

Abstract
In addition to contractile function, muscle provides a metabolic buffer by degrading protein in times of organismal need. Protein is also degraded during adaptive muscle remodeling upon exercise, but extreme degradation in diverse clinical conditions can compromise function(s) and threaten life. Here, we show how two independent signals interact to control protein degradation. In striated muscles of Caenorhabditis elegans, reduction of insulin-like signaling via DAF-2 insulin/IGF receptor or its intramuscular effector PtdIns-3-kinase (PI3K) causes unexpected activation of MAP kinase (MAPK), consequent activation of pre-existing systems for protein degradation, and progressive impairment of mobility. Degradation is prevented by mutations that increase signal downstream of PI3K or by disruption of autocrine signal from fibroblast growth factor (FGF) via the FGF receptor and its effectors in the Ras–MAPK pathway. Thus, the activity of constitutive protein degradation systems in normal muscle is minimized by a balance between directly interacting signaling pathways, implying that physiological, pathological, or therapeutic alteration of this balance may contribute to muscle remodeling or wasting.
Keywords: growth factors, muscle, nematode, protein degradation, signal transduction
Introduction
Human skeletal muscle is a multifunctional tissue containing up to half the total protein content of the body (Rooyackers and Nair, 1997), a metabolic buffer that can be mobilized by protein catabolism in times of organismal need. The existence of specialized ‘heater' cells (Block and Franzini-Armstrong, 1988) and specialized fiber types with different contractile and metabolic properties (Spangenburg and Booth, 2003) suggests that muscle functions are interrelated along a continuum, but the mechanisms that maintain and adjust the relationships among the contractile, endocrine (Smith and Muscat, 2005), heat production, and metabolic buffer roles of muscle are poorly understood. Muscle remodeling in response to increased contractile demand incurred with exercise (Tipton and Wolfe, 2001) provides an example of how adjustment of this interrelation occurs in response to functional demand, and requires protein degradation. However, the protein degradation required to mobilize the metabolic buffer may come at the expense of other functions. For example, loss of lean mass in response to burn injury is probably adaptive but comes at the expense of increased healing time (Pereira et al, 2005). Although many clinical conditions and extramuscular factors trigger protein degradation in muscle, the intramuscular signaling mechanisms are incompletely understood (Szewczyk and Jacobson, 2005).
Under conditions of stress, mammalian cells degrade bulk protein, via both the proteasome and lysosome, as a way to mobilize amino acids (Vabulas and Hartl, 2005). Protein degradation in muscle is traditionally measured by release of labeled amino acids from total cellular protein or by loss of muscle mass rather than by focusing on the degradation of a well-defined substrate (Thompson and Palmer, 1998; Attaix et al, 2001). The inherent difficulty in studying muscle atrophy in live animals has produced a heavy reliance on cell and tissue culture methods to elucidate the intramuscular regulation of muscle atrophy (Thompson and Palmer, 1998; Attaix et al, 2001; Szewczyk and Jacobson, 2005). However, cultured cells lack the innervation required to prevent muscle degradation and consequently the regulation of proteolytic activity does not appear the same in cell culture as in live animals (Attaix et al, 2001; Szewczyk and Jacobson, 2005). To circumvent the experimental limitations of studying muscle protein degradation in mammals and to engage the power of genetic approaches, transgenic lines of Caenorhabditis elegans have been established as a simple model system for studying protein catabolism in vivo (Zdinak et al, 1997; Szewczyk et al, 2000, 2002; Fostel et al, 2003; Szewczyk and Jacobson, 2003, 2005). The rationale is to simplify the substrate problem, such that the degradation or inactivation of single proteins can be followed unambiguously in vivo. Transgenic animals offer this possibility, with the additional ability to control the cellular, subcellular, and/or developmental specificity of expression. For example, a fusion of green fluorescent protein (GFP) to full-length myosin heavy chain has been used to follow the degradation of myofibers, whereas GFP or β-galactosidase expressed from a myosin promoter, but not fused to full-length myosin, allows the degradation of soluble cytosolic proteins to be followed (Zdinak et al, 1997; Fostel et al, 2003). This approach also permits detection of differential regulation of protein degradation; for example, starvation provokes proteasome-mediated degradation of cytosolic but not myofibrillar or nuclear reporter proteins in C. elegans (Szewczyk et al, 2000; Fostel et al, 2003). These proteins appear to report on physiologically relevant protein degradation, insofar as the degradation of two native cytosolic muscle enzymes required for ATP homeostasis (adenylate kinase and arginine kinase) parallels that of the cytosolic reporter proteins (Fostel et al, 2003). Although overexpression of transgene-encoded polyglutamine-containing proteins in C. elegans can be used as a model for studying the polyglutamine pathogenesis (Brignull et al, 2006), there is no indication that transgene-coded fusion proteins are preferentially degraded by virtue of being foreign proteins, nor that fusion proteins trigger an unfolded protein response (Zdinak et al, 1997; Szewczyk et al, 2000, 2002; Fostel et al, 2003; Szewczyk and Jacobson, 2003, 2005). On the contrary, reporter proteins such as GFP are extremely resistant to degradation; so their use in studies of the regulation of protein degradation has been advocated (Dantuma et al, 2000; Andreatta et al, 2001; Bence et al, 2001). Because these reporter proteins are completely stable in well-fed wild-type animals (Zdinak et al, 1997), do not normally exist in wild-type animals, and are degraded under physiological conditions that also evoke the degradation of endogenous muscle proteins (Fostel et al, 2003), these proteins likely report on bulk protein degradation rather than protein-specific degradation.
Using such reporter proteins in C. elegans, it was recently shown that activation of the Ras–MAP kinase (MAPK) pathway downstream of the fibroblast growth factor receptor (FGFR) provokes proteasome-independent degradation of cytosolic muscle protein in response to FGF (Szewczyk and Jacobson, 2003). Surprisingly, at least some of the FGF required to produce this response is produced constitutively by the muscle itself (Bulow et al, 2004). Together, these results imply that C. elegans muscle is poised to undergo constitutive protein degradation, yet such degradation of muscle proteins does not normally occur in well-fed, wild-type animals (Zdinak et al, 1997), implying the existence of countervailing inhibitory signaling within the muscle. Here, we report that insulin-like signaling via the DAF-2 receptor and its effectors PI3K and Akt act to oppose FGFR-controlled constitutive protein degradation in C. elegans muscle, most likely by direct inhibitory phosphorylation of Raf by protein kinase Akt.
Results
Insulin-like signaling opposes proteolysis in C. elegans muscle
In mammalian muscle, insulin and insulin-like growth factor (IGF) promote protein accumulation, whereas low insulin signaling (as in untreated diabetes) conversely leads to muscle wasting, complementary results of effects on both protein degradation and protein synthesis. In C. elegans, an insulin-like signaling pathway is known to control lipid metabolism, longevity, and the choice between normal and dauer larva development (Wolkow et al, 2000).
To test if this insulin-like pathway regulates proteolysis in C. elegans muscle, we constructed strains containing mutations affecting elements of this pathway, along with a transgene-coded reporter of proteolysis in muscle. The reporter is the protein product of the integrated transgene ccIs55(unc-54∷lacZ), a fusion of a small N-terminal fragment of the major myosin heavy-chain UNC-54 with β-galactosidase from Escherichia coli (Fire and Waterston, 1989). As a result of transgene expression from the unc-54 promoter and enhancer, the protein is continually synthesized until adulthood specifically in 95 body wall and eight vulval muscle cells. In well-fed wild-type animals, the reporter protein is completely stable for at least 72 h after the animals reach mid-adulthood, even if additional protein synthesis is blocked pharmacologically (Zdinak et al, 1997). This reporter and other soluble proteins in muscle cytosol are degraded, using pre-existing signaling proteins and proteolytic mechanisms, upon starvation (Zdinak et al, 1997; Fostel et al, 2003), disruption of cholinergic input to muscle (Szewczyk et al, 2000), or in response to Ras activation either by mutation (Szewczyk et al, 2002) or indirectly by FGFR (Szewczyk and Jacobson, 2003).
Animals homozygous for a temperature-sensitive reduction-of-function mutation in the IGF receptor daf-2(m41) (Gems et al, 1998) synthesize normal levels of reporter protein, but unlike wild-type animals degrade the reporter when shifted to 25°C after reaching adulthood (Figure 1A). This degradation is time-dependent and can be followed (Figure 1B and C) by immunoblot with monoclonal anti-β-galactosidase (Zdinak et al, 1997) or by fluorimetric assay of β-galactosidase activity (Zdinak et al, 1997); the two methods agree quantitatively (Figure 1C). As the temperature shift occurs after reporter synthesis has essentially ceased, the ensuing net degradation must be the result of increased protein degradation alone. This experimental design makes irrelevant any effects of DAF-2 on muscle protein synthesis. As muscle is fully differentiated before temperature shift, protein degradation results from acute loss of DAF-2 IGFR activity and not from any associated developmental irregularities. Addition of the protein synthesis inhibitor cycloheximide (CHx), which has been verified to shut down protein synthesis in developing C. elegans muscle (Zdinak et al, 1997), 6 h before temperature shift does not prevent the onset of degradation, indicating that degradation is triggered by a pre-existing signaling pathway and uses pre-existing proteases. Similar experiments (data not shown) with a second temperature-sensitive reduction-of-function allele, daf-2(e1370), confirm that protein degradation does not depend on any allele-specific peculiarity of daf-2(m41).
Figure 1.
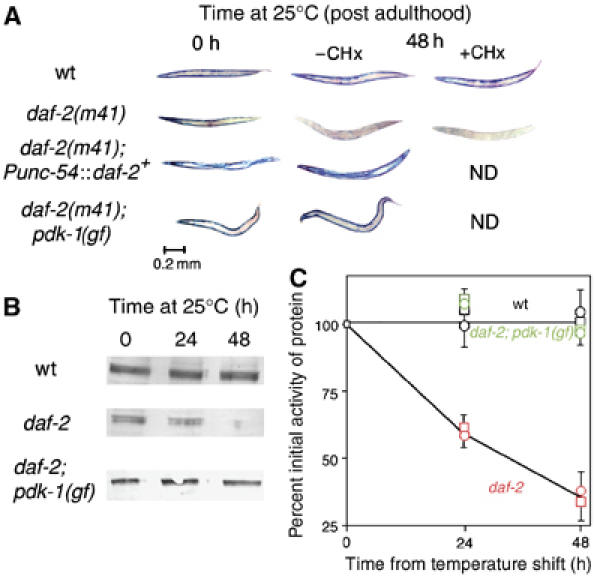
Intramuscular DAF-2 signaling opposes protein degradation in muscle. Animals were grown to mid-adulthood at 15°C and then shifted to 25°C for an additional 48 h either without or with the protein synthesis inhibitor CHx (400 μg/ml) added 6 h before temperature upshift. (A) Histochemical stain for β-galactosidase activity in muscle (blue). Reporter activity in wild-type animals (top row) remains stable after temperature shift even when treated with CHx. daf-2(m41) animals (second row) show a loss of reporter activity after temperature upshift; this is not prevented by CHx pretreatment. The loss of activity in daf-2(m41) animals is rescued by muscle-specific expression of wild-type DAF-2 (Punc-54∷daf-2+, third row) or by a gain-of-function mutation pdk-1(mg142) (bottom row). (B) Immunoblot (Zdinak et al, 1997) of 146 kDa myosin-β-galactosidase fusion protein (using monoclonal anti-β-galactosidase antibody) in 30-worm lysates confirms that the loss of staining seen in (A) corresponds to degradation of the reporter protein. (C) Quantitation (NIH Image software) of protein levels in (B) and of β-galactosidase activity by fluorimetric assay (Zdinak et al, 1997) of 10-worm lysates (means±s.d. of three independent determinations) confirms that protein degradation (squares) and loss of activity (circles) proceed at similar rates in daf-2 animals (red) whereas no degradation occurs in wild-type (black) or daf-2; pdk-1 (gf) animals (green).
The daf-2(m41) mutants shifted to 25°C also showed a progressive decline in movement rate (Figure 2A), an integrative measure of muscle function. This decline paralleled the time-dependent reporter degradation, consistent with the view that LacZ protein is an appropriate indicator of degradation of physiologically relevant muscle proteins. Quantitatively similar decline in movement was also observed in animals lacking the LacZ reporter protein, showing that its presence does not significantly alter the underlying physiology.
Figure 2.
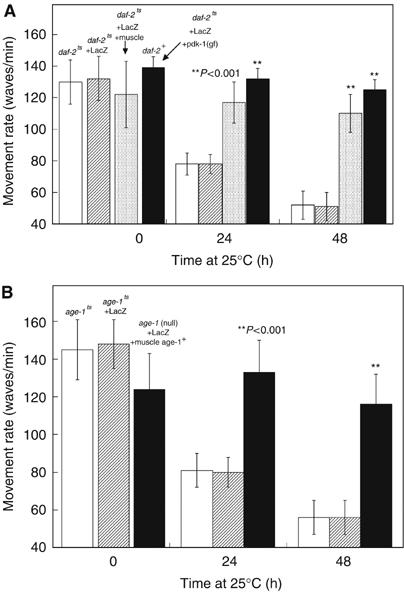
Intramuscular DAF-2 and AGE-1 signaling opposes loss of movement. Animals were grown to mid-adulthood at 15°C (0 h) and then an additional 24 or 48 h at 25°C. Measurements of animal movement rate (Szewczyk et al, 2002) were made for each strain (10 replicates on each of 10 animals of each genotype) at each time point. (A) daf-2(m41ts) mutant animals without (white) or with (striped) the reporter protein display a time-dependent loss of mobility at a rate comparable to reporter protein loss (Figure 1C). Like reporter protein loss, loss of mobility is rescued in animals expressing wild-type daf-2+ only in muscle (shaded) and prevented by gain-of-function mutation in pdk-1 (black). (B) age-1 animals without (white) or with (striped) the reporter protein display a time-dependent loss of movement comparable to daf-2 animals (A). Like reporter protein loss (Figure 3), mobility loss is rescued in animals expressing wild-type age-1+ only in muscle (black). Differences between rescued or suppressed animals compared with unrescued mutant animals were significant (**) at P<0.001 by two-tailed t-test.
To determine whether DAF-2 signals intramuscularly, we rescued DAF-2 activity in muscle cells of daf-2(m41) mutant animals by expression of wild-type daf-2+ from the muscle-specific unc-54 promoter (Wolkow et al, 2000) and found that daf-2+ expression only in muscle is sufficient to prevent protein degradation (Figure 1A). Furthermore, the progressive decline in the mobility of daf-2(m41) at 25°C was significantly rescued (Figure 2A), implying that the decline in mobility results from intramuscular events. These data are in sharp contrast to the observation (Wolkow et al, 2000) that the same Punc-54∷daf-2+ transgene did not rescue the excess intestinal lipid, long lifespan, or constitutive dauer-formation phenotypes of daf-2(e1370).
The effectors of DAF-2 in controlling development are AGE-1 (Morris et al, 1996), PDK-1 (Paradis et al, 1999), and AKT-1 (Paradis and Ruvkun, 1998), and there is evidence that PtdIns-3-kinase mediates the effects of insulin and IGF-1 on protein degradation in rat muscle (Dardevet et al, 1996). A direct demonstration of the role of AGE-1 in opposing protein degradation was made in three ways. First, animals with a temperature-sensitive reduction-of-function mutation in age-1(hx546ts) show a progressive loss of spontaneous movement at 25°C that parallels that in daf-2(m41) animals (Figure 2); this was also true in animals without the LacZ reporter. Second, blockage of AGE-1 activity with the PtdIns-3-kinase inhibitor LY-294002 (Babar et al, 1999) provokes reporter degradation in wild-type adults (Figure 3); this is not prevented by pretreatment with CHx. Third, animals with a temperature-sensitive reduction-of-function mutation in age-1(hx546ts) degrade reporter when shifted to 25°C after adulthood (Figure 3), but not when maintained at 15°C.
Figure 3.

Intramuscular AGE-1 (PtdIns-3-kinase) signaling opposes protein degradation. Animals were grown to mid-adulthood at 15°C (left) or for an additional 48 h at 25°C either without (middle) or with (right) the PtdIns-3-kinase inhibitor LY-294002 (LY, 160 μM). Wild-type animals (top) degrade the reporter protein when treated with LY; this is not prevented by CHx pretreatment. age-1(hx546ts) (second row) mutants degrade the reporter protein upon shift to 25°C even in the absence of LY. Reporter degradation in age-1(mg44) null-mutant animals is prevented by muscle-specific expression of wild-type AGE-1 from a transgene (Punc-54∷age-1+) (third row), but this rescue can be overcome by treatment with LY. A null mutation of the forkhead transcription factor gene daf-16(mgDf50) does not suppress LY-induced protein degradation (fourth row).
Expression of wild-type age-1+ from the muscle-specific unc-54 promoter (Wolkow et al, 2000) in an age-1 null-mutant background prevented loss of mobility (Figure 2B) and protein degradation (Figure 3), confirming inferences from the muscle-specific daf-2+ rescue. Muscle-specific rescue of AGE-1 activity was reversed by treatment with LY-294002, showing that it is the restoration of AGE-1 activity and not some other feature of the rescued animals that is responsible for reporter protein stability. A loss-of-function mutation of the lipid phosphatase DAF-18 (Ogg and Ruvkun, 1998), which normally removes the PtdIns-P3 product of AGE-1 action, is also sufficient to prevent protein degradation upon loss of AGE-1 activity (Figure 4), indicating that PtdIns-P3 mediates signaling.
Figure 4.
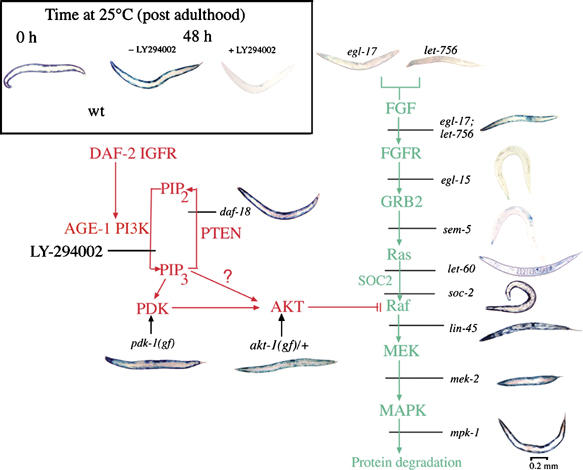
DAF-2 signaling (red pathway) opposes degradation-promoting signal from FGFs (green pathway). All animals were grown to mid-adulthood at 15°C, then treated for 48 h at 25°C with the PtdIns-3-kinase inhibitor LY-294002 (160 μM). This causes LacZ degradation in wild-type animals (Inset). Mutations that increase signal downstream of AGE-1 (pdk-1(mg142), daf-18(e1375), or akt-1(mg144), animals at left) block protein degradation triggered by LY treatment. Mutations that decrease signal at Raf (lin-45(sy96)), MEK (mek-2(ku114)), or MAPK (mpk-1(n2521)) also block protein degradation during LY treatment (lower right), but degradation is not prevented by reduction-of-function mutations affecting FGFR (egl-15(n1783)), GRB-2 (sem-5(n1779 or n1619)), or Ras (let-60(n2021)). Degradation is also blocked in a double mutant (egl-17(n1377); let-756(s2613)) deficient in both FGF homologs, but not in either single mutant. Quantitative data for rates of degradation in FGFR pathway mutants have been published (Szewczyk and Jacobson, 2003; Szewczyk et al, 2002).
PtdIns-P3 signal activates protein kinases PDK (Paradis et al, 1999) and Akt (Paradis and Ruvkun, 1998). A gain-of-function allele of pdk-1 suppresses not only protein degradation (Figure 1) and loss of mobility (Figure 2) in response to a loss of DAF-2 function, but also degradation in response to LY-294002 treatment (Figure 4). This confirms that PDK-1 lies downstream of both DAF-2 and AGE-1, suggesting that DAF-2 signals via AGE-1 and PDK-1 to inhibit protein degradation. Mutational activation of AKT-1 (Paradis and Ruvkun, 1998) also suppresses the protein degradation induced by loss of AGE-1 activity (Figure 4). Of note, a reduction-of-function mutation in pdk-1(sa680) does not provoke reporter degradation (data not shown), suggesting that although activated PDK-1 can replace DAF-2 signaling, PDK-1 itself is not essential to transduce DAF-2 signal. This may reflect the possibility that PtdIns-P3 can directly activate the downstream effector AKT-1 as it does in mammals (Stocker et al, 2002), or it may be that the mutation pdk-1(sa680) does not completely eliminate PDK-1 function (Paradis and Ruvkun, 1998; Paradis et al, 1999). A recent study in C. elegans (Gami et al, 2006) found that some age-1-dependent phenotypes require both pdk-1 and akt-1, whereas some require only the product of one or neither of these genes. Further experiments are required to define more precisely the role of pdk-1 in inhibition of muscle protein degradation in C. elegans.
Insulin opposes degradation via inhibition of FGFR signaling
Insulin-like signaling opposes protein degradation in mammalian muscle at least in part by blocking increased transcription of degradation-related genes (Glass, 2005). However, our CHx experiments indicate that de novo gene expression is not required to trigger degradation; so it is consistent that a loss-of-function (internal deletion) allele of the daf-16 transcription factor gene does not prevent the protein degradation induced by loss of DAF-2 or AGE-1 activity (Figure 3). Thus, in contrast to the situation in control of C. elegans longevity and dauer larva formation (Lin et al, 1997), for control of protein degradation DAF-16 is not the relevant target of AKT-1. Our results also suggest that transcriptional outputs of AKT-1 (Glass, 2005) may not be the only relevant mechanism by which insulin-like signaling opposes proteolysis in muscle.
We therefore investigated what might be the target of AKT-1 in C. elegans muscle. Activation of the Ras–Raf–MEK–MAPK pathway, by mutational activation of either EGL-15 FGFR, LET-60 Ras, or MPK-1 MAPK, promotes degradation of this β-galactosidase reporter and other nonmyofibrillar proteins in muscle (Szewczyk et al, 2002; Szewczyk and Jacobson, 2003). The downstream effectors of MAPK have not yet been identified, although they are proteasome-independent (Szewczyk et al, 2002) and thus distinct from those downstream of acetylcholine receptor, which regulates proteasome-mediated reporter degradation in C. elegans muscle (Szewczyk et al, 2000). Inhibitory phosphorylation of mammalian Raf by Akt has been reported (Zimmermann and Moelling, 1999), serine-to-alanine mutation of consensus Akt phosphorylation sites in LIN-45 Raf promotes ectopic vulval development in C. elegans (Chong et al, 2001) (indicative of Raf activation), and we had noted in daf-2(m41) animals a low-frequency multivulva phenotype, indicative of Ras pathway activation in hypodermis.
To test the possibility that DAF-2 negatively regulates protein degradation induced by EGL-15 FGFR or LET-60 Ras activation of Raf, we tested animals with reduction-of-function mutations in the FGFR–Ras–Raf–MEK–MAPK pathway for their ability to degrade protein in response to inhibition of AGE-1 (Figure 4). Reduction-of-function mutations in the Ras- and Raf-binding protein SOC-2 (Li et al, 2000), or in Raf, MEK, or MAPK were sufficient to block protein degradation in response to inhibition of AGE-1 signaling. Furthermore, inhibition of PI3K activity induces the appearance of the diphosphorylated (activated) form of MPK-1 MAPK (Figure 5), which is sufficient to provoke muscle proteolysis (Szewczyk et al, 2002). By contrast, reduction-of-function mutations in EGL-15, in the GRB2 homolog SEM-5 (Clark et al, 1992), or in LET-60 (Ras) did not block the protein degradation induced by LY-294002 (Figure 4). Given previous demonstrations that FGF signaling in C. elegans uses the EGL-15 FGFR and is transduced by LET-60 Ras, the failure of the egl-15(n1783) and let-60(n2021) mutations to prevent proteolysis may reflect residual function in the mutant products of these particular alleles; null alleles of both genes are lethal (Han and Sternberg, 1990; DeVore et al, 1995). We also found (data not shown) that treatment of let-60(n2021) animals with the farnesyltransferase inhibitor manumycin (Hara and Han, 1995), a combination that is sufficient to block induction of protein degradation by EGL-15 activation (Szewczyk and Jacobson, 2003), did not prevent LY-294002-induced degradation. Thus, we cannot exclude the possibility that there is a Ras-independent route for FGF signaling to Raf (Chang et al, 2000; Schutzman et al, 2001), which becomes obvious only when inhibitory signal from the DAF-2 pathway to LIN-45 Raf is strongly depressed.
Figure 5.

MPK-1 MAPK is diphosphorylated (activated) after inhibition of PI3K. Adult animals were treated with 160 μM LY-294002 for the indicated times at 25°C. Immunoblotting of pTpY-MPK-1 after electrophoresis on 12% SDS–polyacrylamide gels was conducted with monoclonal anti-pTpY-ERK antibody (Szewczyk and Jacobson, 2003). Each lane contained a 50-worm lysate. The untreated animals (0 h) show no immunoreactive pTpY-MPK-1.
Together, these results imply that DAF-2 negatively regulates protein degradation by activating AKT-1 (via AGE-1 and perhaps PDK-1) to phosphorylate Raf and inhibit Raf signaling (Figure 4). If this model is correct, then a reduction-of-function mutation affecting DAF-18, which increases signal downstream of AGE-1, should inhibit protein degradation and the appearance of the diphosphorylated (activated) form of MPK-1 in response to acute FGFR activation; this is in fact the case (Figure 6). Furthermore, a gain-of-function allele of pdk-1 does not block protein degradation in animals with mutationally activated MAPK (not shown), as expected if the IGFR–PI3K–Akt pathway acts to inhibit at the level of Raf.
Figure 6.
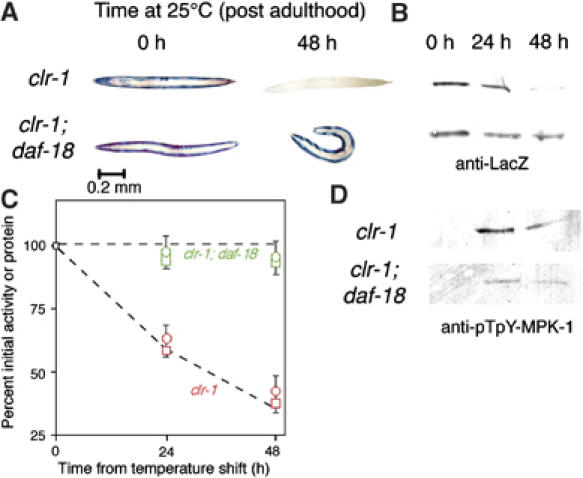
Increased PtdIns-P3 signal blocks protein degradation triggered by activated FGFR. Animals were grown to mid-adulthood at 15°C or for an additional 48 h at 25°C. (A) Histochemical stain for β-galactosidase activity in muscle (blue). Activation of FGFR at 25°C (clr-1(e1745ts) animals, top row) triggers the loss of reporter activity (Szewczyk and Jacobson, 2003). This can be prevented by a mutation (daf-18(e1375) animals, bottom) that increases the level of PtdIns-P3 (see pathway, Figure 4). (B) Immunoblot of 146 kDa β-galactosidase fusion protein in 30-worm lysates confirms that the loss of staining seen in (A) corresponds to degradation of the reporter protein. (C) Quantitation (NIH Image software) of protein levels in (B) and of β-galactosidase activity by fluorimetric assay (Zdinak et al, 1997) of 10-worm lysates (means±s.d. of three independent determinations) confirms that protein degradation (squares) and loss of activity (circles) proceed at similar rates in clr-1 animals (red) whereas little degradation is observed in clr-1; daf-18 animals (green). To facilitate comparison, the dashed lines are taken from Figure 1C and the same scale is used. (D) Immunoblot of pTpY-MPK-1 (30-worm lysate per lane) confirms that MPK-1 is activated in clr-1 animals, and that daf-18 mutation greatly reduces MPK-1 activation (37% as much pTpY-MPK-1 at 24 h and 43% as much at 48 h). Samples were run on the same gel and detected on the same membrane to permit direct comparison of pTpY-MPK-1 levels.
To provide confirming evidence that inhibitory interaction between the IGFR–PI3K–Akt pathway and the Ras–Raf–MEK–MAPK pathway involves LIN-45 Raf as a target of Akt, we eliminated putative Akt phosphorylation sites homologous to S364 and T439 in mammalian B-Raf (Chong et al, 2001), by means of a mutant construct (lin-45AA) in which LIN-45 residues S312 and S451 are replaced by alanines. This construct is driven by a heat-inducible promoter. Induction of the mutant LIN-45AA provokes muscle protein degradation and the appearance of diphosphorylated (activated) MPK-1 (Figure 7), suggesting that Raf activity is normally constrained by phosphorylation of these sites, and implying that all other necessary conditions for Raf activation (e.g., Ras activation) must be satisfied in these muscle cells.
Figure 7.
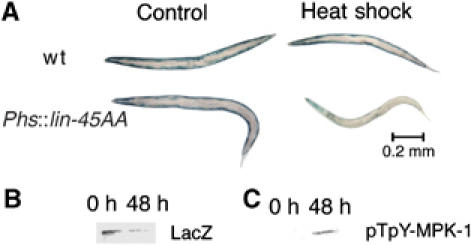
Induced expression of the lin-45AA transgene provokes protein degradation. The transgene, driven by a heat-shock promoter, is carried on an extrachromosomal array marked with a GFP transgene constitutively expressed in gut cells (see Materials and methods). Only animals that expressed the GFP marker were used. Young adult animals were heat-shocked for 40 min at 37°C where indicated, then returned to 20°C for 48 h. (A) Histochemical stain for β-galactosidase activity in muscle (blue). (B) Immunoblot of 146 kDa myosin-LacZ fusion protein in 30-worm lysates confirms that the loss of staining seen in (A) corresponds to degradation of the reporter protein, with 27% of reporter protein remaining at 48 h. (C) Immunoblot of pTpY-MPK-1 (30-worm lysate per lane) confirms that MPK-1 is activated in heat-shocked lin-45AA animals.
Past evidence shows that FGFR-induced protein degradation in C. elegans muscle does not require proteasome activity (Szewczyk et al, 2002; Szewczyk and Jacobson, 2003). It was therefore as expected that proteasome inhibitors also did not block the protein degradation induced by loss of function of daf-2 or age-1 (not shown). A link between insulin-like signaling and autophagy has recently been established in C. elegans (Melendez et al, 2003) and a reduction-of-function mutation that blocks autophagy (unc-51(e369)) does block protein degradation in response to inhibition of AGE-1 activity with LY-294002 or mutational activation of MPK-1 (J Hozella, N Szewczyk and L Jacobson, unpublished results). However, further experiments are required to distinguish whether IGFR and FGFR signaling are controlling autophagy-mediated proteolysis via UNC-51, or if UNC-51 acts in some other way to modulate FGFR signaling in C. elegans. A UNC-51-like kinase appears to regulate signaling downstream of FGFR1 in cultured mouse P19 cells (Avery et al, 2007).
Constitutive FGFR and IGFR signals are balanced in C. elegans muscle
The fact that acute reduction of AGE-1 or DAF-2 function triggers protein degradation carries the surprising implication that in healthy, wild-type, adult C. elegans, there is signal passing down the Ras pathway in body wall muscle cells. Previous studies (Szewczyk and Jacobson, 2003) imply that this signal derives from either of two FGF-like ligands, EGL-17 (Burdine et al, 1997) or LET-756 (Roubin et al, 1999), acting redundantly to activate the EGL-15 FGF receptor. LET-756 FGF is constitutively expressed from body wall muscle itself (Bulow et al, 2004). To confirm that constitutive FGF signaling is required for low insulin signaling to trigger proteolysis, we examined reduction-of-function mutations in egl-17 and let-756. Neither single mutation prevented LY-294002 from provoking protein degradation (Figure 4), but degradation was effectively suppressed in the egl-17;let-756 double mutant. Thus, the degradation-promoting signal in normal muscle emanates from these two FGF ligands, which have at least the potential to act redundantly, and there is no other major source of signal to Ras or Raf under such conditions. Constitutive, autocrine LET-756 signal is consistent with the constitutive Ras signal in muscle that is predicted by our AGE-1 and DAF-2 acute reduction-of-function experiments.
We infer that there is little degradation of reporter protein in the muscles of normal, healthy C. elegans because the constitutive degradation-promoting signal in the FGFR–Ras–RAF–MEK–MAPK pathway is inhibited by signal from the IGFR–PI3K–Akt pathway. The fact that reduction of signal from DAF-2 or AGE-1 is sufficient to trigger protein degradation indicates that there is normally such signal passing through DAF-2 and AGE-1. There are about 38 insulin-like ligands encoded in the C. elegans genome, and the identity and cellular source of the ligand(s) that controls DAF-2 signaling in muscle is presently unknown (Pierce et al, 2001). One interesting possibility is that the insulin-like signal is also autocrine in nature. ‘Mechano growth factor' is an IGF that is expressed in mammalian muscle, and which appears to have a role in muscle repair and adaptation (Goldspink, 2005). The key point is that the reporter protein is not degraded in the muscles of well-fed, healthy adults; so the degradation-promoting signal induced by the FGF ligands must be balanced or overbalanced by the opposing signal from insulin-like ligand(s). It is not easy to evaluate whether the DAF-2 IGFR signal in normal muscle is greatly in excess of the level required to keep in check the FGFR signal to the Raf–MEK–MAPK module, or whether the two pathways are more precariously equipoised. We do know that the decrement of DAF-2 signal required to permit protein degradation is comparable to that which shifts development toward dauer larva formation, because we have observed that daf-2(m41) homozygotes show some protein degradation and some dauer larva formation at 20°C, whereas daf-2(e1370) homozygotes show neither at 20°C and both at 25°C. Thus, as the decision between following normal development and branching to dauer larva formation is a physiologically relevant response to environmental conditions (Golden and Riddle, 1984), it follows that the activity of the DAF-2 pathway can vary within a physiologically significant range to modulate muscle protein degradation. The fact that the degradation-promoting effect of mutational hyperactivation of FGFR can be suppressed in the daf-18;clr-1 double mutant (Figure 6) shows that balance between these signaling pathways can also be established outside the normal physiological range. It is noteworthy that although most developmental phenotypes are induced, most physiological phenotypes are negatively regulated. This raises the possibility that balanced signaling pathways may be common in control of physiological phenotypes.
It is intriguing that distinct and apparently independent outputs from Akt, via DAF-16 to transcription and via LIN-45 Raf to proteolysis, produce biological responses in the same range of signal. One possibility is that protein degradation is important to the tissue remodeling involved in forming a dauer larva, which has a significantly different body shape from that of non-dauer larvae (Cassada and Russell, 1975). We emphasize that we have not investigated whether protein synthesis as well as protein degradation is controlled by this pathway, and have not intensively studied whether the connection between these signals and protein degradation is limited to muscle. It is also possible, of course, that these signaling pathways act predominantly as binary on–off switches rather than as continuously variable stimulus–response mediators, in which case no particular significance can be attached to the coincidence of output sensitivities.
Discussion
It has been reported (Bodine et al, 2001) that Akt in mouse muscle is upregulated during hypertrophy and downregulated during atrophy, and that mutational activation of Akt induces hypertrophy and prevents atrophy. Furthermore, mutational activation of Raf promotes an ‘atrophic' phenotype in cultured mouse myotubes (Rommel et al, 1999). These two observations have not been directly connected with respect to protein degradation, in part because of the difficulty of deconvoluting effects on protein synthesis and degradation in mammalian models, but our data open the question of whether these two highly conserved signaling pathways may be set in opposition in normal mammalian muscle as they are in C. elegans. This would be pertinent not only to pathological conditions ensuing from perturbation of the balance between the two pathways (e.g., low insulin signaling and consequent excess protein degradation in untreated diabetes; Rooyackers and Nair, 1997), but perhaps also to the normal physiological adaptations. We have suggested elsewhere (Szewczyk et al, 2002; Szewczyk and Jacobson, 2003) that FGFR signaling via Ras might be involved in muscle remodeling. The present demonstration that protein degradation is regulated by the balance between two signaling pathways suggests that modulation of both pathways and their interaction should be taken into account in investigations of signals regulating developmental or exercise-induced muscle remodeling and muscle repair (Tipton and Wolfe, 2001; Widegren et al, 2001). The presence of balanced signals also opens the possibility that abnormalities in one signaling pathway might be treated by therapeutic intervention in the balanced pathway. The likelihood that these same signals control protein degradation through both transcriptional and non-transcriptional mechanisms emphasizes that studying changes in transcription of genes involved in protein degradation (Lecker et al, 2004) may be too narrow a focus, and that a single signaling pathway may act via multiple mechanisms to achieve modulation of the activities of protein-degrading systems under adaptive or pathological conditions. Adjusting the activities of pre-existing proteolytic systems can immediately change the rate of protein degradation, whereas adjustment in synthesis of system components is more suitable for long-term adaptations. This dual mode control of protein degradation was recently demonstrated for cultured mammalian cells subject to serum starvation (Vabulas and Hartl, 2005).
Reduction-of-function mutations in the DAF-2–AGE-1–Akt pathway increase longevity (Vanfleteren and Braeckman, 1999) and in some respects (Duhon and Johnson, 1995), but not all (Herndon et al, 2002), slow physiological aging. Expression of daf-2+ or age-1+ in neurons, but not in muscle, reverses the long-lived phenotype (Wolkow et al, 2000). However, physiological functions that have well-behaved changes with chronological age (both in populations and in individuals) often correlate poorly with longevity (Bolanowski et al, 1981). There are age-related dysfunctional changes in the muscles of C. elegans (Herndon et al, 2002) that in some respects model age-related sarcopenia in mammals (Fisher, 2004), and it will be a matter of some interest to determine if the increased protein degradation in the muscles of daf-2 or age-1 mutant animals can be causally related to slower physiological aging in muscle.
Materials and methods
Nematode strains were maintained, grown and roughly age-synchronized as described (Zdinak et al, 1997), except that strains containing unsuppressed temperature-sensitive mutations were routinely kept at 16°C.
Genetic markers used in this study were: on LG I: daf-16(mgDf50), mek-2(ku114), unc-13 (e51); on LG II: age-1(mg44), age-1(hx546ts), clr-1(e1745ts), daf-2(m41ts), daf-2(e1370), sqt-1(sc13); on LG III: mpk-1(n2521), let-756(s2613), unc-32(e189); on LG IV: cha-1(p1182ts), daf-18(e1375), dpy-13(e184), gaIs37(PEF1α∷D-mek(gf) Phs∷mpk-1(wt)), soc-2(n1774), soc-2(ku167), lin-45(sy96), unc-24 (e138), him-8(e1489), let-60(n2021); on LG V: unc-51(e369), akt-1(mg144), ccIs55 (unc-54∷lacZ), dpy-11(e224); on LG X: egl-15 (n1783), egl-17(n1377), iwIs16(Pact-4∷lacZ), pdk-1(mg142), pdk-1(sa680), sem-5(n1619), sem-5(n1779), unc-1(e719).
Strains containing unlinked mutations along with the reporter transgene ccIs55(unc-54∷lacZ), as well as double mutant strains containing the transgene, were constructed by conventional genetic methods. For muscle-specific rescue of age-1 mutation, a strain containing mgEx499 (Punc-54∷age1+; Pmec-7∷GFP) in a genetic background containing both the null allele age-1(mg44) and ccIs55(unc-54∷lacZ) was constructed as follows: ccIs55 males were crossed into a strain (kindly provided by C Wolkow and G Ruvkun) carrying mgEx499 in a background of age-1(mg44) sqt-1(sc13) II and selecting from the F2 progeny those that were sqt-1 homozygotes, showed neuronal GFP fluorescence and stained for β-galactosidase activity (Zdinak et al, 1997), then recloning to ensure homozygosity for ccIs55. For muscle-specific rescue of a daf-2 mutation, a strain that expresses Punc-54∷daf-2+ and unc-54∷lacZ in muscle in a genetic background with daf-2(m41ts) was constructed as follows: males containing ccIs55(unc-54∷lacZ) were crossed at 15°C with hermaphrodites (kindly provided by C Wolkow and G Ruvkun) homozygous for daf-2(e1370) and containing njEx38 (Punc54∷daf2+; Pgoa-1∷GFP; rol-6(st1006)). These animals are temperature-sensitive dauer constitutives, as the extrachromosomal array njEx38 does not rescue daf-2(m41) for the dauer-forming phenotype. The plates were moved to 25°C and cross-progeny (Rol adults) cloned at 25°C, then Rol adult F2 stained for β-galactosidase, and recloned to obtain a line that never produced dauers at 25°C (thus daf-2+), was homozygous for ccIs55, and maintained GFP fluorescence in all or nearly all progeny. Weakly roller males of this line were then crossed at 15°C with hermaphrodites (ccIs55(unc-54∷lacZ) V; daf-2(m41) III), and F2 with strong fluorescence were cloned at 15°C, then shifted to 25°C to identify clones which produced 100% dauer F3. These were allowed to recover at 15°C and confirmed to be homozygous for ccIs55 and to have reliable transmission of both GFP in muscles and the roller phenotype. A strain containing the extrachromosomal array csEx52 (Phs∷lin-45AA+sur-5∷GFP) was obtained from M Sundaram and the array crossed into a genetic background containing ccIs55(unc-54∷lacZ) V. The resulting strain loses the array at significant frequency; so only those individuals carrying the GFP marker were used for experiments.
Methods for gel electrophoresis, immunoblotting with monoclonal anti-β-galactosidase, and fluorimetric assay of β-galactosidase activity were as described (Zdinak et al, 1997). Immunoblotting of MPK-1 after electrophoresis on 12% SDS–polyacrylamide gels used monoclonal anti-pTpY-ERK (Sigma M-8159 for Figure 5 and Cell Signaling no. 9101 for Figures 6 and 7) at 1:2000 for 18 h at 10°C, with detection by peroxidase-labeled donkey anti-mouse and TMB substrate (KPL Labs) (Szewczyk and Jacobson, 2003). Animals were stained for β-galactosidase activity with 5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-gal) as described (Zdinak et al, 1997) for 1–2 h at room temperature, governed by visual examination of stained control animals (wild type or mutants at permissive temperature) included with every experiment. Stained animals were photographed under bright-field illumination. The metered exposure is dominated by the bright-field background, depends little on the staining intensity of the animals being photographed, and is thus approximately constant from experiment to experiment. Images were postprocessed with Adobe Photoshop to construct composite images and to equalize illumination quality and color among images acquired over several years.
Movement rates were measured as described (Szewczyk et al, 2002).
Acknowledgments
We thank C Wolkow, G Ruvkun, and M Sundaram for the gift of C. elegans strains and K Curto for determining that Cell Signaling antibody no. 9101 detects C. elegans pTpY-MPK-1. Many strains used in this work were obtained from the Caenorhabditis Genetics Center, funded by the NIH National Center for Research Resources. SJB was supported by a Barry M Goldwater scholarship. This work was supported by NSF grants MCB-0090734 and MCB-0542355 and by a gift from Regeneron Pharmaceuticals Inc.
References
- Andreatta C, Nahreini P, Hovland B, Kumar J, Edwards-Prasad J, Prasad K (2001) Use of short-lived green fluorescent protein for the detection of proteasome inhibition. BioTechniques 30: 656–660 [DOI] [PubMed] [Google Scholar]
- Attaix D, Combaret L, Pouch MN, Taillandier D (2001) Regulation of proteolysis. Curr Opin Clin Nutr Metab Care 4: 45–49 [DOI] [PubMed] [Google Scholar]
- Avery AW, Figueroa C, Vojtek AB (2007) UNC-51-like kinase regulation of fibroblast growth factor receptor substrate 2/3. Cell Signal 19: 177–184 [DOI] [PubMed] [Google Scholar]
- Babar P, Adamson C, Walker GA, Walker DW, Lithgow GJ (1999) P13-kinase inhibition induces dauer formation, thermotolerance and longevity in C. elegans. Neurobiol Aging 20: 513–519 [DOI] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, Kopito RR (2001) Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292: 1552–1555 [DOI] [PubMed] [Google Scholar]
- Block BA, Franzini-Armstrong C (1988) The structure of the membrane systems in a novel muscle cell modified for heat production. J Cell Biol 107: 1099–1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3: 1014–1019 [DOI] [PubMed] [Google Scholar]
- Bolanowski MA, Russell RL, Jacobson LA (1981) Quantitative measures of aging in the nematode Caenorhabditis elegans. I. Population and longitudinal studies of two behavioral parameters. Mech Ageing Dev 15: 279–295 [DOI] [PubMed] [Google Scholar]
- Brignull HR, Morley JF, Garcia SM, Morimoto RI (2006) Modeling polyglutamine pathogenesis in C. elegans. Methods Enzymol 412: 256–282 [DOI] [PubMed] [Google Scholar]
- Bulow HE, Boulin T, Hobert O (2004) Differential functions of the C. elegans FGF receptor in axon outgrowth and maintenance of axon position. Neuron 42: 367–374 [DOI] [PubMed] [Google Scholar]
- Burdine RD, Chen EB, Kwok SF, Stern MJ (1997) egl-17 encodes an invertebrate fibroblast growth factor family member required specifically for sex myoblast migration in Caenorhabditis elegans. Proc Natl Acad Sci USA 94: 2433–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassada RC, Russell RL (1975) The dauerlarva, a post-embryonic developmental variant of the nematode Caenorhabditis elegans. Dev Biol 46: 326–342 [DOI] [PubMed] [Google Scholar]
- Chang C, Hopper NA, Sternberg PW (2000) Caenorhabditis elegans SOS-1 is necessary for multiple RAS-mediated developmental signals. EMBO J 19: 3283–3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong H, Lee J, Guan KL (2001) Positive and negative regulation of Raf kinase activity and function by phosphorylation. EMBO J 20: 3716–3727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SG, Stern MJ, Horvitz HR (1992) C. elegans cell-signalling gene sem-5 encodes a protein with SH2 and SH3 domains. Nature 356: 340–344 [DOI] [PubMed] [Google Scholar]
- Dantuma NP, Lindsten K, Glas R, Jellne M, Masucci MG (2000) Short-lived green fluorescent proteins for quantifying ubiquitin/proteasome-dependent proteolysis in living cells. Nat Biotechnol 18: 538–543 [DOI] [PubMed] [Google Scholar]
- Dardevet D, Sornet C, Vary T, Grizard J (1996) Phosphatidylinositol 3-kinase and p70 s6 kinase participate in the regulation of protein turnover in skeletal muscle by insulin and insulin-like growth factor I. Endocrinology 137: 4087–4094 [DOI] [PubMed] [Google Scholar]
- DeVore DL, Horvitz HR, Stern MJ (1995) An FGF receptor signaling pathway is required for the normal cell migrations of the sex myoblasts in C. elegans hermaphrodites. Cell 83: 611–620 [DOI] [PubMed] [Google Scholar]
- Duhon SA, Johnson TE (1995) Movement as an index of vitality: comparing wild type and the age-1 mutant of Caenorhabditis elegans. J Gerontol A Biol Sci Med Sci 50: B254–B261 [DOI] [PubMed] [Google Scholar]
- Fire A, Waterston RH (1989) Proper expression of myosin genes in transgenic nematodes. EMBO J 8: 3419–3428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher AL (2004) Of worms and women: sarcopenia and its role in disability and mortality. J Am Geriatr Soc 52: 1185–1190 [DOI] [PubMed] [Google Scholar]
- Fostel JL, Coste LB, Jacobson LA (2003) Degradation of transgene-coded and endogenous proteins in the muscles of Caenorhabditis elegans. Biochem Biophys Res Commun 312: 173–177 [DOI] [PubMed] [Google Scholar]
- Gami MS, Iser WB, Hanselman KB, Wolkow CA (2006) Activated AKT/PKB signaling in C. elegans uncouples temporally distinct outputs of DAF-2/insulin-like signaling. BMC Dev Biol 6: 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gems D, Sutton AJ, Sundermeyer ML, Albert PS, King KV, Edgley ML, Larsen PL, Riddle DL (1998) Two pleiotropic classes of daf-2 mutation affect larval arrest, adult behavior, reproduction and longevity in Caenorhabditis elegans. Genetics 150: 129–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass DJ (2005) Skeletal muscle hypertrophy and atrophy signaling pathways. Int J Biochem Cell Biol 37: 1974–1984 [DOI] [PubMed] [Google Scholar]
- Golden JW, Riddle DL (1984) The Caenorhabditis elegans dauer larva: developmental effects of pheromone, food, and temperature. Dev Biol 102: 368–378 [DOI] [PubMed] [Google Scholar]
- Goldspink G (2005) Mechanical signals, IGF-I gene splicing, and muscle adaptation. Physiology (Bethesda) 20: 232–238 [DOI] [PubMed] [Google Scholar]
- Han M, Sternberg PW (1990) Let-60, a gene that specifies cell fates during Caenorhabditis elegans vulval induction, encodes a ras protein. Cell 63: 921–931 [DOI] [PubMed] [Google Scholar]
- Hara M, Han M (1995) Ras farnesyltransferase inhibitors suppress the phenotype resulting from an activated ras mutation in Caenorhabditis elegans. Proc Natl Acad Sci USA 92: 3333–3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herndon LA, Schmeissner PJ, Dudaronek JM, Brown PA, Listner KM, Sakano Y, Paupard MC, Hall DH, Driscoll M (2002) Stochastic and genetic factors influence tissue-specific decline in ageing C. elegans. Nature 419: 808–814 [DOI] [PubMed] [Google Scholar]
- Lecker SH, Jagoe RT, Gilbert A, Gomes M, Baracos V, Bailey J, Price SR, Mitch WE, Goldberg AL (2004) Multiple types of skeletal muscle atrophy involve a common program of changes in gene expression. FASEB J 18: 39–51 [DOI] [PubMed] [Google Scholar]
- Li W, Han M, Guan KL (2000) The leucine-rich repeat protein SUR-8 enhances MAP kinase activation and forms a complex with ras and Raf [In Process Citation]. Genes Dev 14: 895–900 [PMC free article] [PubMed] [Google Scholar]
- Lin K, Dorman JB, Rodan A, Kenyon C (1997) daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science 278: 1319–1322 [DOI] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301: 1387–1391 [DOI] [PubMed] [Google Scholar]
- Morris JZ, Tissenbaum HA, Ruvkun G (1996) A phosphatidylinositol-3-OH kinase family member regulating longevity and diapause in Caenorhabditis elegans. Nature 382: 536–539 [DOI] [PubMed] [Google Scholar]
- Ogg S, Ruvkun G (1998) The C. elegans PTEN homolog, DAF-18, acts in the insulin receptor-like metabolic signaling pathway. Mol Cell 2: 887–893 [DOI] [PubMed] [Google Scholar]
- Paradis S, Ailion M, Toker A, Thomas JH, Ruvkun G (1999) A PDK1 homolog is necessary and sufficient to transduce AGE-1 PI3 kinase signals that regulate diapause in Caenorhabditis elegans. Genes Dev 13: 1438–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Ruvkun G (1998) Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev 12: 2488–2498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira C, Murphy K, Jeschke M, Herndon DN (2005) Post burn muscle wasting and the effects of treatments. Int J Biochem Cell Biol 37: 1948–1961 [DOI] [PubMed] [Google Scholar]
- Pierce SB, Costa M, Wisotzkey R, Devadhar S, Homburger SA, Buchman AR, Ferguson KC, Heller J, Platt DM, Pasquinelli AA, Liu LX, Doberstein SK, Ruvkun G (2001) Regulation of DAF-2 receptor signaling by human insulin and ins-1, a member of the unusually large and diverse C. elegans insulin gene family. Genes Dev 15: 672–686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommel C, Clarke BA, Zimmermann S, Nunez L, Rossman R, Reid K, Moelling K, Yancopoulos GD, Glass DJ (1999) Differentiation stage-specific inhibition of the Raf-MEK-ERK pathway by Akt. Science 286: 1738–1741 [DOI] [PubMed] [Google Scholar]
- Rooyackers OE, Nair KS (1997) Hormonal regulation of human muscle protein metabolism. Annu Rev Nutr 17: 457–485 [DOI] [PubMed] [Google Scholar]
- Roubin R, Naert K, Popovici C, Vatcher G, Coulier F, Thierry-Mieg J, Pontarotti P, Birnbaum D, Baillie D, Thierry-Mieg D (1999) let-756, a C. elegans fgf essential for worm development. Oncogene 18: 6741–6747 [DOI] [PubMed] [Google Scholar]
- Schutzman JL, Borland CZ, Newman JC, Robinson MK, Kokel M, Stern MJ (2001) The Caenorhabditis elegans EGL-15 signaling pathway implicates a DOS-like multisubstrate adaptor protein in fibroblast growth factor signal transduction. Mol Cell Biol 21: 8104–8116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, Muscat GE (2005) Skeletal muscle and nuclear hormone receptors: implications for cardiovascular and metabolic disease. Int J Biochem Cell Biol 37: 2047–2063 [DOI] [PubMed] [Google Scholar]
- Spangenburg EE, Booth FW (2003) Molecular regulation of individual skeletal muscle fibre types. Acta Physiol Scand 178: 413–424 [DOI] [PubMed] [Google Scholar]
- Stocker H, Andjelkovic M, Oldham S, Laffargue M, Wymann MP, Hemmings BA, Hafen E (2002) Living with lethal PIP3 levels: viability of flies lacking PTEN restored by a PH domain mutation in Akt/PKB. Science 295: 2088–2091 [DOI] [PubMed] [Google Scholar]
- Szewczyk NJ, Hartman JJ, Barmada SJ, Jacobson LA (2000) Genetic defects in acetylcholine signalling promote protein degradation in muscle cells of Caenorhabditis elegans. J Cell Sci 113: 2003–2010 [DOI] [PubMed] [Google Scholar]
- Szewczyk NJ, Jacobson LA (2003) Activated EGL-15 FGF receptor promotes protein degradation in muscles of Caenorhabditis elegans. EMBO J 22: 5058–5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szewczyk NJ, Jacobson LA (2005) Signal-transduction networks and the regulation of muscle protein degradation. Int J Biochem Cell Biol 37: 1997–2011 [DOI] [PubMed] [Google Scholar]
- Szewczyk NJ, Peterson BK, Jacobson LA (2002) Activation of Ras and the MAP kinase pathway promotes protein degradation in muscle cells of Caenorhabditis elegans. Mol Cell Biol 22: 4181–4188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MG, Palmer RM (1998) Signalling pathways regulating protein turnover in skeletal muscle. Cell Signal 1998 10: 1–11 [DOI] [PubMed] [Google Scholar]
- Tipton KD, Wolfe RR (2001) Exercise, protein metabolism, and muscle growth. Int J Sport Nutr Exerc Metab 11: 109–132 [DOI] [PubMed] [Google Scholar]
- Vabulas RM, Hartl FU (2005) Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 310: 1960–1963 [DOI] [PubMed] [Google Scholar]
- Vanfleteren JR, Braeckman BP (1999) Mechanisms of life span determination in Caenorhabditis elegans. Neurobiol Aging 20: 487–502 [DOI] [PubMed] [Google Scholar]
- Widegren U, Ryder JW, Zierath JR (2001) Mitogen-activated protein kinase signal transduction in skeletal muscle: effects of exercise and muscle contraction. Acta Physiol Scand 172: 227–238 [DOI] [PubMed] [Google Scholar]
- Wolkow CA, Kimura KD, Lee MS, Ruvkun G (2000) Regulation of C. elegans life-span by insulinlike signaling in the nervous system [In Process Citation]. Science 290: 147–150 [DOI] [PubMed] [Google Scholar]
- Zdinak LA, Greenberg IB, Szewczyk NJ, Barmada SJ, Cardamone Rayner M, Hartman JJ, Jacobson LA (1997) Transgene-coded chimeric proteins as reporters of intracellular proteolysis: starvation-induced catabolism of a lacZ fusion protein in muscle cells of Caenorhabditis elegans. J Cell Biochem 67: 143–153 [PubMed] [Google Scholar]
- Zimmermann S, Moelling K (1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286: 1741–1744 [DOI] [PubMed] [Google Scholar]