

Abstract
Transforming growth factor-β (TGF-β) family members are multifunctional growth factors involved in regulating diverse biological processes. Despite the critical role for TGF-β in regulating cell proliferation, differentiation, migration and development, its role in regulating NF-κB-dependent inflammatory response still remains unclear. Here, we show that TGF-β1 induces acetylation of NF-κB p65 subunit to synergistically enhance bacterium nontypeable Haemophilus influenzae-induced NF-κB activation and inflammatory response in vitro and in vivo. The TGF-β1-induced acetylation of p65 is mediated via a Smad3/4-PKA-p300-dependent signaling pathway. Acetylation of p65 at lysine 221 by TGF-β1 is critical for synergistic enhancement of bacteria-induced DNA-binding activity, NF-κB activation, NF-κB-dependent transcription of TNF-α and IL-1β and interstitial polymorphonuclear neutrophil infiltration in vitro and in vivo. These studies provide new insights into the novel regulation of NF-κB by TGF-β signaling.
Keywords: NF-κB, p65 acetylation, PKA, Smad, TGF-β1
Introduction
Transforming growth factor-β (TGF-β) family members are multifunctional growth factors involved in regulating diverse biological processes. Despite the critical role for TGF-β in regulating cell proliferation, differentiation, migration and development, its role in regulating NF-κB-dependent inflammation still remains elusive (Wahl, 1994; ten Dijke and Hill, 2004; Feng and Derynck, 2005; Massague et al, 2005). Targeted disruption of the mouse TGF-β1 gene results in excessive inflammatory responses (Shull et al, 1992). Systemic TGF-β administration suppresses inflammation (Wahl, 1994). Consistent with these in vivo findings, TGF-β1 inhibits LPS-induced NF-κB activation in both intestinal epithelial cells and microglial cells (Haller et al, 2003; Le et al, 2004). These data thus suggest that TGF-β acts as a suppressor for NF-κB-dependent inflammatory response. However, there is now growing evidence that TGF-β induces and promotes inflammatory response via activation of NF-κB. For instance, TGF-β1 overexpression in keratinocytes in transgenic mice results in inflammatory skin lesions (Li et al, 2004); local TGF-β administration promotes inflammation (Wahl, 1994). In addition, in vitro studies also indicate that TGF-β–Smad signaling mediates activation of NF-κB in human airway epithelial cells (Jono et al, 2002; Mikami et al, 2006). The molecular mechanisms underlying TGF-β-mediated NF-κB-dependent inflammatory responses remain totally unknown.
TGF-β initiates signaling through the ligand-dependent activation of a heteromeric complex of type II and type I receptors (Feng and Derynck, 2005; Massague et al, 2005). The type II receptor kinase then phosphorylates the type I receptor in a conserved glycine-serine domain, resulting in activation of the type I receptor. The activated type I receptor subsequently recognizes and phosphorylates the Smad subgroup known as receptor-activated Smads (R-Smad), including Smad 2 and 3. This causes dissociation of R-Smad from the receptor, stimulates the assembly of a heteromeric complex between the phosphorylated R-Smad and the Co-Smad, Smad4, and then induces the translocation of the Smad complex to the nucleus, where the Smad complex regulates the expression of target genes. In addition to its direct interaction with Smad DNA-binding element, growing evidence suggests that Smads also regulate gene transcription by direct interaction and functional cooperation with other transcription factors, such as NF-κB.
NF-κB is known to be activated via phosphorylation and degradation of IκB by IκB kinases (IKKs), which in turn leads to the nuclear translocation of NF-κB and the subsequent transcription of NF-κB-dependent genes such as TNF-α (Bonizzi and Karin, 2004; Hayden and Ghosh, 2004). Recently, interesting studies have suggested that degradation of IκBα and nuclear translocation of NF-κB are not sufficient to promote a maximal NF-κB transcriptional activity. Rather, the NF-κB complex must undergo additional post-translational modification involving site-specific phosphorylation and acetylation (Chen et al, 2001, 2002, 2005). The p65 subunit of NF-κB is a principal target of phosphorylation by various kinases. These kinases function both in the cytoplasm and in the nucleus and are differentially induced by various stimuli including LPS and TNF-α. Both the Rel-homology domain and the carboxy-terminal transactivation domain of p65 contain key phosphoacceptor sites that are specifically targeted by various kinases. The most physiologically inducible phosphorylation sites reported for p65 occurs at serine 536, serine 276 and serine 529. Serine 536 is phosphorylated by IKK complex (Sakurai et al, 1999), whereas Ser 529 was phosphorylated by casein kinase II (Wang et al, 2000). In addition, phosphorylation of Ser 276 is mediated by catalytic protein kinase A (PKA) subunit or mitogen- and stress-activated protein kinase 1 (Zhong et al, 1998; Vermeulen et al, 2003). Additional p65 phosphorylation events have been described. Their functional significance, however, remains unclear.
Acetylation, like phosphorylation, is also important for regulation of the nuclear function of NF-κB (Chen et al, 2002, 2005; Kiernan et al, 2003; Hoberg et al, 2006). Endogenous p65 is acetylated in a stimulus-coupled manner after activation of cells with TNF-α or other stimuli at multiple sites. The p300 and CBP appear to play a major role in acetylation of p65. The p300/CBP possesses a histone acetyltransferase (HAT) enzymatic activity that regulates gene expression in part through acetylation of the N-terminal tails of histones (Ogryzko et al, 1996; Chen et al, 2002). In addition to modifying histones, p300 and CBP also directly acetylates p65. Acetylation of NF-κB leads to changes in its biological activity, such as alterations in DNA-binding activity and transcriptional activity (Ogryzko et al, 1996; Kouzarides, 2000; Chen et al, 2002; Gu et al, 2004). Three main acetylation sites have been identified within p65—lysines 218, 221 and 310. Site-specific acetylation of p65 regulates discrete biological action of the NF-κB complex. For example, acetylation of lysine 221 increases the DNA-binding affinity of p65 for κB site, whereas acetylation of lysine 221 alone or in combination with lysine 218 impairs the assembly of p65 with IκBα. Acetylation of lysine 310 of p65 is required for full transactivation by NF-κB complex. The relationship between p65 phosphorylation and acetylation has been also explored recently. There is evidence indicating that the acetylation of p65 is importantly regulated by prior phosphorylation of serine 276 and 536 (Chen et al, 2005; Hoberg et al, 2006). Such phosphorylated and acetylated forms of p65 display enhanced transcriptional activity. Given that TGF-β plays a critical role in regulating inflammatory response via NF-κB in infectious diseases, it is still unclear whether TGF-β regulates NF-κB-dependent inflammatory response via either phosphorylation or acetylation of NF-κB.
The Gram-negative bacterium nontypeable Haemophilus influenzae (NTHi) is an important human pathogen in both adults and children (Foxwell et al, 1998; Murphy, 2000). In adults, it exacerbates chronic obstructive pulmonary diseases (COPD), the fourth leading cause of death in the United States (Murphy, 2006), whereas in children, it causes otitis media, the most common childhood infection and the leading cause of conductive hearing loss (Murphy, 2000). Like most other bacterial infections, NTHi infections are characterized by inflammation. We have previously shown that NTHi activates NF-κB via a Toll-like receptor 2-dependent NIK–IKKα/β–IκBα signaling pathway (Shuto et al, 2001). Based on the essential involvement of NF-κB in NTHi-induced inflammatory responses and TGF-β, an important regulator of inflammation, is highly upregulated in airways of COPD patients (de Boer et al, 1998), we hypothesized that TGF-β signaling may play a critical role in regulating NTHi-induced inflammation via modulation of NF-κB activity. Here, we show that TGF-β1 induces acetylation of NF-κB p65 subunit at lysine 221 to synergistically enhance NTHi-induced NF-κB activation and inflammatory response via a Smad3/4-PKA-dependent mechanism in vitro and in vivo. These studies may provide novel insights into the regulation of NF-κB by TGF-β signaling.
Results
TGF-β1 synergizes with bacterium NTHi to induce NF-κB activation and inflammatory response in vitro and in vivo
To determine whether TGF-β1 regulates NTHi-induced NF-κB activation and inflammatory response, we first measured NF-κB-dependent promoter activity by using luciferase reporter plasmid in a variety of human epithelial cells. As shown in Figure 1A, TGF-β1 enhanced NTHi-induced NF-κB activation in a synergistic manner in human epithelial HeLa cells, airway epithelial A549 cells and middle ear epithelial HMEEC-1 cells as well as human primary bronchial epithelial NHBE cells. Because of the important role for NF-κB in regulating a variety of key inflammatory mediators, we next determined whether TGF-β1 synergistically enhances NTHi-induced transcription of TNF-α and IL-1β by performing real-time quantitative PCR (Q-PCR) analysis. As shown in Figure 1B, TGF-β1 synergistically enhanced NTHi-induced expression of TNF-α and IL-1β in HeLa cells. Similar result was also observed in A549 and primary NHBE cells. We further confirmed whether TGF-β1 also enhances NTHi-induced expression of TNF-α and IL-1β in vivo. As shown in Figure 1C, TGF-β1 synergistically enhanced induction of TNF-α and IL-1β by NTHi in the lungs of mice. Consistent with this result, TGF-β1 also synergistically enhanced polymorphonuclear neutrophil (PMN) accumulation in broncho-alveolar lavage (BAL) fluids from the lungs of the NTHi-inoculated mice (Figure 1D and Supplementary Figure S1). Collectively, these data demonstrate that TGF-β1 synergistically enhances bacterium NTHi-induced NF-κB-dependent inflammatory response in vitro and in vivo.
Figure 1.
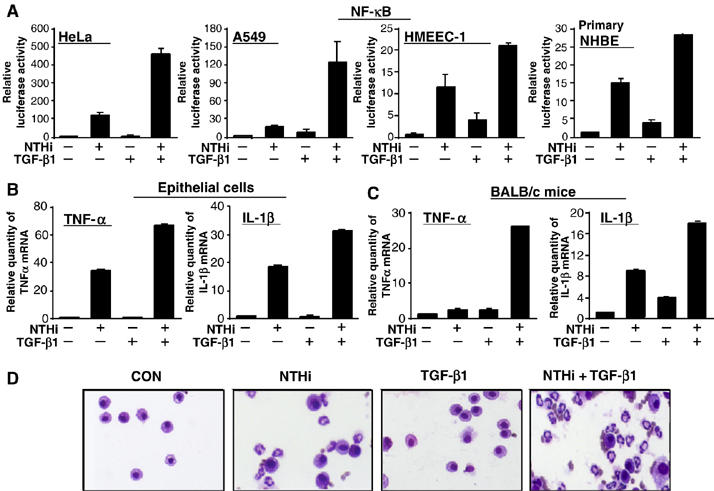
TGF-β1 synergizes with bacterium NTHi to induce NF-κB activation and inflammatory response in vitro and in vivo. (A) TGF-β1 synergistically enhanced NTHi-induced NF-κB-dependent promoter activity in human HeLa, airway A549, middle ear HMEEC-1 and primary bronchial epithelial NHBE cells, as assessed by NF-κB-dependent promoter assays. (B) TGF-β1 synergistically enhanced NTHi-induced expression of TNF-α and IL-1β in HeLa cells, as assessed by real-time Q-PCR analysis. Similar results were also observed in A549 and primary NHBE cells. (C) TGF-β1 synergistically enhanced NTHi-induced expression of TNF−α and IL-1β in the lung of BALB/c mice in vivo. Values are means±s.d. (n=5). (D) TGF-β1 synergistically enhanced NTHi-induced PMN infiltration in the lung of BALB/c mice in vivo. Data are representative of three independent experiments.
TGF-β1 synergistically enhances NTHi-induced NF-κB activation via a mechanism dependent on increased DNA-binding activity, but independent of p65 nuclear translocation
Because phosphorylation and degradation of IκBα and nuclear translocation of NF-κB are critical for NF-κB activation, we next sought to determine whether TGF-β1 synergistically enhances NTHi-induced NF-κB activation by increasing phosphorylation and degradation of IκBα. As shown in Figure 2A, TGF-β1 did not enhance NTHi-induced IκBα phosphorylation and degradation. We then determined whether TGF-β1 enhances NTHi-induced NF-κB activation by causing delay in the cytoplasmic reappearance of IκBα. No further delay in the cytoplasmic reappearance of IκBα was observed with TGF-β1 treatment (Figure 2B). Therefore, it is clear that TGF-β1-induced enhancement of NF-κB activation by NTHi does not occur at the level of IκBα. We next determined whether TGF-β1 enhances NTHi-induced NF-κB activation by inducing its nuclear translocation of p65 by performing Western blot analysis using nuclear extract. As shown in Figure 2C, p65 was markedly translocated to the nucleus when the cells were stimulated with NTHi alone. Simultaneous treatment with TGF-β1 and NTHi resulted in no synergistic enhancement of p65 translocation. Likewise, TGF-β1 also did not prolong nuclear presence of p65 (Figure 2D). These results thus suggest that TGF-β1-induced synergistic enhancement of NF-κB activation did not occur at the level of p65 nuclear translocation. Because the DNA-binding activity of the NF-κB complex is critical for NF-κB to exert its transcriptional activity, we investigated the effect of TGF-β1 on NTHi-induced DNA-binding activity of NF-κB by performing electrophoretic mobility shift assay (EMSA). As shown in Figure 2E, TGF-β1 treatment resulted in synergistic enhancement of DNA binding of NF-κB in cells treated with NTHi. Further analysis using supershift assay reveals that p65 and p50 appear to be the major subunits of the NF-κB band that was synergistically enhanced by TGF-β1 treatment (Figure 2F). To further confirm whether TGF-β enhances p65 DNA binding to an NF-κB-dependent gene promoter such as TNF-α promoter in the context of chromatin, chromatin immunoprecipitation (ChIP) assays were performed. As shown in Figure 2G, TGF-β indeed synergistically enhanced NTHi-induced DNA binding of p65 to the TNF-α promoter. Together, these data suggest that TGF-β1 synergistically enhances NTHi-induced NF-κB activation via a mechanism dependent on increased DNA-binding activity, but independent of p65 nuclear translocation.
Figure 2.
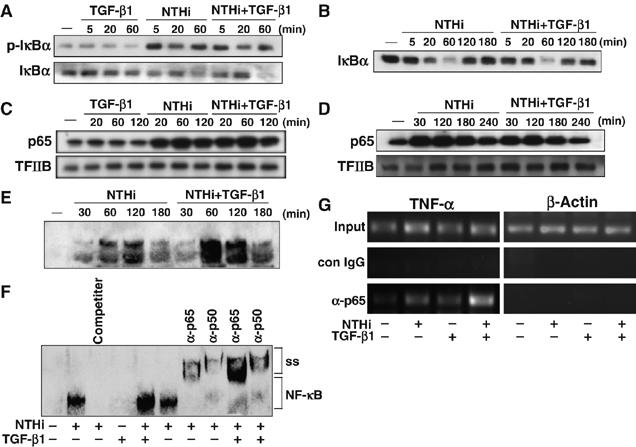
TGF-β1 synergistically enhances NTHi-induced NF-κB activation via a mechanism dependent on increased DNA-binding activity, but independent of p65 nuclear translocation. (A) TGF-β1 did not enhance NTHi-induced phosphorylation and degradation of IκBα in HeLa cells, as assessed by Western blot analysis. (B) TGF-β did not affect cytoplasmic reappearance of IκBα after NTHi treatment in HeLa cells. (C) TGF-β did not enhance NTHi-induced p65 nuclear translocation in HeLa cells. (D) TGF-β did not alter prolonged intranuclear retention of p65 after translocation to the nucleus in HeLa cells. (E) TGF-β1 enhanced NTHi-induced DNA-binding activity of NF-κB, as assessed by performing EMSA in HeLa cells. (F) p65 and p50 appear to be the major subunits of the NF-κB band that was synergistically enhanced by TGF-β1 60 min after treatment, as assessed by supershift assays. For competition experiments, nonlabeled probes (200 times) were used. IgG was used as a control. (G) TGF-β synergistically enhanced NTHi-induced DNA binding of p65 to the TNF-α promoter as assessed by performing ChIP assays. Isotype-matched control IgG was used as a control. Data are representative of three or more independent experiments.
TGF-β1 synergistically enhances NTHi-induced NF-κB activation via induction of p65 acetylation at lysine 221
Having identified the synergistic enhancement of TGF-β1 on NTHi-induced DNA-binding activity of NF-κB, still unknown is how TGF-β1 enhances NTHi-induced NF-κB activation by increasing DNA-binding activity. Post-translational modifications, particularly phosphorylation and acetylation, have been shown to play a critical role in NF-κB activation by enhancing the DNA-binding activity of p65 to the κB site (Chen et al, 2005). Thus, we hypothesized that these modification of p65 may be involved in mediating the enhancement of NTHi-induced DNA-binding activity of NF-κB by TGF-β1. We tested our hypothesis by assessing the effect of TGF-β1 on NTHi-induced p65 phosphorylation and p65 acetylation. As shown in Figure 3A, TGF-β1 enhanced NTHi-induced p65 acetylation but not phosphorylation. It should be noted that TGF-β, like NTHi, also induced phosphorylation of p65 at Ser276 (middle panel). Because trichostatin A (TSA), an inhibitor of histone deacetylase (HDAC), broadly inhibits the action of the HDACs function and results in hyperacetylation of the core histones and nonhistone proteins (Adam et al, 2003; Kiernan et al, 2003), we next evaluated the effect of TSA on p65 acetylation induced by NTHi and TGF-β1. As expected, p65 acetylation was enhanced by TSA treatment (Figure 3B, left panel). Consistent with this result, TSA treatment further enhanced the synergistic activation of NF-κB induced by NTHi and TGF-β (Figure 3B, right panel).
Figure 3.
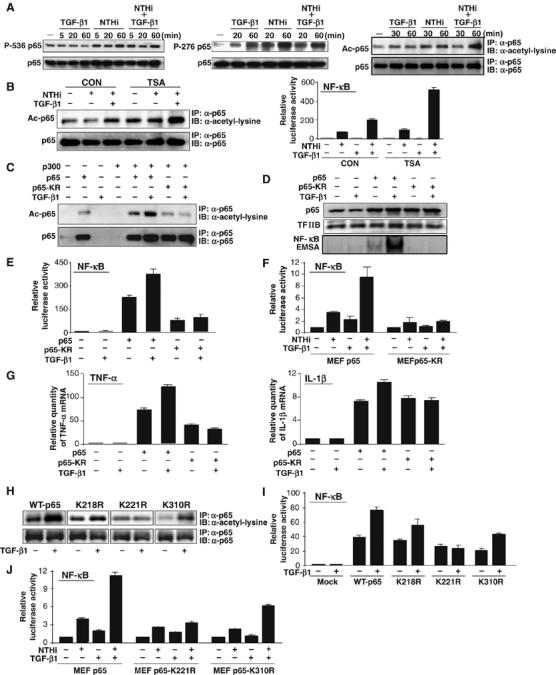
TGF-β1 synergistically enhances NTHi-induced NF-κB activation via induction of p65 acetylation at lysine 221. (A) Synergistic enhancement of p65 acetylation was observed in HeLa cells treated with both NTHi and TGF-β1 (1 ng/ml; right panel), whereas TGF-β1 induced p65 phosphorylation at Ser276 but not Ser536 (left and middle panels). Acetylation of p65 was detected by immunoblotting (IB) of the anti-p65 (α-p65) immunoprecipitates (IP) with anti-acetylated lysine antibodies. Levels of p65 present in each of the lysates are shown in the lower panel. (B) TSA enhanced NTHi-induced p65 acetylation and NF-κB activation. (C) TGF-β1 enhanced p65 acetylation induced by coexpressing WT p300 with WT p65, but not with p65-KR mutant (lysine 218/lysine 221/lysine 310 acetylation site mutant) in HeLa cells. (D) TGF-β1 enhanced DNA-binding activity of NF-κB in HeLa cells transfected with WT p65 but not with p65-KR mutant. (E) TGF-β1 enhanced NF-κB activation in HeLa cells transfected with WT p65 but not with p65-KR mutant. (F) TGF-β1 enhanced NTHi-induced NF-κB activation in p65−/− MEFs reconstituted with WT p65 but not in p65-KR-reconstituted p65−/− MEFs. (G) TGF-β1 enhanced TNF-α and IL-1β expression in HeLa cells transfected with WT p65 but not with p65-KR. (H) Mutation of lysine 221, but not 218 or 310, markedly decreased p65 acetylation in response to TGF-β compared to WT p65. (I) TGF-β1 enhanced NF-κB-dependent transcriptional activity in cells transfected with p65-K218R and p65-K310R, but not with p65-K221R mutant compared to cells transfected with WT p65. (J) Synergistic NF-κB activation was observed in p65-K310R-reconstituted MEFs but not in p65-K221R-reconstituted MEFs in response to NTHi and TGF-β1 compared to p65−/− MEFs reconstituted with WT p65. Values are means±s.d. (n=3). Data are representative of three or more independent experiments.
Owing to the important role of lysines 218, 221 and 310 as major acetylation sites in p65 (Chen et al, 2002), we next examined whether mutation of all three of these lysine residues (p65KR (p65K218/221/310R)) alters p65 acetylation, DNA binding and NF-κB activation by TGF-β1 and NTHi. As shown in Figure 3C, coexpressing wild-type (WT) p65, but not p65KR mutant, with WT p300 together with TGF-β1 treatment markedly induced p65 acetylation. Consistent with this result, TGF-β1 also enhanced NF-κB DNA-binding activity and NF-κB-dependent transcriptional activity in cells transfected with WT p65 but not with p65-KR mutant (Figure 3D and E). To further confirm the functional involvement of these three acetylation sites in synergistic activation of NF-κB by NTHi and TGF-β1, we assessed the synergistic induction of NF-κB in p65−/− MEFs that were reconstituted with either WT p65 expression plasmid or p65-KR mutant. As shown in Figure 3F, synergistic NF-κB activation was observed in WT p65-reconstituted MEFs but not in p65-KR-reconstituted MEFs in response to NTHi and TGF-β1. To confirm whether these three acetylation sites are also involved in synergistic induction of TNF-α and IL-1β, we evaluated induction of TNF-α and IL-1β in cells transfected with either WT p65 or p65-KR mutant upon TGF-β1 treatment. As shown in Figure 3G, TGF-β1 synergistically enhanced induction of TNF-α and IL-1β in cells transfected with WT p65 but not with p65-KR mutant. Taken together, our results suggest that these three acetylation sites are critical for mediating synergistic enhancement of p65 acetylation by TGF-β1, which, in turn, leads to enhancement of DNA-binding activity and NF-κB-dependent transcription of proinflammatory cytokines TNF-α and IL-1β.
We next determined which individual lysine residue is acetylated in response to TGF-β. As shown in Figure 3H, mutation of lysine 221, but not 218 or 310, markedly decreased p65 acetylation in response to TGF-β compared to WT p65. Consistent with this result, TGF-β1 synergistically enhanced NF-κB activation in cells transfected with WT p65, p65-K218R and p65-K310R, but not with p65-K221R mutant (Figure 3I). To further confirm the functional involvement of lysine 221 in synergistic activation of NF-κB by NTHi and TGF-β1, we assessed the synergistic induction of NF-κB in p65−/− MEFs that were reconstituted with either WT p65 expression plasmid or p65-K221R mutant or p65-K310R mutant. As shown in Figure 3J, synergistic NF-κB activation was observed in p65-K310R-reconstituted MEFs but not in p65-K221R-reconstituted MEFs compared to p65−/− MEFs reconstituted with WT p65. Thus, it is evident that lysine 221 residue is critical for mediating synergistic enhancement of p65 acetylation by TGF-β1 and the subsequent NF-κB DNA-binding and transcriptional activity.
p300 is involved in enhancement of p65 acetylation by TGF-β1
Based on recent studies that p300 acetyltransferase plays a major role in acetylation of p65 (Chen et al, 2002, 2005; Kiernan et al, 2003; Hoberg et al, 2006), we next determined whether p300 is involved in synergistic enhancement of p65 acetylation by TGF-β1. As shown in Figure 4A, TGF-β1 enhanced p65 acetylation in epithelial cells cotransfected with WT p65 and WT p300, but not in cells cotransfected with WT p65 and p300 HAT mutant. This result indicates that synergistic enhancement of p65 acetylation by TGF-β1 may involve the HAT activity of p300. To confirm whether p300 HAT activity is also involved in synergistic NF-κB activation by NTHi and TGF-β1, we evaluated the effect of overexpressing of WT p300 and p300 HAT mutant on NTHi- and TGF-β1-induced NF-κB activation. As shown in Figure 4B, overexpression of WT p300 enhanced NTHi- and TGF-β1-induced NF-κB activation, whereas overexpressing p300 HAT mutant inhibited synergistic activation of NF-κB by NTHi and TGF-β1. Thus, these data suggest that p300 is involved in synergistic NF-κB activation by NTHi and TGF-β1 via a mechanism dependent on p65 acetylation.
Figure 4.
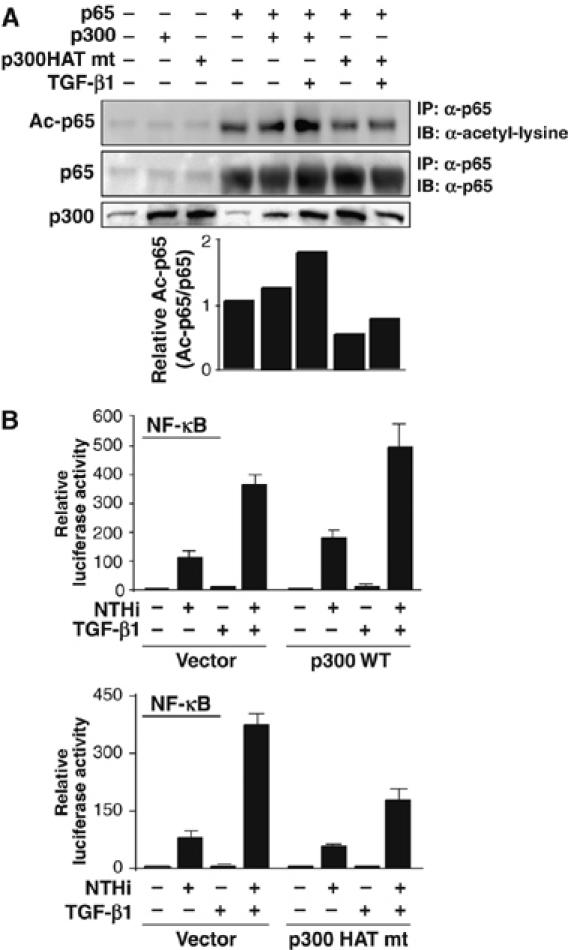
p300 is involved in enhancement of p65 acetylation by TGF-β1. (A) TGF-β1 enhanced p65 acetylation in HeLa cells co-transfected with WT p65 and WT p300 but not p300 HAT mutant. Data are representative of three independent experiments. (B) WT p300 enhanced NTHi-induced NF-κB activation, whereas p300 HAT mutant inhibited synergistic NF-κB activation by NTHi and TGF-β1 in HeLa cells. Values are means±s.d. (n=3).
Smad3 and Smad4 are required for enhanced NF-κB activation by TGF-β1 via a mechanism independent of direct interaction with NF-κB
We next sought to determine the involvement of Smad3 and Smad4 in TGF-β1-induced synergistic enhancement of NF-κB activation. As shown in Figure 5A, overexpression of a dominant-negative mutant of either Smad3 or Smad4 and Smad3 or Smad4 knockdown using siRNA inhibited synergistic activation of NF-κB by NTHi and TGF-β1. It should be noted that Smad4 DN did not inhibit TNF-α-induced NF-κB activation, suggesting that the inhibitory effect of Smad4 DN is specific for NTHi-induced NF-κB activation (Supplementary Figure S2). Consistent with these results, no synergistic enhancement of NF-κB activation was observed in Smad3 null cells and Smad4-deficient MDA-MB-468 cells (Figure 5C and D), whereas cotransfecting Smad3 null cells with WT Smad3 and cotransfecting MDA-MB-468 cells with WT Smad4 expression plasmid rescued the responsiveness to TGF-β1. We further investigated whether Smad3 and Smad4 are also required for the synergistic induction of p65 acetylation by TGF-β and NTHi. As shown in Figure 5E, Smad3 or Smad4 knockdown inhibited p65 acetylation induced by TGF-β and NTHi. We next determined whether there is a direct interaction between NF-κB components and Smads by performing supershift assay with specific antibodies. As shown in Figure 5F, no supershifted band was observed by anti-Smad3 or anti-Smad4. Given that often the interaction of Smads with other transcription factors on DNA is DNA context-dependent, we thus used TNF-α promoter containing an NF-κB-binding site with surrounding sequence to perform Smad supershift assay. As shown in Figure 5G, no supershifted band was observed by anti-Smad3 or anti-Smad4 antibodies. Taken together, these results suggest that Smad3 and Smad4 are required for synergistic induction of p65 acetylation and NF-κB activation by TGF-β1 via a mechanism independent of direct interaction with NF-κB.
Figure 5.
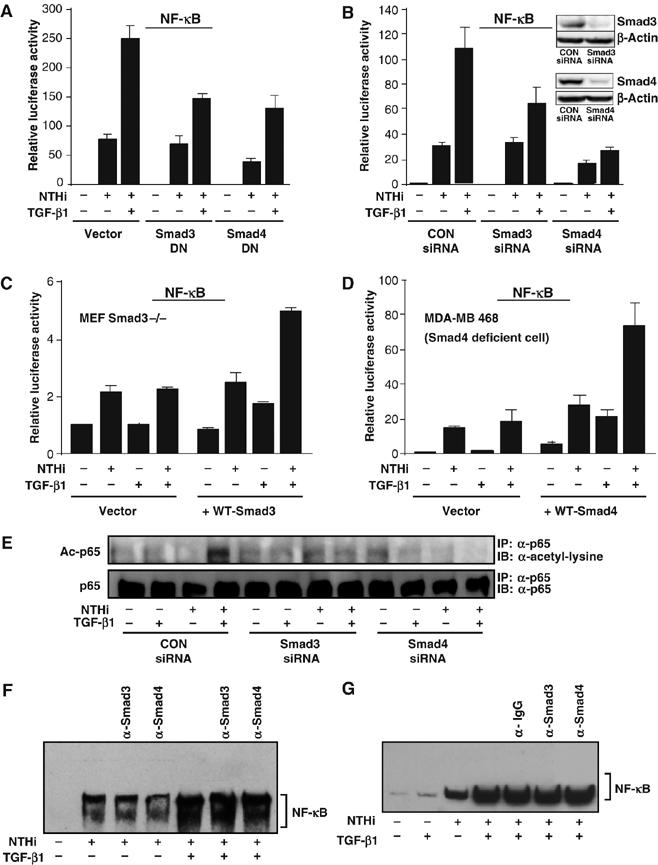
Smad3 and Smad4 are required for enhanced NF-κB activation by TGF-β via a mechanism independent of direct interaction with NF-κB. Overexpression of dominant-negative mutant of Smad3 or Smad4 (A) and Smad3 or Smad4 knockdown using siRNA (B) inhibited synergistic enhancement of NTHi-induced NF-κB activation by TGF-β in HeLa cells. Similar result was also observed in A549 cells. (C, D) TGF-β1 did not enhance NTHi-induced NF-κB activation in Smad3 null cells (C) and Smad4-deficient MDA-MB 468 cells (D). Transfecting Smad3 null cells with WT Smad3 and MDA-MB 468 cells with WT Smad4 rescued the responsiveness to TGF-β-1. (E) Smad3 or Smad4 knockdown inhibited p65 acetylation induced by TGF-β and NTHi in HeLa cells. (F) Smad3 and Smad4 does not appear to directly interact with NF-κB DNA-binding complex as assessed by supershift assay in HeLa cells. (G) No supershifted band was observed by anti-Smad3 or anti-Smad4 antibodies when TNF-α promoter containing an NF-κB-binding site with surrounding sequence was used as a probe to perform Smad supershift assay. IgG was used as a control in (F) and (G). Values are means±s.d. (n=3 for A–D). Data are representative of three independent experiments.
Synergistic enhancement of NTHi-induced p65 acetylation by TGF-β1 is mediated by PKA in vitro and in vivo
Having identified the requirement of Smad3/4 and p300 in mediating p65 acetylation by TGF-β1, which, in turn, leads to enhancement of DNA-binding activity and NF-κB-dependent transcriptional activation, it is still unknown which intermediate signaling molecule links TGF-β–Smad3/4 signaling to p300-mediated p65 acetylation. Among a variety of signaling transducers, PKA has been shown to promote the recruitment of the coactivator p300 via phosphorylation of CREB at Ser133 (Johannessen et al, 2004). There are also strong evidences that PKA enhances acetylation of histone and nonhistone protein in a p300-dependent manner (Salvador et al, 2001; Chang et al, 2005; Kim et al, 2005). Moreover, TGF-β1 can activate PKA without elevating intracellular cAMP level (Zhang et al, 2004). To determine the possible involvement of PKA in TGF-β1-induced synergistic enhancement of p65 acetylation, we first determined whether TGF-β1 activates PKA kinase activity by performing PKA kinase assay. As shown in Figure 6A, TGF-β1 activated PKA activity. The TGF-β1-induced PKA activity was inhibited by PKI, a specific PKA inhibitor peptide that contains a PKA pseudo-substrate sequence and specifically inhibits the catalytic subunits of PKA by binding to the substrate-binding site (Figure 6B; Zhang et al, 2004). We further assessed the effect of TGF-β1 on PKA activation using an siRNA approach. We first confirmed the efficiency of PKA catalytic unit-α-specific siRNA (siRNA-PKAc) in reducing endogenous PKAc expression in HeLa cells transfected with siRNA-PKAc or control siRNA. As expected, the PKAc protein was markedly reduced by PKAc knockdown using siRNA-PKAc. (Figure 6C, left panel). We then assessed the effect of PKAc knockdown on TGF-β1-induced PKA activation. As shown in Figure 6C (right panel), TGF-β1-induced PKA was abrogated by siRNA-PKAc. We next sought to determine whether PKA is involved in synergistic enhancement of NTHi-induced p65 acetylation by TGF-β1. Figure 6D shows that induction of p65 acetylation by NTHi and TGF-β1 was inhibited by PKI treatment. Consistent with these data, expressing WT PKA enhanced p65 acetylation in HeLa cells cotransfected with WT p65, but not with p65-KR mutant (Figure 6E). Thus, it is evident that TGF-β1 enhances NTHi-induced p65 acetylation through PKA activation. To further determine whether TGF-β1-induced PKA activation is also required for synergistic enhancement of DNA-binding activity by NTHi and TGF-β1, we examined the effect of siRNA-PKAc on DNA-binding activity. As shown in Figure 6F, siRNA-PKAc inhibited synergistic enhancement of NTHi-induced DNA-binding activity by TGF-β1. In accordance with these results, coexpressing WT PKA enhanced DNA-binding activity of NF-κB in cells co-transfected with WT p65 but not with p65-KR mutant (Figure 6G). These data indicate that PKA is involved in synergistic enhancement of DNA-binding activity of NF-κB by NTHi and TGF-β1 via induction of p65 acetylation. To further confirm the functional involvement of PKA in synergistic activation of NF-κB by NTHi and TGF-β1, we evaluated the effect of PKI and siRNA-PKAc on NF-κB activation. As expected, synergistic NF-κB activation was inhibited by either PKI or siRNA-PKAc (Figure 6H). Consistently, coexpressing WT PKA enhanced NF-κB activation in cells cotransfected with WT p65 but not with p65-KR mutant (Figure 6I). Finally, PKI also greatly inhibited synergistic enhancement of NTHi-induced expression of TNF-α and IL-1β (Figure 6J). To further investigate the physiological relevance of TGF-β-mediated induction of p65 acetylation by PKA, we assessed the in vivo effect of PKI on synergistic induction of inflammatory response by TGF-β1 and NTHi. As shown in Figure 6K, PKI inhibited synergistic enhancement of NTHi-induced expression of TNF-α in the lungs of mice. Consistent with this result, PKI also markedly inhibited the TGF-β-mediated enhancement of PMN accumulation in BAL fluids from the lungs of the NTHi-inoculated mice (Figure 6L and Supplementary Figure S3), thereby demonstrating the physiological relevance of TGF-β-mediated induction of p65 acetylation by PKA in vivo. Taken together, we concluded from these data that PKA is indeed involved in synergistic enhancement of NTHi-induced NF-κB activation by TGF-β1 through PKA-mediated p65 acetylation in vitro and in vivo.
Figure 6.
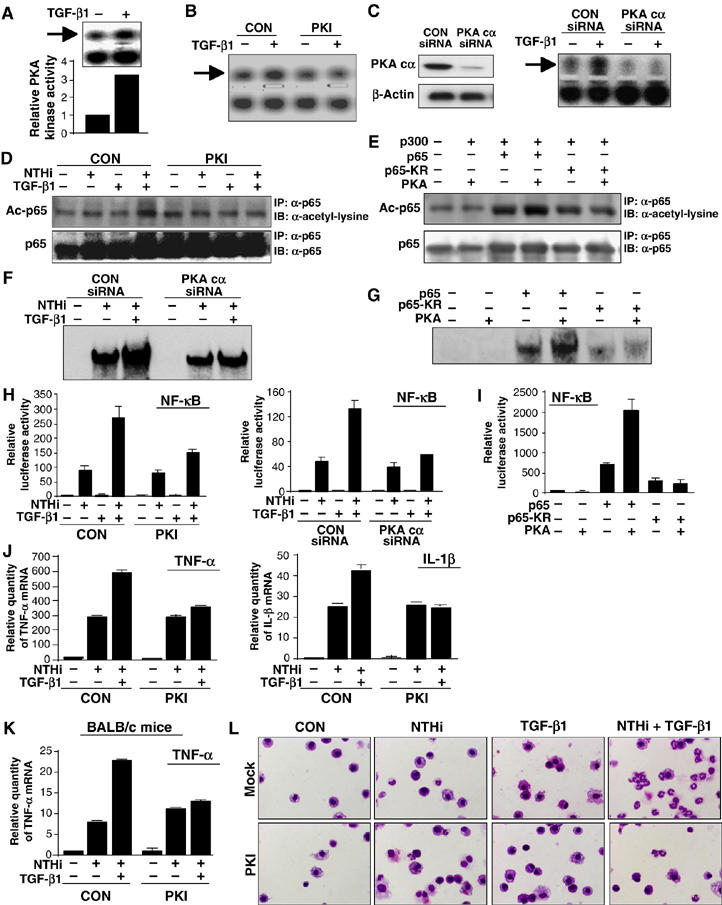
Synergistic enhancement of NTHi-induced p65 acetylation by TGF-β1 is mediated by PKA in vitro and in vivo. (A) TGF-β1 induced PKA activation in HeLa cells. TGF-β1-induced PKA activation was inhibited by either 1 μM PKI (B) or PKAc knockdown using siRNA-PKAc (C). (D) Synergistic p65 acetylation by NTHi and TGF-β1 was inhibited by PKI in HeLa cells. (E) Coexpression of WT PKA and p300 with WT 65 but not with p65-KR mutant markedly induced p65 acetylation in HeLa cells. (F) PKAc knockdown using siRNA-PKAc inhibited synergistic enhancement of NTHi-induced DNA-binding activity by TGF-β1 in HeLa cells. (G) WT PKA enhanced DNA-binding activity in HeLa cells transfected with WT 65 but not with p65-KR mutant. (H) Synergistic NF-κB activation by NTHi and TGF-β1 was inhibited by either PKI (left) or siRNA-PKAc in HeLa cells (right). (I) WT PKA enhanced NF-κB activation in HeLa cells transfected with WT p65, but not with p65-KR. (J) Synergistic enhancement of NTHi-induced TNF-α and IL-1β expression by TGF-β1 was inhibited by PKI in HeLa cells. (K) PKI inhibited synergistic enhancement of NTHi-induced expression of TNF-α in the lungs of mice. (L) PKI markedly inhibited the TGF-β-mediated enhancement of PMN accumulation in BAL fluids from the lungs of the NTHi-inoculated mice. Values are means±s.d. (n=3 for H–K). Data are representative of three independent experiments.
PKA acts downstream of TGF-β–Smad3/4 signaling pathway in mediating TGF-β1-induced synergistic enhancement of NF-κB activation
We have shown that PKA is indeed involved in synergistic enhancement of NF-κB activation by NTHi and TGF-β1. However, it is still unclear whether PKA acts downstream of the TGF-β–Smad signaling pathway. As we have already demonstrated that both Smad3 and Smad4 are required for synergistic NF-κB activation by NTHi and TGF-β1, we next directly assessed the effects of overexpression of dominant-negative mutant of Smad3 or Smad4 and Smad3 or Smad4 knockdown on TGF-β1-induced PKA activation. As shown in Figure 7A and B, TGF-β1-induced PKA activation was inhibited by both treatments. Consistent with these results, TGF-β1-induced PKA activation was not observed in Smad4-deficient MDA-MB-468 cells, whereas overexpression of WT Smad4 conferred responsiveness to TGF-β1 (Figure 7C). To further confirm whether PKA acts downstream of Smad3 and Smad4 in mediating NF-κB activation, we evaluated the effect of PKAc knockdown using siRNA-PKAc on NF-κB activation induced by overexpression of WT Smad3 or WT Smad4. As expected, PKAc knockdown inhibited NF-κB activation induced by overexpression of WT Smad3 or Smad4 (Figure 7D). Together, our data suggest that PKA indeed acts downstream of TGF-β–Smad signaling in mediating activation of NF-κB by TGF-β1.
Figure 7.
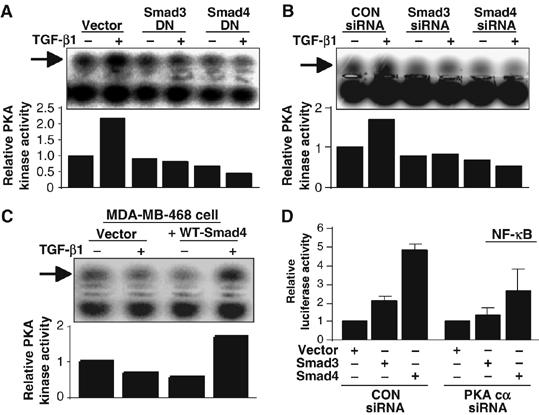
PKA acts downstream of TGF-β–Smad3/4 signaling pathway in mediating TGF-β1-induced synergistic enhancement of NF-κB activation. Overexpression of dominant-negative mutant of either Smad3 or Smad4 (A) and Smad3 or Smad4 knockdown (B) inhibited TGF-β1-induced PKA activation in HeLa cells. (C) TGF-β1 induced PKA activation in Smad4-deficient MDA-MB-468 reconstituted with WT Smad4 as indicated. Data (A–C) are representative of three independent experiments. (D) PKAc knockdown using siRNA-PKAc reduced NF-κB activation induced by overexpression of WT Smad3 and Smad4 in HeLa cells. Values are means±s.d. (n=3).
Discussion
In the present study, we provided direct evidence that TGF-β1 synergizes with Gram-negative bacterium NTHi to induce NF-κB activation and NF-κB-dependent transcription of TNF-α and IL-1β in a variety of human epithelial cells including the primary bronchial epithelial cells. TGF-β-mediated enhancement of NTHi-induced inflammatory responses was also confirmed in the mouse lung in vivo. Moreover, we found that TGF-β1 synergistically enhanced NF-κB activation by NTHi via Smad3/4–PKA–p300-dependent p65 acetylation at lysine 221, which, in turn, leads to enhancement of DNA-binding activity, and NF-κB-dependent transcription of inflammatory mediators (Figure 8).
Figure 8.
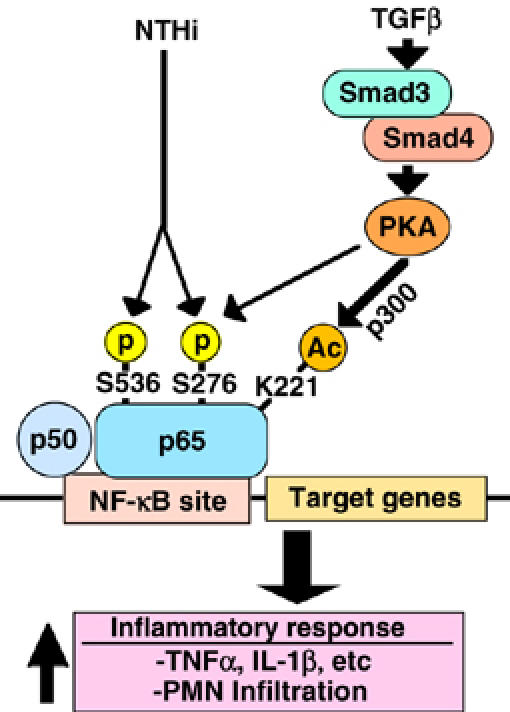
Schematic representation of TGF-β1-induced synergistic NF-κB activation via Smad3/4–PKA–p300-dependent p65 acetylation in human epithelial cells. As indicated, TGF-β induces p65 acetylation at lysine 221 via a Smad3/4–PKA–p300-dependent mechanism, which in turn leads to enhancement of DNA-binding activity, NF-κB activation and NF-κB-dependent inflammatory response in response to bacterium NTHi.
Of particular interest in this study is the direct evidence for TGF-β1 to induce p65 acetylation that in turn leads to enhancement of bacteria-induced NF-κB activation and inflammatory response. Among all known post-translational modifications of p65, p65 acetylation has been shown to play a critical role in controlling the duration and strength of NF-κB and regulating various biological functions of NF-κB including DNA binding, transactivation and the association with the inhibitor IκBα (Bonizzi and Karin, 2004; Hayden and Ghosh, 2004; Chen et al, 2005). Although several studies suggest that TGF-β induces inflammation through mechanisms dependent on NF-κB activation (Wahl et al, 1987), there has been no report showing that TGF-β signaling enhances NF-κB-dependent inflammatory response via induction of p65 acetylation. In the present study, we showed for the first time that TGF-β1 enhances bacteria-induced NF-κB activation via induction of p65 acetylation through a Smad3/4–PKA–p300-dependent mechanism. This result, although rather unexpected, may provide novel insights into the role of TGF-β in regulating NF-κB and NF-κB-dependent inflammatory response in bacterial infection. It should be noted that TGF-β1 induces greater p65 acetylation in the presence of NTHi than in its absence (Figure 3A). Thus it appears that certain bacteria-induced NF-κB activity, for example, phosphorylation, may be required for TGF-β-induced acetylation. Indeed, recent study demonstrated that phosphorylation of p65 regulates p65 acetylation (Chen et al, 2005). As shown in Figure 3A, bacterium NTHi induced phosphorylation of p65 at Ser536 and Ser276 sites. In addition, TGF-β1 alone also induced phosphorylation of p65 at Ser276 site. Thus, p65 phosphorylation appears to be critical for TGF-β1 to induce acetylation of p65. Previous studies have identified three acetylation sites as the main acetylation sites within p65—lysines 218, 221 and 310 (Chen et al, 2002; Kiernan et al, 2003). Site-specific acetylation of p65 regulates discrete biological action of the NF-κB complex. For example, acetylation of lysine 221 increases the DNA-binding affinity of p65 for κB enhancer, whereas acetylation of lysine 221 alone or in combination with lysine 218 impairs the assembly of p65 with IκBα. As evidenced by the data shown in Figure 3C–G, mutation of all three of these lysine residues (p65KR (p65K218/221/310R)) reduced p65 acetylation, DNA binding and NF-κB activation by TGF-β1 and NTHi. Given that TGF-β1 also synergistically enhanced DNA-binding activity induced by NTHi, it is possible that TGF-β1 may enhance NTHi-induced DNA-binding activity by inducing acetylation of p65 at lysine 221. Indeed, further analysis using site-specific mutant of each of these three lysine residues 218, 221 and 310 and p65−/− MEFs that were reconstituted with either WT p65 or p65-K310R mutant confirmed that lysine 221 residue is critical for mediating synergistic enhancement of p65 acetylation by TGF-β1 and NF-κB-dependent transcriptional activity. Also interesting to note is that mutation of K218 and K310 also appears to partially diminish the acetylation and transactivation function of p65. Given that lysine 218 is located adjacent to lysine 221, it is possible that mutation of lysine 218 may interfere with the function of lysine 221 owing to a possible conformational change, thereby resulting in partial diminishment of acetylation and the synergistic enhancement of the transcriptional activity of p65 in response to TGF-β1. Moreover, as acetylation of lysine 310 was previously shown to be required for full transcriptional activity of p65 (Chen et al, 2002), it is anticipated that mutation of lysine 310 would affect the transcriptional activity of p65. This may explain a decrease in both basal activity and the TGF-β-induced transcriptional activity of p65 in cells transfected with p65-K310R (Figure 3I) as well as the reduction of NF-κB activity induced by NTHi alone or in conjunction with TGF-β in MEF p65-K310R (Figure 3J). However, it should be noted that fold induction of the TGF-β-induced synergistic enhancement of transcriptional activity of p65 in either case still remains unchanged. Similarly, the fold induction of p65 acetylation in cells transfected with p65-K310R also remains unchanged (2.1-fold in K310R versus 2.3-fold in WT-p65) (Figure 3H). Nonetheless, it is evident that lysine 221 is critical for mediating synergistic enhancement of p65 acetylation and NF-κB-dependent transcriptional activity in response to TGF-β1. Our findings are in line with previous reports. PKAc is known to directly phosphorylate p65 at S276 (Zhong et al, 1997) and modulate the interaction between p65 and CBP/p300 (Zhong et al, 1998). Furthermore, phosphorylation of p65 at S276 was recently shown to be coupled to its stimulation-induced acetylation (Chen et al, 2005). In the present study, we showed that TGF-β alone also induced phosphorylation of p65 at S276 and synergized with NTHi to induce acetylation of p65 at lysine 221 via a Smad3/4–PKA-dependent mechanism. Taken together, our data reveal a novel molecular mechanism by which TGF-β–Smad3/4 signaling pathway potentiates NF-κB activation through induction of p65 acetylation via a PKA-dependent signaling pathway.
Another important finding in the present study is that TGF-β enhances bacteria-induced NF-κB-dependent inflammatory response in lung bacterial infections in vitro and in vivo. Despite the critical role for TGF-β in regulating cell proliferation, differentiation, migration and development, its role in regulating inflammatory response still remains largely unknown. Different effects (stimulatory or inhibitory) of TGF-β have been reported on the regulation of cytokine induction and inflammatory response. For instance, targeted disruption of the mouse TGF-β1 gene results in excessive inflammatory responses (Shull et al, 1992), whereas TGF-β1 overexpression in keratinocytes in transgenic mice also results in inflammatory skin lesions (Li et al, 2004). The molecular mechanisms underlying the distinct role of TGF-β in regulating inflammation still remain elusive. There is accumulating evidence to suggest that the consequences of regulation of cytokines and inflammation by TGF-β are likely context-dependent and cell-type-specific and may also depend on the concentration of TGF-β itself. Studies from monocytes indicate that during the early stage of inflammation, TGF-β locally acts as a proinflammatory agent by recruiting and activating resting monocytes. As the recruited monocytes are activated and differentiated, they lose responsiveness to TGF-β possibly due to the decreased expression of TGF-β receptor, and specific immunosuppressive actions of TGF-β predominate, eventually leading to the resolution of inflammation (McCartney-Francis and Wahl, 1994; Ashcroft, 1999; Li et al, 2006). Moreover, studies from various cell types indicate that the role of TGF-β in regulating cytokine and inflammation is likely also cell-type-dependent. For instance, TGF-β inhibits the production of and response to cytokines associated with both Th1 and Th2 cells (Li et al, 2006). There is also evidence that TGF-β inhibits E-selectin expression to block adhesion and targeting of leukocytes to the site of inflammation in endothelial cells (Gamble et al, 1993). In addition, recent study also suggests that TGF-β may exert a protective role in CNS diseases characterized by microglial cell activation by proinflammatory stimulants (Le et al, 2004). Furthermore, the effect of TGF-β on cytokines and inflammation may also be dose-dependent. Systemic administration of TGF-β leads to an inhibition of inflammation and reduced tissue destruction (Wahl, 1994). However, a marked increase (greater than 6–8-fold) in circulating TGF-β in TGF-β overexpressing mice leads to renal inflammation (Ashcroft, 1999). Thus, it is evident that the consequence of TGF-β on cytokines and inflammation depends on multiple factors. In the present study, we showed that TGF-β alone did not induce potent NF-κB activation but synergistically enhanced bacteria-induced NF-κB activation and NF-κB-dependent inflammatory response in vitro and in vivo. Given the critical role that inflammatory responses play in host defense, our findings may have some important implication in TGF-β-mediated host defense against bacterial pathogens. The role of TGF-β signaling in infectious diseases remains controversial (Reed, 1999). Most studies have focused on pathogens that infect host macrophages such as Trypanosoma cruzi and a variety of Leishmania species. These studies have demonstrated that excessively produced TGF-β upon infection inhibits macrophage activation, thereby favoring virulence. In certain situations, however, there are also evidences that TGF-β has been correlated with enhanced resistance to microbes such as Candida albicans, thus benefiting the host. Despite these distinct observations that mainly focused on macrophages, little is known about how TGF-β regulates host defense and inflammatory responses in the mucosal epithelial cells of airway. Therefore, our study may bring new insights into the novel role of TGF-β signaling in potentiating host defense and inflammatory response to respiratory bacterial pathogens.
Finally, interesting evidence was also provided for the involvement of PKA in synergistic NF-κB activation by NTHi and TGF-β1. Our data showed that TGF-β1-induced PKA activation, which in turn, leads to enhancement of p65 acetylation, DNA-binding activity and NF-κB activation by NTHi. Because PKA phosphorylates CREB, which in turn promotes recruitment of the coactivator p300 and CBP, and PKA has also been shown to activate acetylation of histone and nonhistone protein (Salvador et al, 2001; Johannessen et al, 2004; Chang et al, 2005; Kim et al, 2005), it is logical that TGF-β1-induced activation of PKA may increase p65 acetylation via p300-mediated mechanism. Our results shown in Figure 4 indeed demonstrate the involvement of p300 in enhancement of p65 acetylation by TGF-β. How does PKA induce p300-mediated p65 acetylation? It is possible that PKA induces p300-mediated p65 acetylation likely through phosphorylation of p65 or p300. It has been shown that PKA induces phosphorylation of p65 on serine 276 (Zhong et al, 1998), and phosphorylation of p65 on serine 276 in turn leads to increased assembly of phospho-p65 with p300, resulting in enhanced acetylation of p65 (Chen et al, 2005). Moreover, there was also evidence that phosphorylation of p300 by PKA is specifically required for proper activation of its HAT activity (Brouillard and Cremisi, 2003). Thus, it is likely that PKA-induced phosphorylation of p300 may also contribute to the increased acetylation of p65. However, our data do not rule out the possible involvement of the other mechanism underlying PKA-mediated p65 acetylation by TGF-β1, such as inhibition of HDAC activity. Sequence analysis of HDAC8 revealed consensus phosphorylation sites for PKA, and it has been shown that HDAC8 is phosphorylated by PKA both in vitro and in vivo, thereby leading to hyperacetylation of histone H3 and H4 (Lee et al, 2004). Further experiments are needed to address this issue.
In conclusion, our present study demonstrated that TGF-β induces p65 acetylation at lysine 221 via a PKA–p300-dependent mechanism, which in turn leads to enhancement of DNA-binding activity, NF-κB activation and NF-κB-dependent inflammatory response in response to bacterium NTHi. This study provides new insights into the novel role of TGF-β signaling in regulating NF-κB activation.
Materials and methods
Bacterial strains
NTHi strain 12 was used in our studies and was cultured as described previously (Shuto et al, 2001) (see Supplementary data for details).
Cell culture
HeLa, A549, Smad3−/− cells and MDA-MB468 (ATCC) were maintained as described (Shuto et al, 2001; Qiu et al, 2003; Mikami et al, 2006). Cell lines stably expressing WT p65, p65KR, K221R and K310R were described previously (Chen et al, 2002). HMEEC-1, NHBE (Cambrex) and mouse embryonic fibroblast (MEF) cells were maintained as described (Jono et al, 2004).
Real-time quantitative RT–PCR analysis of TNF-α and IL-1β
Real-time Q-PCR was performed using an ABI 7700 sequence detection system as described (Jono et al, 2004) (see Supplementary data for details).
Plasmids, transfection and luciferase assay
The reporter construct NF-κB luc was generated as described (Shuto et al, 2001). Smad3DN and WT, Smad4DN and WT were previously described (Jono et al, 2002). WT-p65, p65-KR, p65-K218R, p65-K221R, p65-K310R, WT-p300 and p300 HAT mutant were previously described (Chen et al, 2002, 2005) (see Supplementary data for details).
siRNA
PKAc, Smad3 and Smad4 siRNA oligonucleotides were purchased from Dharmacon. The siRNA was transfected into HeLa cells using RNAifect transfection reagent (QIAGEN) following the manufacturer's instructions (Mikami et al, 2005).
Western blot analysis and immunoprecipitation
See Supplementary data for details.
PKA kinase assays
PKA activity was measured on HeLa whole-cell extracts by using a fluorescent kemptide assay (PepTag® Assay for Non-Radioactive Detection of Protein Kinase A; Promega), by following the supplier's instructions. The relative PKA activity was quantitated by densitometry analyses.
Electrophoretic mobility shift assay
See Supplementary data for details.
Chromatin immunoprecipitation assays
ChIP was performed using an EZ ChIP kit (Upstate Biotechnology) as described (Taggart et al, 2005) (see Supplementary data for details).
Mouse and animal experiments
Animal studies were performed as described (Mikami et al, 2006) (see Supplementary data for details).
Supplementary Material
Supplementary data
Acknowledgments
This work was supported by National Institutes of Health grants DC005843 and DC004562 (to JD Li), CA108454 and GM063773 (to XH Feng), and HL077789 (to C Yan). LFC is the recipient of an Arthritis Foundation Investigator Award.
References
- Adam E, Quivy V, Bex F, Chariot A, Collette Y, Vanhulle C, Schoonbroodt S, Goffin V, Nguyen TLA, Gloire G, Friguet B, de Launoit Y, Burny A, Bours V, Piette J, Lint CV (2003) Potentiation of tumor factor-induced NF-κB activation by deacetylase inhibitors is associated with a delayed cytoplasmic reappearance of IκBα. Mol Cell Biol 23: 6200–6209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft GS (1999) Bidirectional regulation of macrophage function by TGF-β. Microbes Infect 1: 1275–1282 [DOI] [PubMed] [Google Scholar]
- Bonizzi G, Karin M (2004) The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25: 280–288 [DOI] [PubMed] [Google Scholar]
- Brouillard F, Cremisi CE (2003) Concomitant increase of histone acetyltransferase activity and degradation of p300 during retinoic acid-induced differentiation of F9 cells. J Biol Chem 278: 39509–39516 [DOI] [PubMed] [Google Scholar]
- Chang CW, Chuan HC, Yu C, Yao TP, Chen H (2005) Stimulation of GCMa transcriptional activity by cAMP/PKA signaling is attributed to CBP- mediated acetylation of GCMa. Mol Cell Biol 25: 8401–8414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Fischle W, Verdin E, Greene WC (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293: 1653–1657 [DOI] [PubMed] [Google Scholar]
- Chen LF, Mu Y, Greene WC (2002) Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J 21: 6539–6548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LF, Williams SA, Nakano H, Duerr JM, Buckbinder L, Greene WC (2005) NF-κB RelA phosphorylation regulates RelA acetylation. Mol Cell Biol 25: 7966–7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer WI, van Schadewijk A, Sont JK, Sharma HS, Stolk J, Hiemstra PS, van Krieken JHJM (1998) TGF-β1 and recruitment of macrophages and mast cells in airways in COPD. Am J Respir Crit Care Med 158: 1951–1957 [DOI] [PubMed] [Google Scholar]
- Feng XH, Derynck R (2005) Specificity and versatility in TGF-β signaling through Smads. Annu Rev Cell Dev Biol 21: 659–693 [DOI] [PubMed] [Google Scholar]
- Foxwell AR, Kyd JM, Cripps AW (1998) NTHi: pathogenesis and prevention. Microbiol Mol Biol Rev 62: 294–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble JR, Khew-Goodall Y, Vadas MA (1993) Transforming growth factor-beta inhibits E-selectin expression on human endothelial cells. J Immunol 150: 4494–4503 [PubMed] [Google Scholar]
- Gu W, Luo J, Brooks CL, Nikolaev AY, Li M (2004) Dynamics of p53 acetylation pathway. Novartis Found Symp 259: 197–205 [PubMed] [Google Scholar]
- Haller D, Holt L, Kim SC, Schwabe RF, Sartor RB, Jobin C (2003) TGF-β 1 inhibits non-pathogenic Gram negative bacteria-induced NF-κB recruitment to the IL-6 gene promoter in intestinal epithelial cells through modulation of histone acetylation. J Biol Chem 278: 23851–23860 [DOI] [PubMed] [Google Scholar]
- Hayden M, Ghosh S (2004) Signaling to NF-κB. Gene Dev 18: 2195–2224 [DOI] [PubMed] [Google Scholar]
- Hoberg JE, Popko AE, Ramsey CS, Mayo MW (2006) IkB kinase α-mediated derepression of SMRT potentiates acetylation of RelA/p65 by p300. Mol Cell Biol 26: 457–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johannessen M, Delghandi MP, Moens U (2004) What turns CREB on? Cell Signal 16: 1211–1227 [DOI] [PubMed] [Google Scholar]
- Jono H, Shuto T, Xu H, Kai H, Lim DJ, Gum JR Jr, Kim YS, Yamaoka SY, Feng XH, Li JD (2002) TGF-β-Smad signaling pathway cooperates with NF-κB to mediate NTHi-induced MUC2 mucin transcription. J Biol Chem 277: 45547–45557 [DOI] [PubMed] [Google Scholar]
- Jono H, Lim JH, Chen LF, Xu H, Trompouki E, Pan ZK, Mosialos G, Li JD (2004) NF-κB is essential for induction of CYLD, the negative regulator of NF-κB. J Biol Chem 279: 36171–36174 [DOI] [PubMed] [Google Scholar]
- Kiernan R, Bres V, Ng RWM, Coudart MP, Messaoudi SE, Sardet C, Jin DY, Emiliani S, Benkirane M (2003) Post-activation turn-off NF-κB-dependent transcription is regulated by acetylation of p65. J Biol Chem 278: 2758–2766 [DOI] [PubMed] [Google Scholar]
- Kim J, Stallcup MR, Coetzee GA (2005) The role of PKA pathway and cAMP responsive element-binding protein in androgen receptor-mediated transcription at the prostate-specific antigen locus. J Mol Endocrinol 34: 107–118 [DOI] [PubMed] [Google Scholar]
- Kouzarides T (2000) Acetylation: a regulatory modification to rival phosphorylation? EMBO J 19: 1176–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Y, Iribarren P, Gong W, Cui Y, Zhang X, Wang JM (2004) TGF-β1 disrupts endotoxin signaling in microglial cells through Smad3 and MARK pathway. J Immunol 173: 962–968 [DOI] [PubMed] [Google Scholar]
- Lee H, Rezai-Zadeh N, Seto E (2004) Negative regulation of histone deacetylase 8 activity by cAMP-dependent PKA. Mol Cell Biol 24: 765–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li AG, Wang D, Feng XH, Wang XJ (2004) Latent TGFβ1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. EMBO J 23: 1770–1781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MO, Wan YY, Sanjabi S, Robertson AK, Flavell RA (2006) Transforming growth factor-beta regulation of immune responses. Annu Rev Immunol 24: 99–146 [DOI] [PubMed] [Google Scholar]
- Massague J, Seoane J, Wotton D (2005) Smad transcription factors. Genes Dev 19: 2783–2810 [DOI] [PubMed] [Google Scholar]
- McCartney-Francis NL, Wahl SM (1994) Transforming growth factor beta: a matter of life and death. J Leukoc Biol 55: 401–409 [DOI] [PubMed] [Google Scholar]
- Mikami F, Gu H, Jono H, Andalibi A, Kai H, Li JD (2005) EGFR acts as a negative regulator for bacterium NTHi-induced TLR2 expression via an Src- dependent p38 MAPK signaling pathway. J Biol Chem 280: 36185–36194 [DOI] [PubMed] [Google Scholar]
- Mikami F, Lim JH, Ishinaga H, Ha UH, Gu H, Koga T, Jono H, Kai H, Li JD (2006) TGF-β–Smad3/4 signaling pathway acts as a positive regulator for TLR2 induction by bacteria via a dual-mechanism involving functional cooperation with NF-κB and MKP-1-dependent negative cross-talk with p38. J Biol Chem 281: 22397–22408 [DOI] [PubMed] [Google Scholar]
- Murphy TF (2000) Bacterial otitis media: pathogenetic considerations. Pediatr Infect Dis J 19: S9–S15 [DOI] [PubMed] [Google Scholar]
- Murphy TF (2006) The role of bacteria in airway inflammation in exacerbations of chronic obstructive pulmonary disease. Curr Opin Infect Dis 19: 225–230 [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y (1996) The transcriptional coactivator p300 and CBP are histone acetyltransferases. Cell 87: 953–959 [DOI] [PubMed] [Google Scholar]
- Qiu P, Feng XH, Li L (2003) Interaction of Smad3 and SRF-associated complex mediates TGF-βeta1 signals to regulate SM22 transcription during myofibroblast differentiation. J Mol Cell Cardiol 35: 1407–1420 [DOI] [PubMed] [Google Scholar]
- Reed SG (1999) TGF-β in infections and infectious diseases. Microb Infect 1: 1313–1325 [DOI] [PubMed] [Google Scholar]
- Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W (1999) IκB kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 274: 30353–30356 [DOI] [PubMed] [Google Scholar]
- Salvador LM, Park Y, Cotton J, Maizels ET, Jones JCR, Schillace RV, Carr DW, Cheung P, Allis CD, Jameson JL, Hunzicker-Dunn M (2001) Follicle- stimulating hormone stimulates protein kinase A-mediated histone H3 phosphorylation and acetylation leading to select gene activation in ovarian granulose cells. J Biol Chem 276: 40146–40155 [DOI] [PubMed] [Google Scholar]
- Shull MM, Ormsby I, Kier AB, Pawlowski S, Diebold RJ, Yin M, Allen R, Sidman C, Proetzel G, Calvin D, Anninziata N, Doetschman T (1992) Targeted disruption of the mouse TGF-β1 gene results in multiple inflammatory disease. Nature 359: 693–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuto T, Xu H, Wang B, Han J, Kai H, Gu XX, Murphy T, Lim DJ, Li JD (2001) Activation of NF-κB by NTHi is mediated by TLR2–TAK-dependent NIK–IKKα/β–IκBα and MKK3/6–p38 MAP kinase signaling pathways in epithelial cells. Proc Natl Acad Sci USA 98: 8774–8779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taggart CC, Cryan SA, Weldon S, Gibbons A, Greene CM, Kelly E, Low TB, O'Neill SJ, McElvaney NG (2005) Secretory leucoprotease inhibitor binds to NF-kappaB binding sites in monocytes and inhibits p65 binding. J Exp Med 202: 1659–1668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Dijke P, Hill CS (2004) New insights into TGF-β–Smad signalling. Trends Biochem Sci 29: 265–273 [DOI] [PubMed] [Google Scholar]
- Vermeulen L, Wilde GD, Damme PV, Berghe WV, Haegeman G (2003) Transcriptional activation of the NF-κB p65 subunit by MSK1. EMBO J 22: 1313–1324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SM (1994) TGF-β: the good, the bad, and the ugly. J Exp Med 180: 1587–1590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl SM, Hunt DA, Wakefield LM, McCartney-Francis N, Wahl LM, Roberts AB, Sporn MB (1987) TGF-β induces monocyte chemotaxis and growth factor production. Proc Natl Acad Sci USA 84: 5788–5792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Westerheide SD, Hanson JL, Baldwin AS Jr (2000) TNF α-induced phosphorylation of RelA/p65 on Ser529 is controlled by casein kinase II. J Biol Chem 275: 32592–32597 [DOI] [PubMed] [Google Scholar]
- Zhang L, Duan CJ, Binkley C, Li G, Uhler MD, Logsdon CD, Simeone DM (2004) TGF-β-induced Smad3/Smad4 complexes directly activates protein kinase A. Mol Cell Biol 24: 2169–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, Ghosh S (1997) The transcriptional activity of NF-κB is regulated by the IκB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 89: 413–424 [DOI] [PubMed] [Google Scholar]
- Zhong H, Voll RE, Ghosh S (1998) Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1: 661–671 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data