al, 2003).
R174Q was identified in FPD/AML, AML M0, and in a patient with classical CCD (Song et al, 1999; Zhou et al, 1999; Preudhomme et al, 2000; Roumier et al, 2003). The R174Q mutation causes a >10 000 fold decrease in DNA binding. However, the R174Q RD was stably folded, as demonstrated in multiple assays performed by ourselves and others, including urea denaturation, NMR spectroscopy, and by its WT affinity for CBFβ. Transient transfection reporter assays performed previously by other laboratories indicated that R174Q as well as several other DNA-contacting mutants (K83N, K83E, R139G, and R139Q) may have dominant interfering activity (Imai et al, 2000; Michaud et al, 2002).
L148F was identified in a patient with classical CCD (Zhou et al, 1999), and moderately perturbs the RD fold, as illustrated in Figure 1B. L148 is located near the CBFβ heterodimerization interface, and is only 14% solvent-exposed, thus is part of the essential hydrophobic core of the RD. A modeled phenylalanine substitution at L148 sterically clashed with adjacent RD residues in all possible rotamer positions (not shown). L148F disrupted DNA binding by 10-fold, CBFβ binding by 100-fold, and destabilized the domain.
Lastly, we introduced an R177X mutation found in FPD/AML (Song et al, 1999) and AML M0 (Osato et al, 1999) that removes the C-terminal DNA-contacting residue in the RD as well as the entire C-terminus of Runx1, including its transactivation and repression domains.
To assess whether the mutated full-length Runx1 proteins were stable, we transiently transfected cDNAs encoding them into COS cells and analyzed protein levels by Western blot (Figure 3D, left and middle panels). All mutant Runx1 proteins, including the truncated R177X protein, accumulated to similar steady-state levels; thus, the decreased thermodynamic stability observed for some of the isolated RDs did not overtly affect the stability of full-length Runx1 proteins.
We did not assess whether the full-length mutant Runx1 proteins were stable in mice. In mice heterozygous for the mutations, the mutant proteins cannot be distinguished from WT Runx1 with any available antibodies. Furthermore, as described below, mice homozygous for several of the mutations produced no hematopoietic cells from which Runx1 protein levels could be analyzed. Mice heterozygous for the R177X allele, though, could be tested for expression of the truncated Runx1 protein. We were unable to detect the truncated R177X protein in nuclear extracts prepared from the thymuses of R177X/+ mice (Figure 3D, right panel), and suspect that the mRNA for R177X is subject to nonsense-mediated decay, as the truncated protein itself is inherently stable in cells.
Animals homozygous for the R174Q, R177X, and L148F mutant Runx1 alleles died at midgestation with a phenotype identical to that for either a Runx1 exon 3 or 4 deletion, as described previously (Okuda et al, 1996; Wang et al, 1996a) (Table II; Figure 3E). On the other hand, T161A/T161A and T149A/T149A mice were born at the expected Mendelian frequencies (Table II). Although T161A/T161A and T149A/T149A animals were the same size at birth as their +/+ and m/+ littermates, they failed to thrive and became severely stunted in growth (Figure 3E). Most T161A/T161A animals did not survive past 2 weeks of age, and none survived until weaning (Table II). Fifty percent of T149A/T149A mice survived past weaning, overcame their early growth disadvantage, and had a normal lifespan.
Table 2.
Viability of progeny from intercrosses of Runx1m/+ mice
| Biophysical properties | Alive at P0 | Alive at P21 | |||||
|---|---|---|---|---|---|---|---|
| Allele | DNA bindinga | CBFβ bindinga | Thermodynamic stability | +/+ | m/+ | m/m | m/m |
| ΔE4b | − | − | − | 44 | 78 | 0 | |
| R177Xc | 10–90-fold decrease | + | Normald | 46 | 66 | 0 | |
| R174Q | >10 000-fold decrease | + | Normal | 98 | 118 | 0 | |
| L148F | 10-fold decrease | 100-fold decrease | Decreased | 30 | 43 | 0 | |
| T161A | + | 40-fold decrease | Decreased | 33 | 51 | 26 | 0 |
| T149A | + | 14-fold decrease | Decreased | 34 | 69 | 26 | 14 |
| K2 and K3 increases from Table I. | |||||||
| From Wang et al (1996a). | |||||||
| No detectable protein in mice. K2 estimated from yeast one- and modified one-hybrid assays. | |||||||
| Deduced from normal CBFβ binding activity by FRET but not directly measured. | |||||||
T161A/T161A neonates (three of the four necropsied) were found to have a suppurative bronchopneumonia with an anterior distribution of neutrophilic infiltrates within major airways (Figure 3F). There was no evidence of neutrophilic infiltrates or increased myeloid cell proliferation in other tissues including bone marrow, indicating that the lesion most likely occurred in response to some unknown, and possibly infectious insult, suggesting that T161A/T161A mice are most likely predisposed to opportunistic infection. The cause of the bronchopneumonia was not determined. No predisposing anatomical, neurological, or immunological defects were evident.
The mutations' effects on hematopoiesis correlated with viability. Previous studies showed that Runx1-deficient fetuses (exon 3- or 4-deleted alleles) had no liver or aorta/gonad/mesonephros (AGM) hematopoietic progenitors at 11.5 d.p.c. (Okuda et al, 1996; Wang et al, 1996b; Mukouyama et al, 2000). Animals homozygous for mutations that disrupted or impaired DNA binding (R174Q, R177X, and L148F) had essentially no fetal liver or AGM colony forming units-culture (CFU-C) (Figure 4A and B), which correlated with the embryonic lethality conferred by these alleles. Animals homozygous for mutations that affected only CBFβ binding, on the other hand, had hematopoietic progenitors. T161A/T161A 11.5 d.p.c. fetuses had no AGM CFU-C, but had detectable albeit significantly reduced numbers of fetal liver CFU-C (Figure 4A and B). T149A/T149A fetuses had significantly fewer CFU-Cs in the AGM and fetal liver than +/+ fetuses (P=0.0002), but more AGM CFU-C than T161A/T161A fetuses. Thus, the T149A and T161A mutations, which impaired CBFβ binding by 14- and 40-fold, respectively, resulted in hypomorphic Runx1 alleles, whose relative strength correlated with the extent to which CBFβ binding was affected. The numbers of AGM CFU-C in +/+ and the control f/f fetuses were not significantly different (Figure 4A), again demonstrating that the presence of Neo in intron 4 did not affect expression from the Runx1 locus.
Figure 4.
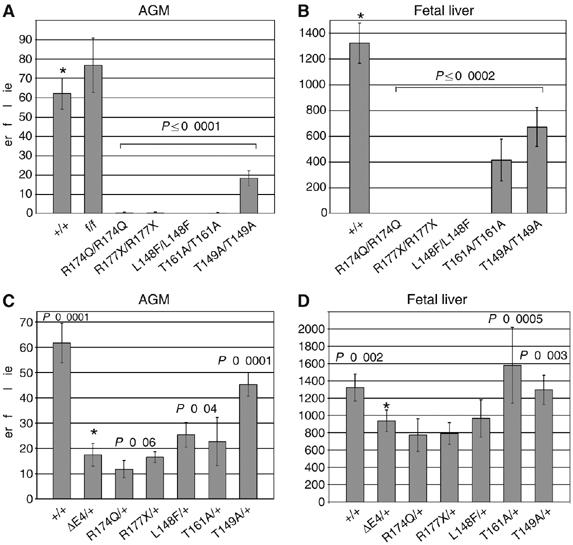
AGM and fetal liver CFU-C assays from 11.5 d.p.c. fetuses. (A) Total number of CFU-Cs (erythroid+granulocyte macrophage+granulocyte erythrocyte monocyte megakaryocyte) per AGM region of Runx1m/m fetuses. Error bars represent 95% confidence intervals. All m/m fetuses were significantly different from +/+ (*). P-values were determined by unpaired two-tailed Student's t-test. n+/+=38; nf/f=9; nR174Q/R174Q=4; nR177X/R177X=3; nL148F/L148F=5; nT161A/T161A=5; nT149A/T149A=10. +/+ values are pooled from all m/+ intercrosses. (B) Total number of fetal liver CFU-Cs (11.5 d.p.c.). All m/m values were significantly different from +/+ (*). n+/+=59; nR174Q/R174Q=3; nR177X/R177X=4; nL148F/L148F=5; nT161A/T161A=5; nT149A/T149A=12. (C) Total number of CFU-Cs per AGM region from Runx1m/+ fetuses. Significant differences from ΔE4/+ (*) are indicated. The difference between R174Q/+ and ΔE4/+ was significant at P=0.06 by unpaired two-tailed Student's t-test, and at P=0.03 by unpaired one-tailed Student's t-test. n+/+=38; nΔE4/+=15; nR174Q/+=14; nR177X/+=10; nL148F/+=8; nT161A/+=10; nT149A/+=39. (D) Total number of CFU-Cs per 11.5 d.p.c. Runx1m/+ fetal liver. Significant differences from ΔE4/+ (*) are indicated (unpaired two-tailed Student's t-test). n+/+=59; nΔE4/+=24; nR174Q/+=13; nR177X/++19; nL148F/+=9; nT161A/+=9; nT149A/+=41.
R174Q mutation creates a weakly dominant-negative Runx1 allele
Runx1 haploinsufficiency in the mouse has been associated with multiple, mild hematopoietic perturbations, including decreased numbers of fetal hematopoietic progenitors, increased CFU-C in the adult mouse bone marrow, a 15% decrease in platelet numbers, and a decrease in the percent of CD4+ cells in the spleen (Wang et al, 1996a, 1996b; Mukouyama et al, 2000; Sun and Downing, 2004). We examined m/+ fetuses and adult mice to ascertain whether any of the mutant Runx1 alleles ameliorated or exacerbated the phenotypes caused by an exon 4-deleted Runx1 allele (ΔE4; Figure 3C).
AGM regions from 11.5 d.p.c. ΔE4/+ fetuses had a greater than three-fold decrease in CFU-C compared to +/+ AGM regions (Figure 4C). The number of CFU-C in R174Q/+ AGM regions was reduced slightly more than in ΔE4/+ fetuses (P=0.06, unpaired two-tailed t-test and P=0.03 by one-tailed t-test), suggesting that the R174Q mutation creates a weak dominant-negative Runx1 allele. R177X/+ fetuses, on the other hand, had similar numbers of AGM CFU-Cs as ΔE4/+ fetuses, and both L148F/+ and T149A/+ AGM regions had significantly more AGM CFU-C than those from ΔE4/+ fetuses (Figure 4C). The difference in AGM CFU-C between R174Q/+ and L148F/+ fetuses was highly significant (P=0.0002). The variability in T161A/+ AGM CFU-C assays was higher than for other genotypes; therefore, significance could not be established. Thus, the AGM CFU-C assay was able to discriminate between nonfunctional (ΔE4 and R177X), strongly hypomorphic (L148F) and weakly dominant-negative (R174Q) Runx1 alleles. The number of fetal liver CFU-C was less sensitive to alterations in Runx1 dosage, with only +/+, T161A/+, and T149A/+ fetuses having significantly more CFU-C than ΔE4/+ fetuses (Figure 4D).
Clinical blood counts (CBCs) identified no significant differences between m/+ and +/+ adult mice (not shown); however, T149A/T149A mice had a 23% decrease in platelet numbers, a decreased total white blood cell count (WBC), a decreased total lymphocyte count, and an increased percentage of monocytes compared with T149A/+ and +/+ littermates (Table III). All m/+ (including ΔE4/+) animals had significant decreases in the percentage of CD4+ splenic T cells and in the CD4+:CD8+ ratio compared to +/+ animals, but no change was observed in the percentage of CD8+ cells (Table IV). R174Q/+ mice had a smaller percentage of CD4+ cells and a lower CD4+:CD8+ ratio than ΔE4/+ animals, again supporting the notion that the R174Q mutation creates a weakly dominant-negative Runx1 allele. The overall cellularity of spleens was not altered in any m/+ mice (not shown). Runx1m/+ animals were not predisposed to leukemia following ethylnitrosurea injection during the 1 year they were observed (not shown).
Table 3.
CBCs of +/+, T149A/+, and T149A/T149A mice
| +/+ (n=23) | T149A/+ (n=25) | T149A/T149A (n=12) | |
|---|---|---|---|
| Hematologic parameter | |||
| RBC count (M/μl) | 10.6±1.6 | 11.3±1.7 | 10.6±2.1 |
| Hemoglobin level (g/dl) | 14.2±1.7 | 14.5±1.0 | 13.5±1.3 |
| Hematocrit level (%) | 54.2±8.4 | 57.1±8.0 | 50.9±9.3 |
| WBC count (K/μl) | 8.4±1.5 | 8.9±3.0 | 5.7±1.6* |
| Lymphocyte count (K/μl) | 6.4±1.3 | 6.7±2.5 | 4.1±1.4* |
| Platelets (K/μl)* | 750±124 | 730±120 | 540±75* |
| Differential of WBC | |||
| Lymphocytes (%) | 76.5±8.7 | 74.8±10.0 | 72. 0±14.5 |
| Neutrophils (%) | 18.9±7.9 | 20.0±10.0 | 20.5±13.4 |
| Monocytes (%) | 4.3±1.7 | 4.8.±1.6 | 7.3±2.9* |
| Mice were analyzed at 6–10 weeks of age. | |||
| *Significantly different from +/+ at P=0.0001. | |||
Table 4.
Percentage of CD4+ and CD8+ cells in spleen
| Genotype | % CD4 Single positivea | % CD8 Single positivea | CD4:CD8 | nb |
|---|---|---|---|---|
| +/+ | 10.7±2.7 | 4.4±1.1 | 2.5 | 99 |
| ΔE4/+ | 7.2±2.0* | 4.3±1.4 | 1.8* | 23 |
| R174Q/+ | 5.9±1.7*§ | 4.1±1.3 | 1.5*§ | 35 |
| R177X/+ | 6.7±2.0* | 3.7±1.0 | 1.8* | 8 |
| L148F/+ | 8.4±2.7* | 4.6±1.6 | 1.9* | 28 |
| T161A/+ | 7.9±1.8* | 4.8±0.5 | 1.6* | 7 |
| T149A/+ | 7.3±2.1* | 4.0±0.7 | 1.8* | 9 |
| Average±s.d. | ||||
| Number of animals analyzed. | ||||
| *P=0.01 compared to +/+. | ||||
| §P=0.01 compared to ΔE4/+. |
Discussion
Three classes of RD mutations contribute to human disease
Mutations in the RD of Runx1 and Runx2 are found in hematopoietic diseases and in CCD, respectively. We have grouped these mutations into three distinct categories based on their DNA and CBFβ-binding activities (Table I and Supplementary Figure 2). Class D mutations, which are found in both hematopoietic disorders and in CCD but predominate in the hematopoietic diseases, affect DNA binding but not CBFβ binding. All but one of these mutations (G138D) altered DNA-contacting residues. The one example of a class D mutation we analyzed in vivo, R174Q, resulted in a weakly dominant-negative Runx1 allele. The dominant-negative activity was seen as subtle but significant decreases in AGM hematopoietic progenitor numbers and in the percentage of splenic CD4+ cells. In addition, we found that mice doubly heterozygous for the R174Q mutation and another very weak hypomorphic Runx1 allele, L148A (R174Q/L148A), suffer from a profound failure to thrive syndrome and the majority died before weaning (unpublished results). On the other hand, mice doubly heterozygous for a Runx1 deletion and the same L148A allele (ΔE4/L148A) were smaller than their littermates but otherwise healthy. Thus, the R174Q mutation exhibited dominant-negative activity in three biological settings: AGM hematopoietic progenitor numbers, CD4+ cells, and viability on a sensitized genetic background.
Class DB (DNA/CBFβ) mutations create RDs that have impaired DNA and CBFβ binding, and are found in both CCD and in hematopoietic diseases. These mutations would be predicted to disrupt the hydrophobic core of the protein (A107P, M124R) or remove specific H bonds critical for the fold (S140N, T169I) resulting in a local or global change in the RD structure. Most DB mutations decreased the RD's thermodynamic stability. An exception was R118Q, which was a borderline DB mutation as it only very moderately decreased CBFβ binding by five-fold. We analyzed one class DB mutation in vivo, L148F, which created a strongly hypomorphic Runx1 allele. We predict that other class DB mutations that more severely disrupt DNA binding than L148F will create completely nonfunctional alleles.
Class B mutations, which are found exclusively in Runx2 in CCD, moderately to severely impaired CBFβ binding, destabilized the RD, but had no measurable effect on DNA binding. When two class B mutations (T149A and T161A) were introduced into the murine Runx1 gene, they resulted in hypomorphic alleles.
One conclusion from our studies is that the majority of the mutations found in CCD are functionally distinct from those found in hematopoietic diseases. The class B mutations found in CCD, in all likelihood, create weak hypomorphic RUNX2 alleles. In contrast, none of the hematopoietic mutations are class B mutations, and many of them are class D mutations.
One possible explanation for the prevalence of hypomorphic RUNX2 alleles in CCD is that many amino-acid substitutions in the RD have the ability to only moderately perturb DNA or CBFβ binding (Li et al, 2003) and, as suggested by others (Yoshida et al, 2002), bone formation is apparently sensitive to mild (less than two-fold) reductions in Runx2-CBFβ dosage. Correspondingly, the lack of hypomorphic class B mutations in FPD/AML and leukemia might reflect a relative insensitivity of hematopoiesis to less than two-fold decreases in Runx1 dosage, and thus only the smaller subset of more severe mutations can cause hematopoietic disease. However, an alternative explanation for the relative ease of finding hypomorphic RUNX2 alleles in CCD could be that there is a clinical detection bias. As minor skeletal malformations are presumably more easily and less invasively diagnosed than small disturbances in hematopoiesis, hypomorphic RUNX2 alleles may be more readily identified. However, it is important to keep in mind that CCD is associated with mutations in RUNX2, whereas hematopoietic disorders are associated with mutations in RUNX1. We analyzed the effect of these mutations on Runx1 function in hematopoiesis. As Runx2 is expressed in different cells, has different targets, and is involved in different pathways, the effect of these mutations on Runx2 function could also be different.
Implications of dominant-negative Runx1 alleles
Hundreds of cancer-associated missense mutations have been identified in the structurally related p53 DNA-binding domain. Some of these mutations involve DNA-contacting residues (‘DNA-contact' mutations), whereas the majority are so-called ‘structural' mutations that affect the stability of the p53 DNA-binding domain (Bullock and Fersht, 2001). p53 binds DNA as a dimer of dimers through interactions between four p53 DNA-binding domains and a separate oligomerization domain (Lee et al, 1994; Jeffrey et al, 1995; Kitayner et al, 2006). Several of the amino acids that are mutation hot spots in the p53 DNA-binding domain support the structure of both the DNA-binding interface and of the dimerization interface (Bullock and Fersht, 2001; Kitayner et al, 2006). Both DNA-contact and structural mutations in the Tp53 gene create dominant-negative or gain-of-function alleles. However, in the case of p53, the structural mutations are thought to create more severe dominant-negative Tp53 alleles than the DNA-contact mutations (Halevy et al, 1990; Hinds et al, 1990; Olive et al, 2004). This may be caused by the effect structural mutations have both on the ability of p53 to form stable tetramers and to interact with other proteins.
In contrast, it was a class D mutation in a DNA-contacting residue that resulted in a non-DNA-binding but correctly folded RD that created a dominant-negative Runx1 allele. This finding predicts that a correctly folded RD that cannot occupy the DNA is sequestering or altering the activity of a protein or proteins that are present in limiting amounts. The obvious candidate that comes to mind is CBFβ, as the correctly folded RDs retain CBFβ binding. However, there is little evidence to suggest that a two-fold reduction in CBFβ levels has a biological impact (Sasaki et al, 1996; Wang et al, 1996b; Talebian et al, 2007). Furthermore, the non-DNA-bound form of the RD has a 10-fold lower affinity for CBFβ than the DNA-bound RD (Tang et al, 2000b); therefore, presumably less than half of the available CBFβ will be sequestered into non-DNA-binding Runx1 complexes. The genetic data predict that another, as yet unidentified protein that binds an intact Runx1-CBFβ heterodimer is present in limiting amounts, and is being sequestered by non-DNA-binding Runx1-CBFβ complexes.
Modeling FPD/AML in the mouse
One of the potential outcomes of these analyses was a mouse model for FPD/AML. FPD/AML is characterized by decreased platelet numbers and abnormal response times to agonists of platelet aggregation. Several patients were shown to have reduced numbers of committed hematopoietic progenitors in their peripheral blood or bone marrow (Song et al, 1999). Although moderately (15%) decreased platelet counts were previously reported in mice haploinsufficient for Runx1 (Sun and Downing, 2004), we were unable to reliably detect this small decrease for reasons that may have to do with the greater variability in the genetic background of our mice. However, we were able to document mild thrombocytopenia in mice homozygous for the hypomorphic T149A mutation. Thus, the T149A/T149A mice may provide a model for the platelet dysfunctions associated with FPD/AML, and suggest that reducing functional Runx1 dosage by slightly more than 50% may provide a strategy for more faithfully modeling at least some aspects of RUNX1 haploinsufficiency in human disease.
Materials and methods
Yeast one- and modified one-hybrid assays
The yeast one-hybrid and modified one-hybrid assays were described previously (Bravo et al, 2001; Li et al, 2003). We detected β-galactosidase expression with filter assays.
Purification of RD and CBFβ proteins
Expression and purification of the RD (aa 41–214) and CBFβ(1–141) for urea denaturation, 15N-1H HSQC spectroscopy, and EMSAs were performed as described previously (Berardi et al, 1999; Zhang et al, 2003). Protein for NMR analysis was labeled in 15N minimal media supplemented with 10% rich media (Bioexpress Cell Growth Media, Cambridge Isotopes CGM-1000-N).
Fluorescent proteins for FRET analyses were produced by fusing an N-terminal Hisx6 tag in-frame to Cerulean (Rizzo et al, 2004) and subcloning it between the NdeI and BamHI sites of pET-22b (Novagen), then inserting codons for Runx1(41–190) downstream of Cerulean between the BamHI and XhoI sites. Venus-CBFβ was cloned by first inserting an N-terminal His tag and EYFP (Clontech) into the pET22b vector between the NdeI and BamHI sites. CBFβ was then inserted between the BamHI and XhoI sites to create an EYFP-CBFβ construct. The five point mutations necessary to convert EYFP to Venus (Nagai et al, 2002) were subsequently introduced to generate the final Venus-CBFβ construct. Plasmids were transformed into Rosetta (DE3) (Novagen) competent cells for expression, and purified by standard Ni affinity chromatography (HisTrap, Pharmacia).
Urea denaturation monitored by fluorescence spectroscopy
Denaturation was monitored using the intrinsic fluorescence of the single tryptophan (W79) in the RD as described previously (Zhang et al, 2003), with the following minor modifications. Fluorescence measurements were made on a Fluoromax-3 spectrofluorometer (Jobian Yvan Horba) at 4°C. Samples were excited at 280 nm using excitation and emission band passes of 2.1 and 5 nm, respectively. Fluorescence in the range of 300–370 nm was recorded with an increment of 2 nm and an integration time of 1 s, and the fluorescence intensity (counts/s) at 340 nm recorded.
NMR spectroscopy
15N-1H HSQC spectra were recorded on a Varian Inova 500 MHz NMR spectrometer equipped with an actively shielded triple resonance probe (Nalorac Corp). Spectra of free and DNA-bound RDs were recorded at 20 and 40°C, respectively. The RD:DNA complex was prepared as described previously (Berardi et al, 1999).
FRET
Cerulean-Runt and Venus-CBFβ fusion proteins were dialyzed into FRET assay buffer (25 mM Tris–HCl (pH 7.5), 150 mM KCl, and 2 mM MgCl2). Protein concentrations were determined by UV absorption at 433 nm for Cerulean-Runt and 513 nm for Venus-CBFβ (ɛ=34 000 and 92 000 M−1 cm−1, respectively). Equal molar amounts (2 μM) of RD and CBFβ in 2 ml FRET buffer plus 0.01% BSA were incubated at 4°C for 1 h in the dark. Samples were serially diluted two-fold and read in a Fluoromax-3 spectrofluorometer (Jobian Yvan Horba). Samples were excited at 433 nm using excitation and emission band passes of 1.3 and 2.0 nm, respectively, and scanned from 440 to 610 nm. The ratio of the emission intensities at 525 and 474 nm was plotted against the log of the protein concentrations. A sigmoidal curve was fit using Origin 5.0 software.
Equilibrium binding constant measurements
EMSAs were performed to measure the equilibrium binding constants of the RD for DNA (K2) and of CBFβ for an RD:DNA complex (K3) as described previously (Tang et al, 2000a; Li et al, 2003; Zhang et al, 2003), using the Core site from the SL3–3 murine leukemia virus enhancer (GGATATCTGTGGTTAAGCA) as the DNA probe.
Generation of mutant Runx1 alleles
Targeting vectors were used to replace the WT exon 4 of Runx1 with a mutated exon 4 containing specific point mutations. Mutations were made in an AvrII–AflII fragment encompassing Runx1 exon 4 subcloned into pBluescript SK+. The mutated AvrII–AflII fragment was excised, AscI linkers added, and the fragment subcloned into an AscI site in polylinker sequence located between the 5′ homology region and the floxed neo gene. The targeting vector was linearized with NotI, electroporated into J1 or R1 ES cells, and mice were generated and screened by standard protocols. Heterozygous animals were maintained on a mixed C57BL/6+129S1/SVImJ background. The presence of a given point mutation in the Runx1 locus was confirmed by PCR amplifying tail DNA prepared from animals heterozygous for the mutation using the primers RDint (5′-GAGTCCCAGCTGTCAATTCC) and Ex4-3int (5′-CTCTTCAGGAAAGA GGCTAAGC), which were complementary to sequences in introns 3 and 4, respectively. The PCR product was subcloned using a Topo TA kit (Invitrogen, Carlsbad, CA), and plasmids from several independent bacterial colonies were sequenced with the primers Ex4-5seq (5′-CTCAATATGTTCTGTTCTGTTTCC) and Ex4-3seq (5′-TTGTGTACACCAGCCCAGACAG) to confirm that 50% of the colonies had the correct point mutations. The Runx1ΔE4 (Runx1tm1Spe) and Runx1f (Runx1tm3Spe) alleles were described previously (Wang et al, 1996a; Growney et al, 2005).
Western blots
COS (3 × 106) cells were transfected with 9 μg Runx1 in pcDNA 3.1 following the manufacturer's protocol (Fugene). Nuclear extracts from COS cells and thymocytes were separated through 12% SDS–PAGE gels. Whole-cell lysates from thymocytes were prepared in RIPA buffer (105 cells/μl) in the presence of complete EDTA-free proteinase inhibitor cocktail tablets (Roche, Basel, Switzerland) and fractionated through NuPAGE 4–12% Novex Bis-Tris Gels (Invitrogen). Blots were probed with mouse monoclonal antibodies to Runx1 (3.2.5 or 3.2.3.1) or CBFβ (141.4.1+141.2.2.5) and either ECL reagents (Amersham, Piscataway, NJ) or SuperSignal West Pico Chemiluminescent reagents (Pierce, Rockford, IL) and quantified using ImageQuant 5.0.
Histology
Two- to four-day-old neonates (four T161A/T161A, two +/+, three T161A/+) were immediately decapitated posteuthanasia, and more than 50 tissues examined by routine histology (fixation in 10% neutral buffered formalin, paraffin embedding and 5 μm sectioning with hematoxylin and eosin staining).
Fetal liver and AGM colony-forming assays
Colony forming assays (CFU-C) were performed as described previously (Wang et al, 1996b). Single-cell suspensions from livers were prepared by passage through 18- and 22-gauge needles (four times each). Single-cell suspensions of AGM regions were prepared as described by Dzierzak and de Bruijn (2002).
FACS analysis
Spleen cells from 4- to 6-week-old animals were stained with the antibodies CD4-allophycocyanin (APC, clone RM4-5) and CD8-fluorescein isothiocyanate (FITC, clone 53-6.7) (Pharmingen), and analyzed using a Becton Dickinson FACS Calibur. Data were pooled from experiments performed on heterozygous and WT littermates on different days.
Clinical blood cell counts
Intra-orbital blood was collected from 6- to 10-week-old mice, transferred to tubes coated with EDTA (Startsedt), and counted on a Drew Scientific Hemavet 850.
ENU mutagenesis
Animals 4–6 weeks of age were injected intraperitoneally with 50 mg/kg of ENU (Sigma N3385). Animals were monitored for visible signs of illness and deaths were noted.
Statistical analysis
An unpaired, two-tailed Student's t-test was performed on data sets unless otherwise noted.
Supplementary Material
Supplementary Figure Legends
Supplementary Figure 1
Supplementary Figure 2
Acknowledgments
This work was funded by R01CA89419 to NAS and JHB. CJM was supported by T32GM08704. Flow cytometry and the transgenic mouse facility were supported in part by the Core Grant of the Norris Cotton Cancer Center (CA23108).
References
- Berardi M, Sun C, Zehr M, Abildgarrd F, Peng J, Speck NA, Bushweller JH (1999) The Ig fold of the core binding factor a Runt domain is a member of a family of structurally and functionally related Ig-fold DNA-binding domains. Structure 10: 1247–1256 [DOI] [PubMed] [Google Scholar]
- Bravo J, Li Z, Speck NA, Warren AJ (2001) The leukemia-associated AML1 (Runx1)–CBFβ complex functions as a DNA-induced molecular clamp. Nat Struct Biol 8: 371–378 [DOI] [PubMed] [Google Scholar]
- Buijs A, Poddighe P, van Wijk R, van Solinge W, Borst E, Verdonck L, Hagenbeek A, Perason P, Lokhorst H (2001) A novel CBFA2 single-nucleotide mutation in familial platelet disorder with propensity to develop myeloid malignancies. Blood 98: 2856–2858 [DOI] [PubMed] [Google Scholar]
- Bullock AN, Fersht AR (2001) Rescuing the function of mutant p53. Nat Rev Cancer 1: 68–76 [DOI] [PubMed] [Google Scholar]
- Corazza A, Pettirossi F, Viglino P, Verdone G, Garcia J, Dumy P, Giorgetti S, Mangione P, Raimondi S, Stoppini M, Bellotti V, Esposito G (2004) Properties of some variants of human beta2-microglobulin and amyloidogenesis. J Biol Chem 279: 9176–9189 [DOI] [PubMed] [Google Scholar]
- Dzierzak E, de Bruijn M (2002) Isolation and analysis of hematopoietic stem cells from mouse embryos. In Methods in Molecular Medicine—Hematopoietic Stem Cell Protocols, Klug CA, Jordan CT (eds.),Vol. 63, p 1 Totawa, NJ: Humana Press Inc. [DOI] [PubMed] [Google Scholar]
- Gorbatyuk VY, Tsai CK, Chang CF, Huang TH (2004) Effect of N-terminal and Met23 mutations on the structure and dynamics of onconase. J Biol Chem 279: 5772–5780 [DOI] [PubMed] [Google Scholar]
- Growney JD, Shigematsu H, Li Z, Lee BH, Adelsperger J, Rowan R, Curley DP, Kutok JL, Akashi K, Williams IR, Speck NA, Gilliland DG (2005) Loss of Runx1 perturbs adult hematopoiesis and is associated with a myeloproliferative phenotype. Blood 106: 494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halevy O, Michalovitz D, Oren M (1990) Different tumor-derived p53 mutants exhibit distinct biological activities. Science 250: 113–116 [DOI] [PubMed] [Google Scholar]
- Harada H, Harada Y, Niimi H, Kyo T, Kimura A, Inaba T (2004) High incidence of somatic mutations in the AML1/RUNX1 gene in myelodysplastic syndrome and low blast percentage myeloid leukemia with myelodysplasia. Blood 103: 2316–2324 [DOI] [PubMed] [Google Scholar]
- Harada H, Harada Y, Takada H, Kimura A, Inaba T (2003) Implications of somatic mutations in the AML1 gene in radiation-associated and therapy-related myelodysplastic syndrome/acute myeloid leukemia. Blood 101: 673–680 [DOI] [PubMed] [Google Scholar]
- Hinds PW, Finlay CA, Quartin RS, Baker SJ, Fearon ER, Vogelstein B, Levine AJ (1990) Mutant p53 DNA clones from human colon carcinomas cooperate with ras in transforming primary rat cells: a comparison of the ‘hot spot' mutant phenotypes. Cell Growth Differ 1: 571–580 [PubMed] [Google Scholar]
- Imai S, Kurokawa M, Izutsu K, Hangaishi A, Takeuchi K, Maki K, Ogawa S, Chiba S, Mitaini K, Hirai H (2000) Mutations of the AML1 gene in myelodysplastic syndrome and their functional implications in leukemogenesis. Blood 96: 3154–3160 [PubMed] [Google Scholar]
- Jeffrey PD, Gorina S, Pavletich NP (1995) Crystal structure of the tetramerization domain of the p53 tumor suppressor at 1.7 angstroms. Science 267: 1498–1502 [DOI] [PubMed] [Google Scholar]
- Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, Shakked Z (2006) Structural basis of DNA recognition by p53 tetramers. Mol Cell 22: 741–753 [DOI] [PubMed] [Google Scholar]
- Lee B, Thirunavukkarasu K, Zou L, Pastore L, Baldini A, Hecht J, Georffroy V, Ducy P, Karsenty G (1997) Missense mutations abolishing DNA binding of the osteoblast-specific transcription factor OSF2/CBFA1 in cleidocranial dysplasia. Nat Genet 16: 307–310 [DOI] [PubMed] [Google Scholar]
- Lee W, Harvey TS, Yin Y, Yau P, Litchfield D, Arrowsmith CH (1994) Solution structure of the tetrameric minimum transforming domain of p53. Nat Struct Biol 1: 877–890 [DOI] [PubMed] [Google Scholar]
- Li Z, Yan J, Matheny CJ, Corpora T, Bravo J, Warren AJ, Bushweller JH, Speck NA (2003) Energetic contribution of residues in the Runx1 Runt domain to DNA binding. J Biol Chem 278: 33088–33096 [DOI] [PubMed] [Google Scholar]
- Michaud J, Wu F, Osato M, Cottles G, Yanagida M, Asou N, Shigesada K, Ito Y, Benson A, Weiss H, Horwitz M, Scott H (2002) In vitro analyses of known RUNX1/AML1 mutations in dominant familial platelet disorder with predisposition to acute myelogenous leukemia: implications for mechanisms of pathogenesis. Blood 99: 1364–1372 [DOI] [PubMed] [Google Scholar]
- Mukouyama Y, Chiba N, Hara T, Okada H, Ito Y, Kanamaru R, Miyajima A, Satake M, Watanabe T (2000) The AML1 transcription factor functions to develop and maintain hematogenic precursor cells in the embryonic aorta-gonad-mesonephros region. Dev Biol 220: 27–36 [DOI] [PubMed] [Google Scholar]
- Mundlos S (1999) Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet 36: 177–182 [PMC free article] [PubMed] [Google Scholar]
- Mundlos S, Otto F, Mundlos C, Mulliken JB, Aylsworth AS, Albright S, Lindhout D, Cole WG, Henn W, Knoll JHM, Owen MJ, Mertelsmann R, Zabel BU, Olsen BR (1997) Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 89: 773–779 [DOI] [PubMed] [Google Scholar]
- Nagai T, Ibata K, Park ES, Kubota M, Mikoshiba K, Miyawaki A (2002) A variant of yellow fluorescent protein with fast and efficient maturation for cell-biological applications. Nat Biotechnol 20: 87–90 [DOI] [PubMed] [Google Scholar]
- Nagata T, Werner MH (2001) Functional mutagenesis of AML1/RUNX1 and PEBP2β/CBFβ define distinct, non-overlapping sites for DNA recognition and heterodimerization by the Runt domain. J Mol Biol 308: 191–203 [DOI] [PubMed] [Google Scholar]
- Nagata T, Gupta V, Sorce D, Kim W-Y, Sali A, Chait B, Shigesada K, Ito Y, Werner MH (1999) Immunoglobulin motif DNA recognition and heterodimerization of the PEBP2/CBF Runt domain. Nat Struct Biol 6: 615–619 [DOI] [PubMed] [Google Scholar]
- Okuda T, van Deursen J, Hiebert SW, Grosveld G, Downing JR (1996) AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 84: 321–330 [DOI] [PubMed] [Google Scholar]
- Olive KP, Tuveson DA, Ruhe ZC, Yin B, Willis NA, Bronson RT, Crowley D, Jacks T (2004) Mutant p53 gain of function in two mouse models of Li–Fraumeni syndrome. Cell 119: 847–860 [DOI] [PubMed] [Google Scholar]
- Osato M (2004) Point mutations in the RUNX1/AML1 gene: another actor in RUNX leukemia. Oncogene 23: 4284–4296 [DOI] [PubMed] [Google Scholar]
- Osato M, Asou N, Abdalla E, Hoshino K, Yamasaki H, Okubo T, Suzushima H, Takatsuki K, Kanno T, Shigesada K, Ito Y (1999) Biallelic and heterozygous point mutations in the Runt domain of the AML1/PEPBP2 αβ gene associated with myeloblastic leukemias. Blood 93: 1817–1824 [PubMed] [Google Scholar]
- Pérez-Alvarado GC, Munnerlyn A, Dyson HJ, Grosschedl R, Wright PE (2000) Identification of the regions involved in DNA binding by the mouse PEBP2α protein. FEBS Lett 470: 125–130 [DOI] [PubMed] [Google Scholar]
- Preudhomme C, Warot-Loze D, Roumier C, Grardel-Duflos N, Garard R, Lai J-L, Dastugue N, Macintyre E, Denis C, Bauters F, Kerckaert JP, Cosson A, Fenaux P (2000) High incidence of biallelic point mutations in the Runt domain of the AML1/PEBP2αB gene in M0 acute myeloid leukemia and in myeloid malignancies with acquired trisomy 21. Blood 96: 2862–2869 [PubMed] [Google Scholar]
- Quack I, Vonderstrass B, Sock M, Aylsworth AS, Becker A, Brueton L, Lee P, Majewski F, Mulliken JB, Suri M, Zenker M, Mundlose S, Otto F (1999) Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am J Hum Genet 65: 1268–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Zhao L, Sivashanmugam A, Miao Y, Korando L, Yang Z, Reardon CA, Getz GS, Brouillette CG, Jerome WG, Wang J (2005) Engineering mouse apolipoprotein A-I into a monomeric, active protein useful for structural determination. Biochemistry 44: 14907–14919 [DOI] [PubMed] [Google Scholar]
- Rizzo MA, Springer GH, Granada B, Piston DW (2004) An improved cyan fluorescent protein variant useful for FRET. Nat Biotechnol 22: 445–449 [DOI] [PubMed] [Google Scholar]
- Roumier C, Eclache V, Imbert M, Davi F, MacIntyre E, Garand R, Talmant P, Lepelley P, Lai J-L, Casasnovas O, Maynadie M, Mugneret F, Bilhou-Naberra C, Valensi F, Radford I, Mozzoconacci MJ, Arnoulet C, Duchayne E, Dastugue N, Cornillet P, Daliphard S, Garnache F, Boudjerra N, Jouault H, Fenneteau O, Pedron B, Berger R, Flandrin G, Fenaux P, Preudhomme C (2003) M0 AML, clinical and biologic features of the disease, including AML1 gene mutations: a report of 59 cases by Groupe Français d'Hématologie Cellularie (GFHC) and the Groupe Français de Cytogénétique Hématologique (GFCH). Blood 101: 1277–1283 [DOI] [PubMed] [Google Scholar]
- Sanderson A, Stott K, Stevens TJ, Thomas JO (2005) Engineering the structural stability and functional properties of the GI domain into the intrinsically unfolded GII domain of the yeast linker histone Hho1p. J Mol Biol 349: 608–620 [DOI] [PubMed] [Google Scholar]
- Sasaki K, Yagi H, Bronson RT, Tominaga K, Matsunashi T, Deguchi K, Tani Y, Kishimoto T, Komori T (1996) Absence of fetal liver hematopoiesis in mice deficient in transcriptional coactivator core binding factor beta. Proc Natl Acad Sci USA 93: 12359–12363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song WJ, Sullivan MG, Legare RD, Hutchings S, Tan X, Kufrin D, Ratajczak J, Resende IC, Haworth C, Hock R, Loh M, Felix C, Roy DC, Busque L, Kurnit D, Willman C, Gewirtz AM, Speck NA, Busweller JH, Li FP, Gardiner K, Poncz M, Maris JM, Gilliland DG (1999) Haploinsufficiency of CBFA2 causes familial thrombocytopenia with propensity to develop acute myelogenous leukaemia. Nat Genet 23: 166–175 [DOI] [PubMed] [Google Scholar]
- Sun C, Downing JR (2004) Haploinsufficiency of AML1 results in a decrease in the number of LTR-HSCs while simultaneously inducing an increase in more mature progenitors. Blood 104: 3565–3572 [DOI] [PubMed] [Google Scholar]
- Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura A, Shiina M, Sato K, Kumasaka T, Yamamoto M, Ishii S, Ogata K (2001) Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFβ. Cell 104: 755–767 [DOI] [PubMed] [Google Scholar]
- Talebian L, Li Z, Guo Y, Gaudet J, Speck ME, Sugiyama D, Kaur P, Pear WS, Maillard I, Speck NA (2007) T lymphoid, megakaryocyte, and granulocyte development are sensitive to decreases in CBFβ dosage. Blood 109: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Shi J, Zhang L, Davis A, Bravo J, Warren AJ, Speck NA, Bushweller JH (2000a) Energetic and functional contribution of residues in the core binding factor β (CBFβ) subunit to heterodimerization with CBFα. J Biol Chem 275: 39579–39587 [DOI] [PubMed] [Google Scholar]
- Tang Y-Y, Crute BE, Kelley JJ III, Huang X, Yan J, Shi J, Hartman KL, Laue TM, Speck NA, Bushweller JH (2000b) Biophysical characterization of interactions between the core binding factor α and β subunits and DNA. FEBS Lett 470: 167–172 [DOI] [PubMed] [Google Scholar]
- Walker LC, Stevens J, Campbell H, Corbett R, Spearing R, Heaton D, Macdonald DH, Morris CM, Ganly P (2002) A novel inherited mutation of the transcription factor RUNX1 causes thrombocytopenia and may predispose to acute myeloid leukemia. Br J Haematol 117: 878–881 [DOI] [PubMed] [Google Scholar]
- Wang Q, Stacy T, Binder M, Marín-Padilla M, Sharpe AH, Speck NA (1996a) Disruption of the Cbfa2 gene causes necrosis and hemorrhaging in the central nervous system and blocks definitive hematopoiesis. Proc Natl Acad Sci USA 93: 3444–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Stacy T, Miller J, Lewis A, Gu T, Huang X, Bushweller J, Bories J, Alt F, Ryan G, Liu P, Wynshaw-Boris A, Binder M, Marin-Padilla M, Sharpe A, Speck N (1996b) The CBFβ subunit is essential for CBFα2 (AML1) function in vivo. Cell 87: 697–708 [DOI] [PubMed] [Google Scholar]
- Yoshida T, Kanegane H, Osato M, Yanagida M, Miyawake T, Ito Y, Shigesada K (2002) Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype–phenotype correlations. Am J Hum Genet 71: 724–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Li Z, Yan J, Pradhan P, Corpora T, Cheney M, Bravo J, Warren AJ, Bushweller JH, Speck NA (2003) Mutagenesis of the Runt domain defines two energetic hotspots for heterodimerization with core binding factor β subunit. J Biol Chem 278: 33097–33104 [DOI] [PubMed] [Google Scholar]
- Zhou G, Chen Y, Zhou L, Thirunavukkarasu K, Hecht J, Chitayat D, Gelb B, Pirinen S, Berry SA, Greenberg CR, Karsenty G, Lee B (1999) CBFA1 mutation analysis and functional correlation with phenotypic variability in cleidocranial dysplasia. Hum Mol Genet 8: 2311–2316 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure Legends
Supplementary Figure 1
Supplementary Figure 2