

Abstract
Our current knowledge of the general factor requirement in transcription by the three mammalian RNA polymerases is based on a small number of model promoters. Here, we present a comprehensive chromatin immunoprecipitation (ChIP)-on-chip analysis for 28 transcription factors on a large set of known and novel TATA-binding protein (TBP)-binding sites experimentally identified via ChIP cloning. A large fraction of identified TBP-binding sites is located in introns or lacks a gene/mRNA annotation and is found to direct transcription. Integrated analysis of the ChIP-on-chip data and functional studies revealed that TAF12 hitherto regarded as RNA polymerase II (RNAP II)-specific was found to be also involved in RNAP I transcription. Distinct profiles for general transcription factors and TAF-containing complexes were uncovered for RNAP II promoters located in CpG and non-CpG islands suggesting distinct transcription initiation pathways. Our study broadens the spectrum of general transcription factor function and uncovers a plethora of novel, functional TBP-binding sites in the human genome.
Keywords: ChIP-on-chip, promoter, TBP, transcription factor profiling
Introduction
The comprehensive mapping of transcription regulatory regions in the genome of higher eukaryotes and the analysis of transcription factors recruited to these sites are major challenges notwithstanding the availability of the entire sequence of many genomes. Current annotations are skewed towards protein-coding genes and the assignment of promoters towards CpG islands. Regulatory regions positioned far away from the transcription start site such as enhancers and locus control regions are difficult to identify. In silico prediction of regulatory regions remains difficult notwithstanding first successes (Xie et al, 2005; Hallikas et al, 2006).
Our knowledge of the organization and factor composition of promoters in higher eukaryotes is based largely on reporter gene assays and in vitro transcription reconstitution studies involving a small number of model promoters. Collectively, these studies identified and characterized general transcription factors and provided valuable insights of the mechanisms of transcription (Lee and Young, 2000; Sims et al, 2004). It has remained unresolved whether general transcription factors are universally involved in transcription or whether they are truly specific for a given RNAP class. Experimental approaches to systematically identify regulatory regions and to characterize their organization and regulation are, therefore, of great importance.
The multitude of general (co)factors, sequence-specific DNA-binding factors, bridging complexes, chromatin modifying and remodeling complexes involved in transcription is staggering and has been estimated to involve up to 6% of the protein coding genes in mammalian genomes (Tupler et al, 2001). Chromatin immunoprecipitation (ChIP) has proven to be a valuable tool in establishing the involvement and chronology of the recruitment of transcription factors and cofactors to a gene or locus. Application of ChIP to large sets of genes, ChIP-on-chip, has added a new dimension to target site identification and transcription factor occupancy profiling. General patterns and principles of gene regulation are currently being uncovered (Ren et al, 2000, 2002; Cam et al, 2004; Kim et al, 2005; Boyer et al, 2006).
Here, we report the identification and annotation of genomic binding sites of the central transcription factor TBP (TATA-binding protein) using sequential ChIP and direct cloning of the DNA fragments. Annotation of the clones revealed unique genomic loci containing known or predicted sites and a surprisingly large proportion of TBP-binding sites in introns or in regions without gene annotation. An experimentally derived TBP target site microarray was used in ChIP-on-chip to obtain binding profiles 26 transcription factors and two histone marks. We show that some transcription factors hitherto reported to regulate transcription by RNA polymerase II (RNAP II) are also recruited to rRNA promoters suggesting cross-regulation between these classes of genes. Furthermore, correlation analysis of ChIP-on-chip data revealed distinct profiles corresponding to CpG and non-CpG island promoters transcribed by RNAP II suggesting distinct mechanisms of transcription initiation.
Results
Identification of in vivo TBP-binding sites
To identify a broad selection of in vivo TBP-binding sites, we used sequential ChIP using the human U2OS cell line, a highly specific monoclonal antibody against the N-terminal part of the TBP (Ruppert et al, 1996) and cloning of the precipitated DNA fragments (Supplementary Figure 1A). We reasoned that targeting TBP, the central factor in transcription, should ensure that promoters of genes transcribed by all three RNA polymerases were obtained. Cloning of ChIP'ed DNA fragments without prior amplification yielded a library of >20K colonies. A representative number of colonies (2000) were randomly picked. The lengths of the cloned fragments ranged from 40 to 500 bp averaging about 160 bp (Supplementary Figure 1A and B).
Inserts larger than 40 bp were annotated using the UCSC genome browser and NCBI BLAST. Highly repetitive sequences and such with less than 90% identity to the genome were eliminated (Figure 1A). The sequence complexity and overlap of the remaining putative TBP-binding sites (1361 clones) were analyzed via genome alignment and sequence comparison using TIGR Assembler (Sutton et al, 1995); about 61% of the target sites (864 clones) were present only once. Overlapping sequences (497 clones) were collapsed into a total of 177 contigs and that mainly comprised promoters of high-copy-number genes such as tRNA and rRNA.
Figure 1.
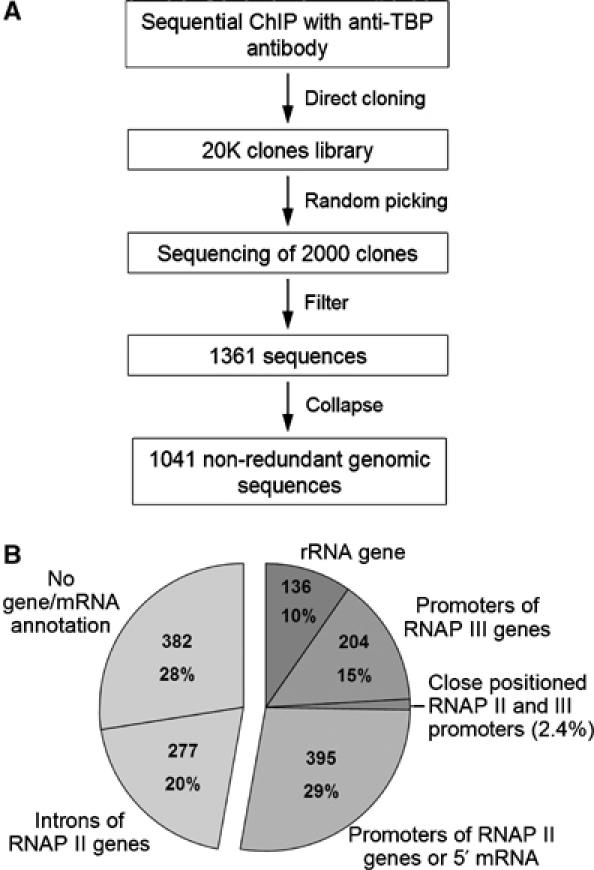
Construction and annotation of the TBP-binding site library. (A) Outline of the strategy for ChIP-cloning and filtering of sequences. ‘Filter': short sequences (<40 bp), highly repetitive sequences and those with less than 90% identity to the genome were eliminated. ‘Collapse': 497 overlapping sequences were collapsed into 177 contigs. (B) Pie diagram of annotation. Transcription-linked features were obtained from UCSC genome browser (HG16) using a 1 kb window centered at the cloned DNA sequences. Annotation of RNAP II genes was based on SWISS-PROT, TrEMBL, RefSeq and mRNA GenBank databases. Identity to rRNA genes was obtained by NCBI BLAST alignment. The number of clones in different categories was determined using the non-collapsed set of 1361 clones.
TBP-binding sites were annotated and sorted on the basis of transcription-linked features such as the presence of known genes and mRNA. An annotation window of 1 kb centered on the cloned sequence was chosen based on the resolution of ChIP experiments. Annotation of the top-ranked hit for each sequence revealed that 29% overlapped with the first exon of annotated genes or with the 5′ end of mRNA (Figure 1B) and mostly located to CpG islands. A remarkably large fraction of targets was located in introns of known genes or in regions lacking a gene or mRNA annotation (20 and 28%, respectively). Fragments corresponding to RNAP III genes (15%) comprised tRNA and other different small structural RNA genes. rDNA sequences accounted for 10% of the cloned fragments.
Validation of TBP-binding sites by ChIP-on-chip
To study the binding sites of TBP by ChIP-on-chip we PCR-amplified inserts from the 2000 randomly picked clones, printed them on glass slides and hybridized DNA from input chromatin and TBP ChIP. The ChIP/input ratios of a set of reference promoters printed on the array showed a highly significant correlation value (r=0.83, P=10−7) with TBP occupancy as determined by single gene quantitative PCR (qPCR) (Supplementary Figure 2). This implies that the data obtained by ChIP-on-chip faithfully reflects TBP occupancy in vivo. To define a threshold value, we computed frequency histograms of the ChIP/input ratios for all targets as well as for annotated promoters of the RNAP I, II and III class. On the vast majority (>95%) of RNAP II promoters, TBP was enriched more than two-fold over negative controls (Figure 2A). RNAP I and III targets displayed a high TBP occupancy ranging from 6- to >30-fold. Applying an arbitrary two-fold cutoff value implies that ∼90% of the targets are significantly enriched for TBP.
Figure 2.
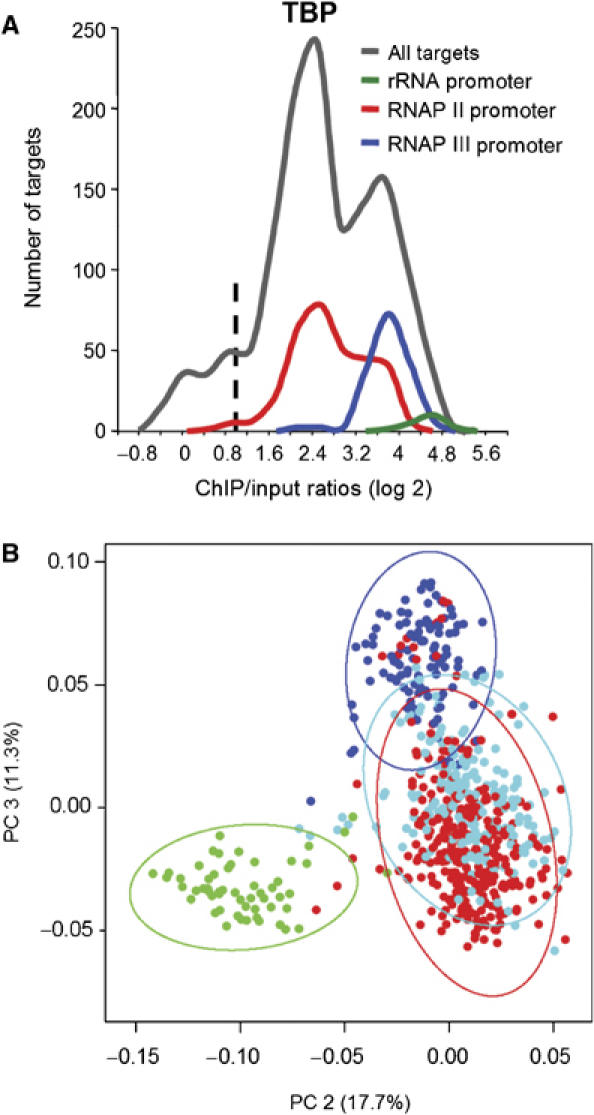
Analysis of ChIP-on-chip data for different classes of promoters. (A) Frequency histograms of TBP ChIP/input ratios (non-collapsed set). Dashed line indicates two-fold threshold. Promoters of RNAP I, II and III genes are colored in green, red and blue, respectively. Normalization controls correspond to ‘0' value on the histograms. (B) Projection of the ChIP-on-chip data set into the space of the second and third PCs. Intronic targets and those without gene/mRNA annotation are highlighted in light blue. The spaces containing 95% of targets are shown as ovals of the RNAP I, II and III targets. The fraction of variance comprised in individual PCs is indicated in brackets.
ChIP-on-chip on the TBP-binding site microarray was also validated by profiling for binding sites of the transcription factors E2F1 and E2F4. We identified 22 targets that were selectively enriched with both E2F1 and E2F4 (Supplementary Table I); most of them corresponding to promoters of previously identified E2F target genes (Ren et al, 2002; Cam et al, 2004). Hence, the microarray can be used to reliably measure transcription factor occupancy.
Principal component analysis
For a comprehensive profiling of general transcription factors and assessment of factors occupancy, ChIP-on-chip experiments were performed with antibodies against 26 different RNAP I, II and III-linked transcription factors and two histone marks that correlate with transcription. The intrinsic structure and complexity of the data set was assessed by principal component analysis (PCA) of the ChIP/input ratios for the different transcription factors. PCA defines a small set of latent orthogonal variables (principal components, PCs) that describe maximal possible variance in the entire data set. Figure 2B shows that targets segregated into three spaces according to the highest variance in their factor profiles; color coding of the known TBP target sites belonging to either of the three gene classes visualized their good separation. A small number of known RNAP II promoters ended up in the RNAP III realm and vice versa; inspection of their genomic organization revealed that these targets contained closely positioned RNAP II and III promoters. Importantly, the majority of non-annotated, novel TBP-binding sites was found in the space assigned to RNAP II. We conclude that these TBP target sites are most likely regulatory regions directing RNAP II-dependent transcription.
Novel TBP-binding sites direct transcription
To further characterize these novel TBP-binding sites, we compared their transcription factor occupancies with those of annotated promoters by computing the frequency histogram on the ChIP/input ratios. The novel TBP-binding sites showed a slightly lower distribution of TBP, TFIIB and RNAP II occupancy compared to annotated promoters (Supplementary Figure 3). To test whether the novel TBP-binding sites can direct transcription, we randomly picked 27 targets for further analysis. The majority of these targets (25/27) showed significant enrichment for TBP and RNAP II in single gene qPCR (data not shown); the qPCR values correlated well with ChIP/input ratios for TBP as determined by microarray analysis (r=0.83).
To assess the competence of these sites to direct transcription two approaches were used. First, the validated novel TBP-binding sites were PCR-amplified from the genome as ∼1 kb fragments and cloned into a promoter-less reporter along with positive and negative controls. The majority of the intronic sites (11 out of 15) activated unidirectional transcription of the reporter gene (Figure 3A). Remarkably high activation (∼250-fold) was found for a site (F11-3-46) located in the first intron of the EGFR gene ∼100 kb downstream of the transcription start site. Transcription activation was collinear with the direction of transcription of the EGFR gene suggesting that this novel site is an alternative promoter. Several intronic TBP target sites, such as F5-4-46 located in the 1st intron of TFIIAαβ genes, displayed promoter activity in the opposite direction suggesting novel antisense transcripts. The majority of the novel TBP-binding sites without gene/mRNA annotation (8 out of 11) displayed significant activation of the promoter-less reporter (Figure 3B). Interestingly, one of the novel TBP target sites comprised the intronic enhancer of GADD45 gene. Consistent with its well-documented enhancer function, this target activated SV-40 promoter in both orientations (Figure 3D). A number of other TBP-binding sites tested in this assay displayed enhancer activity (Figure 3D) suggesting that a fraction of the cloned TBP-binding sites may comprise enhancers. Promoters of five housekeeping genes identified in our screen were used as positive controls in this assay and they displayed on average stronger activation potential than the novel TBP-binding sites (Figure 3C). Eight randomly chosen genomic regions displayed little to no transcription activation (Supplementary Figure 4).
Figure 3.
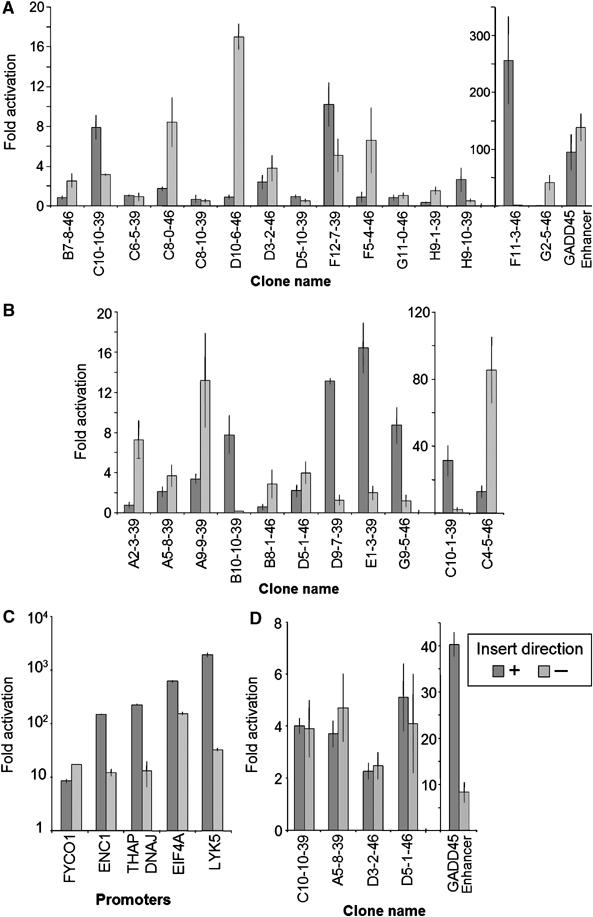
Functional analysis of novel TBP-binding sites. Genomic DNA fragments containing novel TBP-binding sites were cloned in both directions in front of promoter-less (A–C) or SV-40 promoter containing (D) reporter-gene plasmid vectors and transfected into U2OS cells; ratios of transcription activity of the reporter gene over empty vector are shown. (A) Novel TBP-binding sites located in introns of RNAP II genes. The ‘+' and ‘–' refer to the direction of transcription of the gene (sense and antisense, respectively). (B) Novel TBP-binding sites lacking gene/mRNA annotation. The ‘+' and ‘–' refer to the direction of the sequence (UCSC genome browser definition) with respect to the reporter gene. (C) Promoters of RNAP II-transcribed genes. (D) Enhancer assay: analysis of the targets in a reporter vector with SV-40 promoter. The ‘+' and ‘–' refer to the direction of the sequence (UCSC genome browser definition) with respect to the reporter gene.
To test the promoter activity of the novel sites in their genomic location in vivo, we used strand-specific RT qPCR (sts-RT qPCR) to identify transcripts originating from the TBP-binding sites. Primers were designed in close proximity (about 500 bp) around TBP-binding sites (Figure 4A). The ratio between relative RNA levels for two probes targeting the same strand (A/C and D/B, respectively) was used to assess the directionality of transcription: high A/C and D/B ratios suggest specific transcription started at novel sites in ‘–' and ‘+' direction, respectively. High ratios for both A/C and D/B would imply bidirectional transcription.
Figure 4.
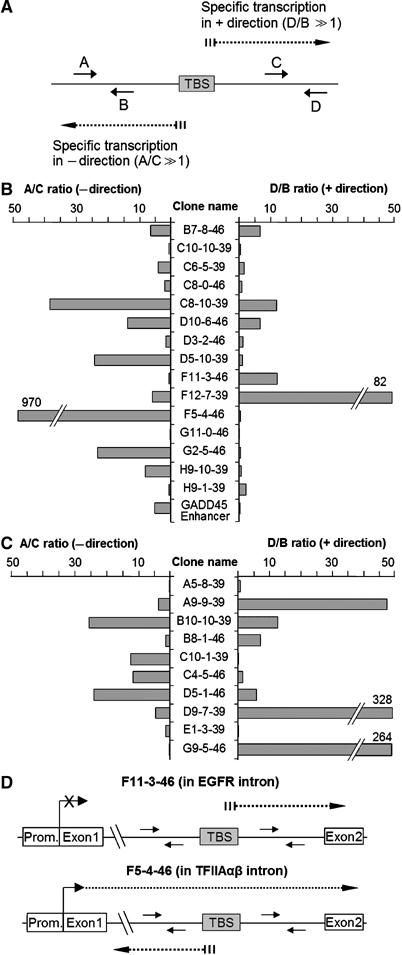
Analysis of strand-specific transcripts at novel TBP-binding sites. (A) Schematic presentation of novel TBP-binding site and location of sts-RT qPCR probes. Dotted lines indicate putative transcripts initiated at the TBP-binding site. The probes named A and C are complementary to transcripts in the ‘–' direction, and probes B and D to transcripts in the ‘+' direction. The A/C and D/B ratios between RNA levels were taken to assess transcription specifically started within the novel TBP-binding sites in ‘–' and ‘+' directions, respectively. (B) The ratios A/C (left part) and D/B (right part) for TBP-binding sites in introns. Transcriptional directions indicated with ‘+' and ‘–' refer to sense and antisense direction. (C) Same as (B) measured for the TBP-binding sites loci lacking a gene annotation. The directions of transcription indicated with ‘+' and ‘–' refer to UCSC genome browser definition. (D) Schematic presentation of transcripts from the EGFR and TFIIAαβ genes.
As presented in Figure 4B and C, about half of the targets (17/26) yielded transcripts originating around the novel TBP-binding sites (ratios >5-fold). A good correspondence to reporter assay was observed for 12 targets. For example, high D/B ratio was obtained at intronic EGFR site (F11-3-46) suggesting that transcription is initiated at the TBP-binding site in sense direction (collinear with the gene) (Figure 4D) corroborating and extending its assignment as an alternative promoter. Similarly, high A/C ratio obtained at the intronic site in TFIIAαβ gene (F5-4-46) underscores the presence of antisense transcription (Figure 4D) as also deduced form the reporter assay. Interestingly, a novel site located in a centromeric satellite region (B10-10-39) displayed both high A/C and D/B ratios suggesting bidirectional transcription.
Collectively, these data provide strong evidence that the majority of the novel TBP-binding sites function as genuine promoters.
Correlation profiling analysis
To gain insight into the occupancy of the TBP-binding sites by general transcription factors in relation to their function in transcription, we performed correlation analyses determining the degree of linear relationship between variables, that is, between ChIP/input ratios for each of the different factors. The correlation values were calculated between every possible pair of factors on all the targets and were then color visualized (Figure 5A). To bring the multitude of values into an order, clustering algorithms were applied to calculate hierarchical dendrogram based on the difference between correlation values (Figure 5A and B); the length of the branches is used as measure of the degree of difference (similarity–dissimilarity). This type of analysis can be used to compare occupancy profiles: factors co-recruited to the same target sets will show a high correlation and will cluster together, whereas factors that do not co-occupy the same target sets will have a low correlation and will be placed more distant from each other.
Figure 5.
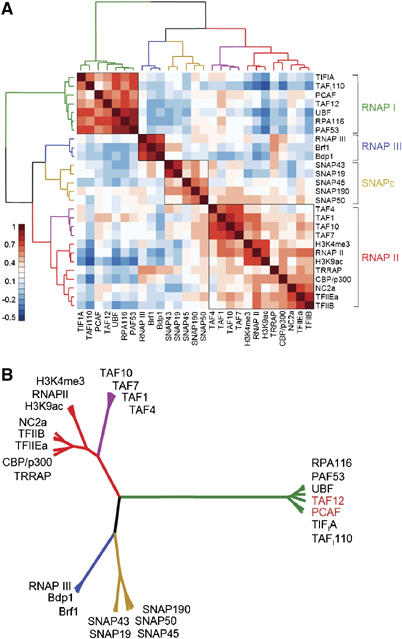
Correlation analyses of ChIP-on-chip data sets. Pearson correlation values were calculated on entire ChIP-on-chip data set (25 antibodies against general transcription factors and two active histone marks) and structured by hierarchical clustering (Ward's). The resulting dendrogram is represented as a cluster (A) and a rooted tree (B). The latter is combined with color-visualized correlation values as depicted. The branches corresponding to the different clusters are color-coded. TBP was excluded from the analysis.
Analysis of the entire ChIP-on-chip data set revealed four major clusters (Figure 5A and B). RNAP III-specific factors such as Bdp1 and Brf1 found in TFIIIB and the RNAP III subunit RPC1 clustered in one branch and showed a negative correlation with RNAP I and II factors. Another branch of the dendrogram consists of subunits of the SNAPc complex that are specifically involved in transcription from small nuclear RNA genes.
The third branch brings together the known RNAP II factors and the two histone marks correlated with active promoters; H3K9ac and H3K4me3 (Berger, 2002; Santos-Rosa et al, 2002). RNAP II closely co-clustered with these histone modifications in line with recent findings (Bernstein et al, 2005; Kim et al, 2005). The transcription coactivator CBP/p300 and the negative cofactor NC2 showed a high correlation with general factors such as TFIIB suggesting that these factors serve general roles in RNAP II transcription. TBP-associated factors (TAFs) were clustered in a distinct sub-branch suggesting that the RNAP II targets are heterogeneous with respect to TAF occupancy.
The RNAP I branch displays short distances between factors (Figure 5A and B) and was the farthest separated and hence the most dissimilar from the other branches which is in good agreement with the PCA analysis (Figure 2B). Surprisingly, the histone acetylase PCAF hitherto known as a subunit of the STAGA/PCAF complex (Vassilev et al, 1998) and TAF12, known as a component of the PCAF and TFIID complexes (Ogryzko et al, 1998), co-clustered with RNAP I-specific factors. The recruitment of these factors—hitherto described as RNAP II-specific—to rDNA units was confirmed by single gene qPCR analysis (Supplementary Figure 5).
Involvement of TAF12 in transcription of rRNA genes
The association of PCAF with rDNA is in accordance with our previous studies showing that PCAF acetylates TAFI68 and stimulates RNAP I transcription in a reconstituted in vitro system (Muth et al, 2001). The presence of an RNAP II-specific TAF at the rDNA promoter was surprising and suggested that TAF12 may play a role in RNAP I transcription. To examine whether TAF12 is associated with the RNAP I-specific TBP-TAFI-complex SL1, we performed GST pull-down assays and measured the interaction of TAF12 with individual subunits of SL1, for example, TBP, TAFI110, TAFI68 and TAFI48. Consistent with published data, TBP was found to associate with GST-TAF12 (Hoffmann and Roeder, 1996). Noteworthy, TAFI48 and TAFI110, but not TAFI68 and the RNAP I transcription factors TIF-IA and UBF, were specifically retained on GST-TAF12 beads, indicating a direct interaction of TAF12 with SL1 (Supplementary Figure 6). The interaction of SL1 and TAF12 was also shown by co-immunoprecipitation experiments. Partially purified SL1 was precipitated with antibodies against TAFI110, and coprecipitated TBP and TAF12 were identified on Western blots. A significant amount of TAF12 coprecipitated with TBP and TAFI110, showing that TAF12 is associated at least with a subpopulation of SL1 in vivo (Figure 6A). Notably, TAF10, another RNAP II-specific TAF, was not detected in the immunoprecipitation.
Figure 6.
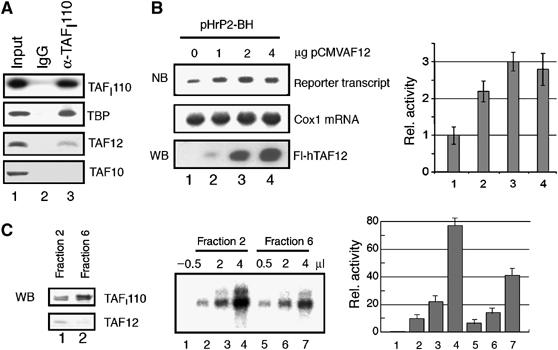
TAF12 associates with SL1 and stimulates rDNA transcription. (A) TAF12 is associated with SL1. HeLa nuclear extracts were fractionated by chromatography on phosphocellulose and SP resins, and SL1 was immunoprecipitated using anti-TAFI110 antibodies (lane 3) or rabbit IgG (lane 2) as a control. The immunoprecipitates were analyzed on Western blots for TBP, TAF12 and TAF10 as indicated. The input (lane 1) contains 50% of the material used for the IP. To monitor the efficiency of TAFI110 precipitation, 10% of the input fraction and 10% of the IP were separated by SDS–PAGE and probed with anti-TAFI110 antibodies (top panel). (B) U2OS cells were cotransfected with 2 μg of the rDNA reporter plasmid pHrP2-BH and increasing amounts of pCMV-FLAG-hTAF12 (indicated on top) in a total amount of 8 μg. Reporter transcripts and cytochrome oxidase 1 (cox 1) mRNA were detected using appropriate 32P-labeled riboprobes and quantified (NB). The expression of Flag-TAF12 was verified on Western blots with anti-FLAG antibodies (WB). The bar diagram represents the relative level of reporter transcripts from three independent experiments. (C) TAF12-containing SL1 fractions stimulate RNAP I transcription in vitro. TAF12 copurifies with transcriptionally active SL1 (left panel). HeLa nuclear extracts were chromatographed on phosphocellulose and S-Sepharose. Individual S-Sepharose fractions (20 μl of fractions 2 and 6, respectively) were probed for the presence of TAFI110 and TAF12 on immunoblots. RNAP I transcription was assayed in a reconstituted system. The reactions were supplemented with SL1 fractions containing detectable amounts of TAF12 (fraction 2) or fractions with trace amounts of TAF12 (fraction 6). In lane 1, no SL1 fraction was added. The bar diagram represents the relative level of transcription from three different experiments.
To directly assess the role of TAF12 in RNAP I transcription, U2OS cells were cotransfected with a human rDNA reporter as well as an expression vector encoding Flag-tagged hTAF12, and the level of reporter transcripts was monitored on Northern blots (Figure 6B). Consistent with TAF12 playing a role in RNAP I transcription, overexpression of Flag-hTAF12 stimulated transcription of the rDNA reporter up to three-fold. Moreover, in vitro transcription assays using an SL1-responsive reconstituted system revealed that SL1 fractions that contain detectable amounts TAF12 supported higher levels of transcription than SL1 fractions without or with low amounts of TAF12 (Figure 6C). These results provide compelling evidence that TAF12—in addition to its established role in RNAP II transcription—serves a function in transcription by RNAP I.
Distinct factor profiles on CpG and non-CpG targets
To assess whether the DNA sequence composition such as CpG content specifies transcription factor occupancy or utilization, we filtered out RNAP I and III targets and sorted remaining targets enriched for TBP into two bins: overlapping or non-overlapping with CpG islands. A small number of closely positioned RNAP II/RNAP III promoters remained in the subsequent analyses. About half of the targets ended up in the CpG island bin in line with estimations of the number of genomic CpG island promoters (56%) (Antequera and Bird, 1994). The vast majority of known, annotated RNAP II promoters (84%) were found in the CpG islands bin (Figure 7A). Besides a small number of annotated RNAP II promoters, the non-CpG island bin contained the majority of TBP target sites located in introns or such lacking a gene annotation. Based on the PCA and functional analysis (Figures 2A, 3 and 4), these TBP-binding sites were classified as RNAP II regulatory regions.
Figure 7.
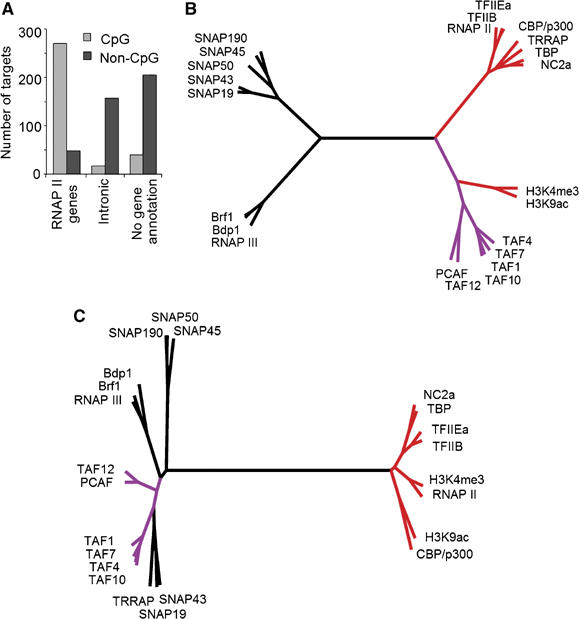
Distinct correlation profiles for CpG and non-CpG island RNAP II targets. (A) Distribution of CpG and non-CpG island targets in the different annotation groups. The CpG islands database was obtained from the UCSC genome browser. (B, C) Rooted trees represent Ward's hierarchical clustering of Pearson correlation values calculated on CpG (B) and non-CpG (C) targets. The branches of TAFs and other general transcription factors are colored in purple and red, respectively.
Correlation analysis of the CpG-island bin revealed a dendrogram with four main branches (Figure 7B): two closely positioned branches containing the general RNAP II factors (marked in red) and the TAFs (marked in purple). The two other branches were placed opposite to the RNAP II factors and they contained clusters of SNAPc proteins and RNAP III factors. These branches were well structured because of the presence of snRNA genes as well as juxtaposed RNAP II and III promoters. The dendrogram calculated for targets in the non-CpG bin revealed two opposing branches: one branch was well structured and contained the general RNAP II factors (Figure 7C). Surprisingly, TAFs did not cosegregate with the RNAP II factors but were placed at a large distance in the opposing branch that was not well structured and contained RNAP III factors and SNAPc proteins. The opposite positioning of TAFs relative to the other RNAP II factors on the non-CpG TBP-binding sites suggests that TAFs are not efficiently recruited to the non-CpG targets.
Collectively, these data suggest different functional interactions of TAFs and other RNAP II factors on non-CpG versus CpG island targets pointing to distinct mechanisms of transcription initiation.
Discussion
In this study, we used ChIP followed by cloning of the precipitated genomic DNA fragments to identify in vivo TBP-binding sites. The vast majority (∼90%) of the cloned and filtered genomic fragments appear to be true in vivo TBP-binding sites (Figure 2A). Sequencing and annotation of these sites revealed that a remarkably large fraction (49%) is located in introns of known genes and in genomic locations lacking a gene annotation (Figure 1B). PCA placed these novel TBP-binding sites in the same space as annotated RNAP II targets.
Functional reporter assays revealed that the majority of these novel TBP-binding sites displayed unidirectional transcriptional activity (Figure 3A and B) providing evidence that these novel sites function are genuine promoters. sts-RT qPCR supported and extended this conclusion showing that transcripts could originate from the novel TBP-binding sites in vivo (Figure 4). Collectively, the data provide strong evidence that the majority of novel TBP-binding sites are true functional promoters.
A number of the cloned TBP-binding sites displayed significant direction-independent activation of SV-40 promoter fulfilling the criteria of enhancers. The fact that the well-known GADD45 enhancer was also among our TBP-binding sites reinforces the notion that our approach also yielded enhancers. The presence of promoter-specific factors such as TBP and RNAP II on enhancers can be explained by DNA looping (Tolhuis et al, 2002) and crosslinking via protein–protein contacts. An alternative and very intriguing explanation is that a subset of general transcription factors may be directly recruited and assembled onto enhancers and subsequently handed over to the promoter or that some ‘enhancers' act as promoters that may help to maintain an open chromatin structure (Szutorisz et al, 2005).
The remarkably large fraction of novel functional TBP-binding sites in our library indicates that the genome contains many more promoters that have not been identified experimentally or by current annotation algorithms. If this proportion holds true for the entire human genome (⩾50%), the number of functional TBP-binding sites may exceed ∼80 000 which is roughly 2 × more than the number of genes annotated to date. Taking into account the multitude of different tissues and developmental stages, the total number of promoters and enhancers is likely to be significantly larger. Our observations corroborate and extend recent transcriptome and ChIP-on-chip studies that reached similar conclusions (Kapranov et al, 2002; Bertone et al, 2004; Cawley et al, 2004; Cheng et al, 2005; Kim et al, 2005).
Integrated analysis of transcription factors binding profiles
We performed a comprehensive ChIP-on-chip study involving 26 general factors and two histone marks on ∼1000 experimentally derived TBP-binding sites. To uncover properties that cannot be extracted from individual subsets of data, we analyzed the ChIP-on-chip data in an integrated manner rather than as a collection (summation) of datasets for individual factors.
PCA revealed a high intrinsic structure in the data set and segregated the TBP-binding sites into three distinct clusters. One of the advantages of PCA for ChIP-on-chip data analysis is that a ‘true–false' threshold does not need to be established for each antibody. The presence of negatives in the data set does not obscure the analysis; on the contrary, it provides a higher level of overall variance that favors segregation of the most similar variables. The segregation of the three major gene classes transcribed by RNAP I, II and III indicates regulation by highly characteristic and distinct combinations of transcription factors.
We also used correlation profiling that calculates the degree of linear relationship between two multitudes of data points, in our case between ChIP/input ratios for different transcription factors. When applied to ChIP-on-chip data, it can be used to determine the degree of similarity/dissimilarity between transcription factors on the basis of their binding profiles on a large number of targets. Like in PCA, a ‘true–false' threshold does not need to be established. To organize the multitude of correlation values of the entire data set, we used hierarchical clustering algorithms to calculate the differences between correlations and to convert them into distances so as to build a cluster dendrogram. Analysis of the entire data set revealed four major branches (Figure 5A and B) corresponding to RNAP I, II, and III and SNAPc target genes providing evidence for the involvement of distinct sets of factors in transcription by the three RNA polymerases in vivo. The branches had compact substructures with the exception of the RNAP II branch. The latter displayed a more open branch structure that likely reflects the broad assortment and heterogeneity of multiprotein complexes involved in transcription initiation by RNAP II (Lee and Young, 2000; Naar et al, 2001) as well as the temporally ordered recruitment of factors to heterogeneous RNAP II promoters (Cosma, 2002). High correlation values were obtained for proteins that simultaneously bind the same genomic locations and make long-lived contacts, such as in biochemically stable multiprotein complexes, because they can be co-crosslinked with high probability and efficiency. For example, RPA116 and PAF53 that are both subunits of RNAP I (Seither et al, 1997) or the Bdp1 and Brf1 subunits of the TFIIIB complex involved in RNAP III transcription (Schramm and Hernandez, 2002) have very high correlation values and are placed at short distances from each other in the dendrogram (Figure 5B). The distance between RPA116/PAF53 and Bdp1/Brf1 is, however, very far because the probability and efficiency of co-crosslinking is low or absent as the proteins are part of functionally unrelated biochemical complexes and their genomic binding site repertoires do not overlap. Extending the same logic to TAF12 and PCAF that tightly cluster in the RNAP I branch implies that they can be part of a stable complex that is distinct from the PCAF/STAGA/TFTC complexes. In line with these observations, PCAF has previously been shown by us to acetylate TAFI68 and stimulate transcription of rDNA gene in a reconstituted transcription system (Muth et al, 2001). Here, we provide evidence that TAF12 is also involved in RNAP I transcription (Figure 6). First, overexpression of TAF12 stimulated RNAP I transcription in a cell-based reporter assay. Second, RNAP I transcription was stimulated after supplementing a reconstituted transcription system with a TAF12-containing SL1 fraction. Finally, TAF12 was found in endogenous SL1 complex and physically bound at rDNA promoter. Note that the cluster analysis performed on the subset of CpG promoters resulted in TAF12 and PCAF cluster together with other RNAP II TAFs in line with their role in TFIID and SAGA (Figure 7B). Thus, our data show that TAF12 and most likely also PCAF have dual functions in RNAP I and II transcription.
Distinct clustering patterns on CpG and non-CpG RNAP II targets
The primary DNA sequence of promoters plays an important role in recruitment of specific transcription factors. Multiple core promoter elements that are specifically bound by general transcription factors during pre-initiation complex formation have been described. Whether a particular factor is involved in transcription of a given gene class has not yet been addressed in a comprehensive manner in higher eukaryotes.
To assess whether the transcription factor occupancy on RNAP II genes involves distinct subsets of general transcription factors, we performed correlation analysis separately on non-CpG targets and on the targets located in CpG islands. Our correlation dendrograms showed a remarkable difference in the clustering and positioning of TAFs (Figure 7B and C); TAFs were placed at a larger distance from other basal RNAP II factors on non-CpG islands but clustered close on CpG island targets. Our data suggest that TAFs and the other general factors are not or very transiently co-recruited to non-CpG promoters and, therefore, are not efficiently co-crosslinked. TAFs appear to be (more) stably recruited to CpG island promoters, perhaps because these promoters are more active. This assumption is in line with our finding that many novel non-CpG sites show slightly reduced RNAP II occupancy. The virtually identical occupancy values for TBP and TFIIB on CpG versus non-CpG targets suggest that these novel TBP-binding sites are occupied with the RNAP II machinery. Reporter assays show that most of the non-CpG targets comprise transcription-competent promoters. Thus, it is likely that in analogy to yeast (Basehoar et al, 2004; Huisinga and Pugh, 2004) at least two major pathways of transcription initiation by RNAP II exist in mammals. It will be interesting to extend these observation genomewide and to perform time-resolved ChIP-on-chip following gene activation to unravel the order of factor recruitment.
Materials and methods
ChIP and ChIP cloning
U2OS cells were crosslinked with 1% formaldehyde for 30 min at room temperature, quenched with 0.125 M glycine and washed at 4°C with three buffers: (i) PBS, (ii) buffer of composition 0.25% Triton X-100, 10 mM EDTA, 0.5 mM EGTA, 20 mM HEPES pH 7.6 and (iii) 0.15 M NaCl in HEG buffer (1 mM EDTA, 0.5 mM EGTA, 20 mM HEPES pH 7.6). Cells were then suspended in ChIP incubation buffer (0.15% SDS, 1% Triton X-100, 150 mM NaCl, HEG) and sheared using a Branson-250 sonicator. Sonicated chromatin was centrifuged for 5 min and then incubated overnight with purified anti-TBP antibody (Diagenode) and protein A/G beads (Santa Cruz). Beads were washed six times with different buffers at 4°C: two times with solution of composition 0.1% SDS, 0.1% DOC, 1% Triton, 150 mM NaCl, HEG, one time with the solution same as before but with 500 mM NaCl, one time with solution of composition 0.25 M LiCl, 0.5% DOC, 0.5% NP-40, HEG and two times with HEG. Precipitated chromatin was eluted with 400 μl of elution buffer (1% SDS, 0.1 M NaHCO3), incubated at 65°C for 4 h in the presence of 200 mM NaCl, phenol extracted and precipitated with 20 μg of glycogen at −20°C overnight. For sequential ChIP, chromatin was eluted with a small volume of elution buffer, diluted to specific incubation conditions and processed same as that of the first IP with the same amount of antibody.
For cloning, ChIP was performed with 108 cells and DNA obtained after the second ChIP was extracted and treated with T4 DNA polymerase to generate blunt ends, purified, ligated into a pBluescript vector and used for transformation of Escherichia coli.
qPCR
ChIP experiments were analyzed by qPCR with specific primers using a SYBR green kit (Applied Biosystems). Efficiency of ChIP was calculated as percentage of input and specificity—as folds over negative controls (transcriptionally silent genomic loci such as promoters and coding regions of β-globin and myoglobin genes). Primers for qPCR were designed with Primer Express and verified by in silico PCR (genome.cse.ucsc.edu/cgi-bin/hgPcr) and by pPCR as amplifying a single specific amplicon. PCR efficiency of primers was calculated with series of 10-times dilutions and accepted when found to be reliable (20.15). Primer sequences are available as Supplementary Table II.
TBP-binding site microarray and ChIP-on-chip
Inserts from the clones obtained in TBP ChIP-cloning procedure were PCR-amplified, purified and used for sequencing and printing on glass slides. Every target was printed six times in different parts of the slide to ensure robustness of the microarray data.
For ChIP-on-chip experiments, ChIP'ed and input DNA was amplified by LM-PCR as described (Ren et al, 2000), labeled with Cy5 and Cy3 using random priming, purified and dissolved in hybridization buffer (33% formamide, 2.5 × SSC, 6.6% dextran sulfate). Hybridization was performed overnight at 45°C. Slides were washed at room temperature for 20 min with 0.1 × SSC, scanned and analyzed. Median values were calculated for six spots printed on array for each target and the ratios from two hybridizations were averaged. Targets with low intensity (below 2SD of local background) were filtered. The data is available from GEO under accession number GSE6738.
ChIP-on-chip data analysis
The ChIP/input ratios were normalized to the median of four reference controls (promoter and coding regions of myoglobin and β-globin genes which were validated as negative by single gene qPCR for the antibodies). PCA and correlation analyses were performed using R software package (www.R-project.org) on data from non-redundant targets enriched for TBP >2-fold. In the final data matrix, all factors were rescaled to have zero mean and unit variance. Up to four PCs were considered in PCA. Pearson correlations were calculated for every pair of transcription factors and hierarchical clustering on these values was performed using Ward's clustering and average linkage. Stability of the clustering dendrograms was established in two ways. First, comparison of structures obtained with Ward's clustering and average linkage revealed significant similarity when calculated at level of 3–5 clusters. Second, leaving each factor out in turn revealed no structural changes in most cases, only for very few factors this resulted in minor changes of the clustering trees.
Promoter/enhancer gene-reporter assays
Genomic fragments of about 1 kb containing the validated TBP-binding sites were PCR-amplified and ligated in front of the reporter gene of pGL3-basic (promoter-less) or pGL3-promoter (SV-40 promoter) vectors. These constructs were transfected into U2OS cells together with pSV2-CAT by calcium phosphate method, gene-reporter activity was measured and normalized to CAT activity. The values were averaged from 2 to 6 replicates. The baseline of reporter gene expression was determined as average of eight transfections of the empty pGL3 vectors.
sts-RT qPCR
One microgram of total RNA isolated from U2OS cells with Trizol reagent (Invitrogen) was treated with DNaseI at 37°C for 20 min followed by inactivation at 80°C for 20 min. Ten picomoles of specific probe was added and denatured at 75°C for 10 min. To obtain high specificity, the reaction was not placed on ice but instead, the temperature was ramped 0.3°C/s down to 60°C and 8 μl of reaction mix prewarmed at 60°C was added (3 μl of 5 × first strand buffer (Invitrogen), 2 μl of 0.1 M DTT, 1 μl of 10 μM each dNTP, 2 μl of water), mixed and incubated for 2 min. Then 1 μl of heat-stable reverse transcriptase (SuperScript III, Invitrogen) was added, the samples were mixed and incubated at 60°C for 40 min. Then the samples were incubated at 95°C for 15 min to inactivate reverse transcriptase, treated with RNaseH+RNaseA at 37°C for 20 min, diluted 2 × and 5 μl from the samples were used for qPCR. The results were normalized (% of GAPDH mRNA). The analysis has been repeated twice with different RNA preparations and the results were averaged.
Functional analysis of TAF12
The cDNA encoding TAF12 was inserted into the plasmids pRc/CMV-Flag (Voit et al, 1999) and pGEX-4T3. For reporter assays, 3 × 105 U2OS cells were cotransfected with a total amount of 8 μg of plasmid DNA including 2 μg of the rDNA reporter plasmid pHrP2-BH, 1 μg of pEGFP, to monitor transfection efficiency at the same level, and different amounts of pRc/CMV-Flag-TAF12. RNA was isolated 40 h after transfection, and 5 μg of total RNA were subjected to Northern blot analysis (Voit et al, 1999). To normalize for RNA loading, the Northern blots were re-hybridized with a riboprobe for cytochrome c oxidase 1 mRNA.
SL1 was partially purified from HeLa nuclear extracts by sequential chromatography on phosphocellulose P11 (1.5 M KCl fraction) and S-Sepharose (700 mM fraction). SL1 was immunoprecipitated with anti-TAFI110 antibodies for 4 h at 4°C in IP buffer (20 mM Tris–HCl, pH 7.9, 150 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, 20% glycerol, 1 mM PMSF, 0.2% NP-40 and protease inhibitors). Mock IP was carried out in the presence of rabbit IgGs (Dianova). Immunoprecipitated proteins were bound to Protein A-Sepharose (1 h at 4°C), and subjected to Western blotting.
For GST pull-down assays, GST and GST-TAF12 were immobilized on GT-Sepharose and incubated with 20 μl of reticulocyte lysates (TNT, Promega) containing in vitro synthesized 35S-labeled transcription factors and 35S-methionine. After incubation in buffer AM-150/0.2% NP-40 (substituted with protease inhibitors) for 4 h at 4°C, beads were washed and eluted proteins were separated by SDS–PAA PAGE and visualized by a PhosphorImager.
In vitro transcription reactions (25 μl) contained 20 ng of linearized template pHrP2-BH, 2 μl of TIF-IA/TIF-IC (Q-Sepharose-fraction), 10 ng of recombinant Flag-UBF1 purified from Sf9 cells (Voit et al, 1999), 0.5–2 μl of SL1 (S-Sepharose fraction), 4 μl of RNA polymerase I (H-400 fraction) and transcription buffer supplemented with ribonucleotides as described (Muth et al, 2001).
Antibodies
TAF4, -7 and -12 antibodies were kindly provided by Irwin Davidson; TAF10 and TRRAP by Laszlo Tora; PCAF by Yoshihiro Nakatani; NC2a by Michael Meisterernst. The other antibodies were purchased: TAF1 (sc-735), CBP/p300 (sc-369), TFIIEa (sc-237), RNAP II N-terminus of RPB1 subunit (sc-899), E2F1 (sc-251), E2F4 (sc-1082) from Santa Cruz; RNAP II CTD (8wg16) and phosphorylated CTD at Ser5 (H14) from BABCO; H3K9ac from Upstate and H3K4me3 from Abcam.
Supplementary Material
Supplementary Figures and Tables
Acknowledgments
We are very grateful to our colleagues Irwin Davidson, Laszlo Tora, Yoshihiro Nakatani and Michael Meisterernst for antibodies. We thank Vera van Noort and Martijn Huynen for help in data analysis. We thank our colleagues for valuable suggestions and discussions. This work was supported by National Scientific Organization (NGI 050-71-016), National Cancer Foundation (KWF-KUN 2005-3347) and HEROIC, an Integrated Project funded by the European Union under the 6th Framework Programme (LSHG-CT-2005-018883).
References
- Antequera F, Bird A (1994) Predicting the total number of human genes. Nat Genet 8: 114. [DOI] [PubMed] [Google Scholar]
- Basehoar AD, Zanton SJ, Pugh BF (2004) Identification and distinct regulation of yeast TATA box-containing genes. Cell 116: 699–709 [DOI] [PubMed] [Google Scholar]
- Berger SL (2002) Histone modifications in transcriptional regulation. Curr Opin Genet Dev 12: 142–148 [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, Bekiranov S, Bailey DK, Huebert DJ, McMahon S, Karlsson EK, Kulbokas EJ III, Gingeras TR, Schreiber SL, Lander ES (2005) Genomic maps and comparative analysis of histone modifications in human and mouse. Cell 120: 169–181 [DOI] [PubMed] [Google Scholar]
- Bertone P, Stolc V, Royce TE, Rozowsky JS, Urban AE, Zhu X, Rinn JL, Tongprasit W, Samanta M, Weissman S, Gerstein M, Snyder M (2004) Global identification of human transcribed sequences with genome tiling arrays. Science 306: 2242–2246 [DOI] [PubMed] [Google Scholar]
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R (2006) Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441: 349–353 [DOI] [PubMed] [Google Scholar]
- Cam H, Balciunaite E, Blais A, Spektor A, Scarpulla RC, Young R, Kluger Y, Dynlacht BD (2004) A common set of gene regulatory networks links metabolism and growth inhibition. Mol Cell 16: 399–411 [DOI] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, Wheeler R, Wong B, Drenkow J, Yamanaka M, Patel S, Brubaker S, Tammana H, Helt G, Struhl K, Gingeras TR (2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 116: 499–509 [DOI] [PubMed] [Google Scholar]
- Cheng J, Kapranov P, Drenkow J, Dike S, Brubaker S, Patel S, Long J, Stern D, Tammana H, Helt G, Sementchenko V, Piccolboni A, Bekiranov S, Bailey DK, Ganesh M, Ghosh S, Bell I, Gerhard DS, Gingeras TR (2005) Transcriptional maps of 10 human chromosomes at 5-nucleotide resolution. Science 308: 1149–1154 [DOI] [PubMed] [Google Scholar]
- Cosma MP (2002) Ordered recruitment: gene-specific mechanism of transcription activation. Mol Cell 10: 227–236 [DOI] [PubMed] [Google Scholar]
- Hallikas O, Palin K, Sinjushina N, Rautiainen R, Partanen J, Ukkonen E, Taipale J (2006) Genome-wide prediction of mammalian enhancers based on analysis of transcription-factor binding affinity. Cell 124: 47–59 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Roeder RG (1996) Cloning and characterization of human TAF20/15 Multiple interactions suggest a central role in TFIID complex formation. J Biol Chem 26: 18194–18202 [DOI] [PubMed] [Google Scholar]
- Huisinga KL, Pugh BF (2004) A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae. Mol Cell 13: 573–585 [DOI] [PubMed] [Google Scholar]
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR (2002) Large-scale transcriptional activity in chromosomes 21 and 22. Science 296: 916–919 [DOI] [PubMed] [Google Scholar]
- Kim TH, Barrera LO, Zheng M, Qu C, Singer MA, Richmond TA, Wu Y, Green RD, Ren B (2005) A high-resolution map of active promoters in the human genome. Nature 436: 876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, Young R (2000) Transcription of eukaryotic protein-coding genes. Annu Rev Gen 34: 77–137 [DOI] [PubMed] [Google Scholar]
- Muth V, Nadaud S, Grummt I, Voit R (2001) Acetylation of TAF(I)68, a subunit of TIF-IB/SL1, activates RNA polymerase I transcription. EMBO J 20: 1353–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naar AM, Lemon BD, Tjian R (2001) Transcriptional coactivator complexes. Annu Rev Biochem 70: 475–501 [DOI] [PubMed] [Google Scholar]
- Ogryzko VV, Kotani T, Zhang X, Schiltz RL, Howard T, Yang XJ, Howard BH, Qin J, Nakatani Y (1998) Histone-like TAFs within the PCAF histone acetylase complex. Cell 94: 35–44 [DOI] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Robert F, Wyrick JJ, Aparicio O, Jennings EG, Simon I, Zeitlinger J, Schreiber J, Hannett N, Kanin E, Volkert TL, Wilson CJ, Bell SP, Young RA (2000) Genome-wide location and function of DNA binding proteins. Science 290: 2306–2309 [DOI] [PubMed] [Google Scholar]
- Ruppert SM, McCulloch V, Meyer M, Bautista C, Falkowski M, Stunnenberg HG, Hernandez N (1996) Monoclonal antibodies directed against the amino-terminal domain of human TBP cross-react with TBP from other species. Hybridoma 15: 55–68 [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411 [DOI] [PubMed] [Google Scholar]
- Schramm L, Hernandez N (2002) Recruitment of RNA polymerase III to its target promoters. Genes Dev 16: 2593–2620 [DOI] [PubMed] [Google Scholar]
- Seither P, Zatsepina O, Hoffmann M, Grummt I (1997) Constitutive and strong association of PAF53 with RNA polymerase I. Chromosoma 106: 216–225 [DOI] [PubMed] [Google Scholar]
- Sims RJ III, Mandal SS, Reinberg D (2004) Recent highlights of RNA-polymerase-II-mediated transcription. Curr Opin Cell Biol 16: 263–271 [DOI] [PubMed] [Google Scholar]
- Sutton G, White O, Adams M, Kerlavage A (1995) TIGR Assembler: a new tool for assembling large shotgun sequencing projects. Genome Sci Technol 1: 9–19 [Google Scholar]
- Szutorisz H, Dillon N, Tora L (2005) The role of enhancers as centres for general transcription factor recruitment. Trends Biochem Sci 30: 593–599 [DOI] [PubMed] [Google Scholar]
- Tolhuis B, Palstra RJ, Splinter E, Grosveld F, de Laat W (2002) Looping and interaction between hypersensitive sites in the active beta-globin locus. Mol Cell 10: 1453–1465 [DOI] [PubMed] [Google Scholar]
- Tupler R, Perini G, Green MR (2001) Expressing the human genome. Nature 409: 832–833 [DOI] [PubMed] [Google Scholar]
- Vassilev A, Yamauchi J, Kotani T, Prives C, Avantaggiati ML, Qin J, Nakatani Y (1998) The 400 kDa subunit of the PCAF histone acetylase complex belongs to the ATM superfamily. Mol Cell 2: 869–875 [DOI] [PubMed] [Google Scholar]
- Voit R, Hoffmann M, Grummt I (1999) Phosphorylation by G1-specific cdk–cyclin complexes activates the nucleolar transcription factor UBF. EMBO J 18: 1891–1899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie X, Lu J, Kulbokas EJ, Golub TR, Mootha V, Lindblad-Toh K, Lander ES, Kellis M (2005) Systematic discovery of regulatory motifs in human promoters and 3 UTRs by comparison of several mammals. Nature 434: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Tables