

Abstract
The exonuclease-based real-time polymerase chain reaction (PCR) exploits 5′→3′ exonuclease activity of Taq polymerase and measures PCR product accumulation as the reaction proceeds through a dual-labeled fluorogenic probe. The utility of this exonuclease-based PCR assay as a rapid alternative to conventional PCR for follicular lymphoma-associated t(14;18)(q32;q21) was evaluated in this study. The specificity of the assay for t(14;18) involving bcl-2 and immunoglobulin heavy-chain joining region (JH) genes was assessed by analyzing DNA from 53 patients (38 B-cell non-Hodgkin’s lymphomas and 15 nonneoplastic proliferations) and correlating the exonuclease PCR data with conventional PCR results. bcl-2/JH fusion sequences were detected by exonuclease-based PCR in 24 of 25 cases shown to be bcl-2 rearranged by conventional PCR. Fusion sequences were not detected in patients who were negative by conventional PCR. The overall concordance between the two assays was 98% (52 of 53 cases concordant positive or negative). In a serial dilution study using t(14;18)-positive cell line DNA, exonuclease-based PCR detected fusion sequences at DNA concentrations of 5 pg, equivalent to 0.6 to 0.8 genomes per reaction. Thus, this study demonstrated that exonuclease-based PCR for t(14;18) is both specific and highly sensitive. The elimination of the post-PCR amplicon detection steps and the ability to quantitate the input target DNA sequences make this assay ideal for routine diagnostics and monitoring minimal residual disease.
The conventional polymerase chain reaction (PCR) assay commonly utilized to detect t(14;18)(q32;q21) associated with follicular lymphoma is a highly sensitive and powerful technique. 1-8 However, post-PCR amplicon detection steps such as gel electrophoresis and Southern blotting are time consuming and tedious and generally involve radioactive material. We describe the application of a recently developed 5′ exonuclease-based PCR assay 9-12 that enables amplicon detection in real time without downstream processing for t(14;18) involving bcl-2 and the immunoglobulin heavy chain joining region genes. This assay exploits the 5′–3′ exonuclease activity of Taq polymerase and integrates fluorogenic PCR with a laser-based instrumentation system, the PRISM 7700 Sequence Detector (PE Applied Biosystems, Foster City, CA), to detect and quantitate specific PCR amplicons as the reaction proceeds. 13-15 In this novel assay, a nonextendable oligonucleotide probe, positioned downstream of one of the primers, is labeled with a reporter fluorescent dye at the 5′ end and a quencher fluorescent dye at the 3′ end and is included in each PCR assay. During amplification, annealing of the probe to its target sequence generates a substrate that is cleaved by the 5′ exonuclease activity of Taq DNA polymerase when the enzyme extends the primer into the region of the probe. When the probe is intact, the reporter dye emission is quenched due to the physical proximity of the reporter and quencher dyes. 10,16 However, the 5′ exonuclease-mediated release of the reporter dye from the probe during the extension phase results in an increase in the fluorescent signal of the reporter dye. This increase in fluorescence intensity is proportional to the amount of amplicon produced and is monitored in real time during PCR amplification using the the PRISM 7700 Sequence Detector. Because of the need for rapid detection techniques in molecular diagnostics, we decided to investigate the utility of this technique in the detection of t(14;18) in lymphoma cases.
Materials and Methods
Specimens from histologically and immunophenotypically characterized B-cell non-Hodgkin’s lymphomas (38 cases) and nonneoplastic proliferations (15 cases) were selected for the exonuclease-based PCR analysis. All samples had been analyzed previously with the conventional PCR assay. High-molecular weight DNA was isolated from fresh or frozen lymph node biopsies, bone marrow, or peripheral blood specimens in accordance with standard proteinase K digestion and organic extraction procedures.
Conventional PCR Assay
The conventional PCR assay for the detection of t(14;18) was performed according to the previously described method, 17 with slight modifications using primers for the bcl-2 major breakpoint region (mbr), the bcl-2 minor cluster region (mcr), and the conserved immunoglobulin heavy-chain joining region (JH) (Table 1) ▶ in a Model 9600 thermal cycler (PE Applied Biosystems). The reaction mix contained 500 ng of genomic DNA, 10 mmol/L of Tris-HCl (pH 8.0), 200 μmol/L of each deoxynucleotide triphosphate, 50 mmol/L KCl, 1.5 mmol/L MgCl2, 0.25 μg of each primer, and 1.25 U of AmpliTaq polymerase in a final volume of 50 μl. After heating at 94°C for 7 minutes, the DNA was subjected to 40 cycles of PCR by denaturing at 94°C for 30 seconds, annealing at 54°C for 30 seconds, and extending at 72°C for 1 minute. The last cycle was followed by a 5-minute elongation step. Eighteen microliters of the amplified product was resolved by electrophoresis on 2% Nusieve agarose gels (FMC Bioproducts, Rockland, ME), stained with ethidium bromide, and visualized under ultraviolet light. The amplification products were subsequently transferred to Sure Blot membranes (Oncor, Gaithersburg, MD) according to the manufacturer’s instructions. The membranes were hybridized with a T4-nucleotide kinase 32P-end-labeled internal oligonucleotide probe specific for either bcl-2 mbr or bcl-2 mcr, and the hybridized fragments were detected by autoradiography.
Table 1.
Primers and Probes
| Conventional PCR | |
|---|---|
| Primers | |
| bcl-2 mbr | 5′-TTAGAGAGTTGCTTACGTGGCCTG-3′ |
| bc-2 mcr | 5′-GACTCCTTTACGTGCTGGTACC-3′ |
| JH | 5′-GGACTCACCTGAGGAGACGGTGACC-3′ |
| Probes | |
| bcl-2 mbr | 5′-CTGTTTCAACACAGACCC-3′ |
| bcl-2 mcr | 5′-ACCAGGTNCCTTGGCCCCA-3′ |
| Exonuclease-based PCR | |
| Primers | |
| bcl-2 mbr | 5′-TTAGAGAGTTGCTTTACGTGGCC-3′ |
| bcl-2 mcr | 5′-CCTGGCTTCCTTCCCTCTGT-3′ |
| JH | 5′-ACTCACCTGAGGAGACGGTGAC-3′ |
| Fluorescent dye-labeled probes | |
| bcl-2 mcr | 5′-FAM-TCTCTGIGGAGGAGTGGAAAGGAAGG-TAMR |
| bcl-2 mbr | 5′-FAM-TTTCAACACAGACCCACCCAGAGCC-TAMRA |
5′ Exonuclease-Based PCR Assay
The 5′ exonuclease-based fluorogenic bcl-2/JH PCR was performed in a PRISM 7700 sequence detecter equipped with a 96-well thermal cycler in the presence of 0.15 μmol/L of a bcl-2-specific (either bcl-2 mbr or bcl-2 mcr) oligonucleotide probe (Table 1) ▶ . The probes were labeled with 6-carboxy fluorescein at the 5′ end and 6-carboxy-tetramethyl rhodamine at the 3′ end. Typically, PCR was carried out in a 50-μl mix containing 35 ng of genomic DNA, 1× TaqMan buffer, 4 mmol/L MgCl2, 400 μmol/L dUTP, 200 μmol/L dATP, 200 μmol/L dCTP, 200 μmol/L dGTP, 125 μmol/L of each primer, 0.5 U of AmpErase uracil-N-glycosylase, and 1.25 U of AmpliTaq Gold. A fluorescent dye, 6-carboxy-X-rhodamine, was included in the TaqMan buffer to serve as an internal reference. The DNA was subjected to 40 cycles of a two-step PCR after denaturation at 95°C for 10 minutes. The AmpErase uracil-N-glycosylase was activated before the denaturation step by heating the mix for 2 minutes at 50°C. Each cycle consisted of a 15-second denaturation step at 95°C and a 1-minute combined annealing/extension step at 60°C. The fluorescence emission data for each sample was available for analysis immediately after the completion of PCR.
Collection and Analysis of Fluorescence Emission Data
The increase in fluorescence signal in each of the 96 wells was monitored in real time during PCR amplification by the 7700 Sequence Detector equipped with a charge-coupled device camera. The on-line software system analyzed the spectral data collected during the extension phase of each cycle and plotted fluorescence intensity versus cycle number. The fluorescence data were expressed as Rn or ΔRn, where Rn, or the normalized reporter signal, is the fluorescence signal of the reporter dye divided by the fluorescence signal of the passive, internal reference dye, and ΔRn is Rn minus the baseline signal established in the first few cycles of PCR. Normalization corrects for fluorescent fluctuations resulting from changes in volume or concentration due to pipetting errors.
Design of the Fluorogenic Probe
bcl-2-specific probes that were devoid of self-complementarity and complementarity to either the reverse or the forward primer were designed using the sequences internal to the bcl-2-specific primers. Because uracil-N-glycosylase was used in the reaction mix to prevent carryover contamination, probes and primers were designed to have a melting temperature (TM) greater than 55°C to avoid degradation of newly synthesized amplicons by the enzyme. Furthermore, to ensure proper hybridization of the probe to the target sequence, oligonucleotides with a TM at least 5°C higher than the actual annealing/extension temperature were chosen as probes.
Reagents and PCR Controls
All PCR reagents, including the primers and fluorescent dye-labeled probes, were obtained from PE Applied Biosystems. DNA extracted from lymphoma samples or from cell lines exhibiting reciprocal translocations involving JH exons and either the bcl-2 mbr or the bcl-2 mcr was used as a positive control for bcl-2 mbr and bcl-2 mcr PCR assays, respectively. Human DNA obtained from nonneoplastic tissue was used as a negative control. The presence of amplifiable DNA in the samples was confirmed by co-amplification of β-globin during conventional PCR.
Results
Figure 1 ▶ schematically illustrates the exonuclease-based cleavage of the probe during the extension phase of the PCR. Essentially, annealing of the bcl-2-specific fluorescent probe to its complementary template strand during the course of the amplification generated a substrate suitable for exonuclease attack. Cleavage of the hybridized probe by the exonuclease activity of Taq polymerase during the extension of the bcl-2-specific primer separated the reporter dye from the quencher dye. This separation abrogated the quencher effect on the fluorescence of the reporter dye. Nucleolytic cleavage also removed the probe from the target strand and enabled primer extension to continue. Thus, the presence of the probe in the reaction did not interfere with the overall PCR process. The detection of a specific, amplified DNA product, however, required hybridization of the bcl-2 probe to the template DNA and its subsequent cleavage by Taq polymerase during the extension phase.
Figure 1.
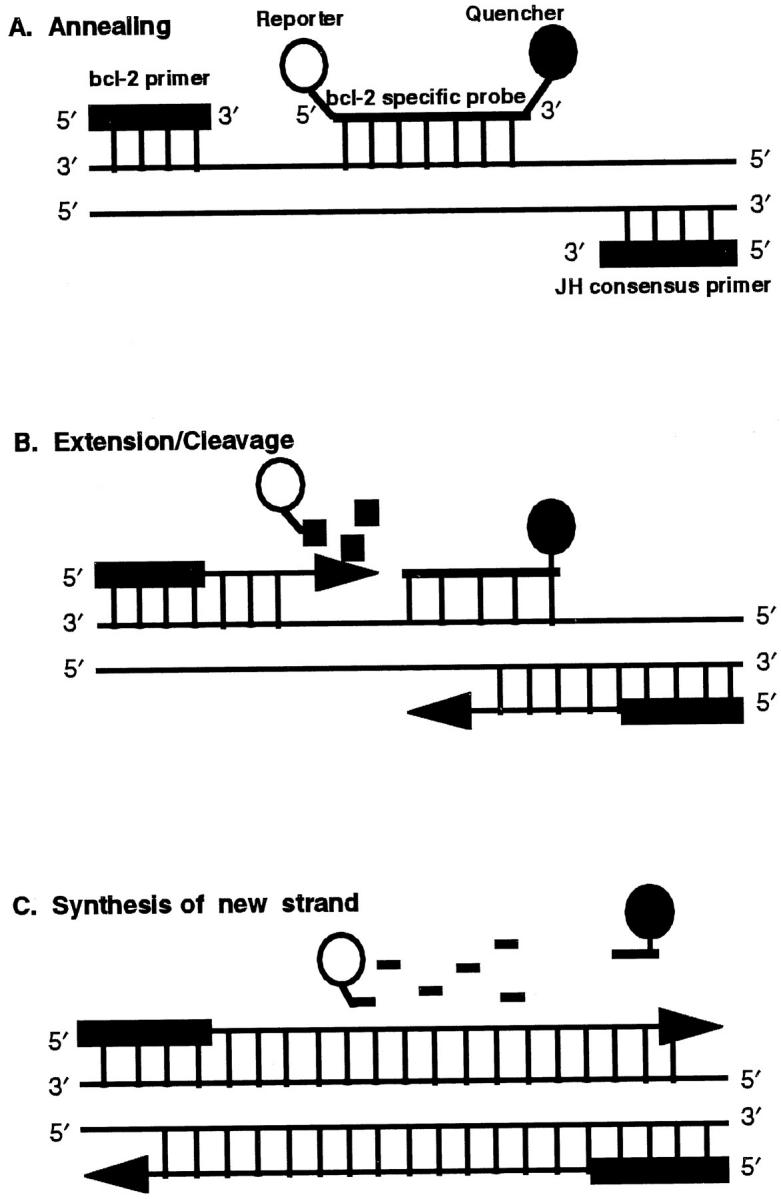
Schematic of sequence-specific annealing and 5′–3′ exonuclease-based cleavage of the fluorescent dye-labeled probe. A: Annealing of the primers and the probe to the target sequence. The probe is labeled with a reporter dye, 6-carboxy fluorescein, at the 5′ end and a quencher dye, 6-carboxy-tetramethyl rhodamine, at the 3′ end. When both dyes are attached to the probe, fluorescence emission of the reporter dye is quenched by the quencher dye. B: Extension of the primer and the initiation of cleavage of the probe at its 5′ end by Taq polymerase. C: Release of the probe from the target strand and completion of the new strand synthesis. The separation of the reporter dye from the quencher dye abrogates the quencher effect and results in an increase in the fluorescence signal of the reporter dye.
Figure 2 ▶ illustrates a typical amplification plot generated using the fluorogenic PCR assay. Initially, DNA from a patient with follicular lymphoma and confirmed t(14;18)(q32;q21) involving the bcl-2 mbr and the JH was analyzed by exonuclease-based PCR. DNA from the patient was diluted with DNA from nonneoplastic tissue to achieve final reaction concentrations of 250, 63, and 15 ng. Reactions with 250 ng of normal DNA served as a negative control. After amplification, the changes in the fluorescence signal of the reporter dye were analyzed and results plotted as ΔRn (y axis) versus cycle number (x axis) (Figure 2) ▶ . No change in the fluorescence signal of the reporter dye was observed in negative control. However, when DNA from a patient with t(14;18) was present, after an initial lag period, an increased fluorescence signal above that of the negative control was observed in all reactions. It became evident from the experiment that the increase in the reporter dye signal caused by the cleavage of the probe during extension of the nonrearranged normal bcl-2 gene (negative control) was below the detection limit of the instrument and thus would not lead to false positive results. Because an exponential increase in fluorescence was observed even at 15 ng of target DNA, we arbitrarily chose to use 35 ng of DNA (unless specified) in subsequent exonuclease-based PCR assays.
Figure 2.

Amplification plot of exonuclease-based PCR assay for bcl-2 mbr/JH sequences generated by the PRISM 7700 Sequence Detector. The graph shows fluorescence emission data (ΔRn) collected during the extension phase of each cycle of the PCR. DNA from a patient with t(14;18)(q32;q21) involving the bcl-2 mbr was subjected to exonuclease-based PCR assay as described in Materials and Methods. The final concentration of patient DNA in PCR reactions is as indicated. DNA (250 ng) derived from nonneoplastic tissue served as the negative control.
The specificity of the assay for the detection of t(14;18) was then tested by analyzing various lymphoma samples and correlating the results with conventional PCR data (Table 2) ▶ . Rearrangements at the bcl-2 mbr were detected in 19 (36%) and 20 (38%) of 53 cases analyzed by exonuclease-based and conventional PCR assays, respectively. Rearrangements involving the bcl-2 mcr were detected in 5 of 5 conventional PCR-positive cases. Conventional PCR-negative cases remained negative by exonuclease-based PCR assay. Review of the conventional PCR results of the one discrepant case revealed that bcl-2 mbr/JH fusion sequences were detected only after Southern blotting and 72-hour exposure, indicating that a low number of initial target sequences were in the sample. This, coupled with the fact that 14 times more DNA (500 versus 35 ng) was used in conventional PCR, may explain the discrepancy in the results of the two assays. Unfortunately, we were unable to repeat the exonuclease-based PCR assay using larger amounts of DNA, as additional DNA was not available from this case.
Table 2.
Comparison of Conventional PCR versus Fluorogenic PCR in the Detection of t(14;18) (q32;q21)
| Diagnosis | No. of cases | Conventional PCR | Fluorogenic PCR | ||
|---|---|---|---|---|---|
| bcl-2 mbr positive | bcl-2 mcr positive | bcl-2 mbr positive | bcl-2 mcr positive | ||
| Reactive lymphoid hyperplasia | 2 | 0 | 0 | 0 | 0 |
| Other nonneoplastic proliferations | 13 | 0 | 0 | 0 | 0 |
| Follicular lymphomas | |||||
| Large cell | 8 | 4 | 2 | 4 | 2 |
| Mixed cell | 7 | 4 | 1 | 3 | 1 |
| Small cleaved cell | 9 | 6 | 1 | 6 | 1 |
| Other lymphomas | |||||
| Small lymphocytic | 4 | 0 | 0 | 0 | 0 |
| Diffuse large cell | 8 | 6 | 1 | 6 | 1 |
| Diffuse mixed cell* | 1 | 0 | 0 | 0 | 0 |
| Unclassified B cell | 1 | 0 | 0 | 0 | 0 |
*Follicle center cell type.
To assess the sensitivity of the exonuclease-based fluorogenic bcl-2/JH PCR assay, 0.5 μg of DNA (corresponding to approximately 70,000 to 80,000 cells) from a cell line with rearrangement at the bcl-2 mcr was serially diluted with normal genomic DNA to achieve 101-fold to 105-fold dilutions. Then the DNA was analyzed simultaneously by both conventional and fluorogenic PCR assays (Figure 3) ▶ . Triplicate PCRs were performed for each DNA dilution in the exonuclease-based PCR assay. As the input target quantity increased, the cycle number at which an increase in fluorescence signal could be noticed decreased, indicating that the assay was sensitive to the starting target concentration (Figure 3, A and B) ▶ . The three stages of product accumulation seen in a typical PCR reaction (ie, exponential, linear, and plateau phases) became apparent when ΔRn was plotted on a log scale (Figure 3B) ▶ . Utilizing this assay, bcl-2 mcr/JH fusion sequences were consistently detected when the DNA was diluted 105-fold. However, as expected from Poisson distribution, at this dilution, which corresponds to 5 pg of DNA (0.6 to 0.8 genomes) per reaction, an exponential increase in fluorescence intensity was observed only in 2 of 3 PCR reactions. A slight increase in fluorescence was seen in negative control probably due to linear amplification of the germline bcl-2 gene. By conventional PCR, bcl-2 mcr/JH amplicons were seen at 103-fold dilution, but not at higher dilutions, after 72 hours of autoradiography (Figure 3C) ▶ .
Figure 3.
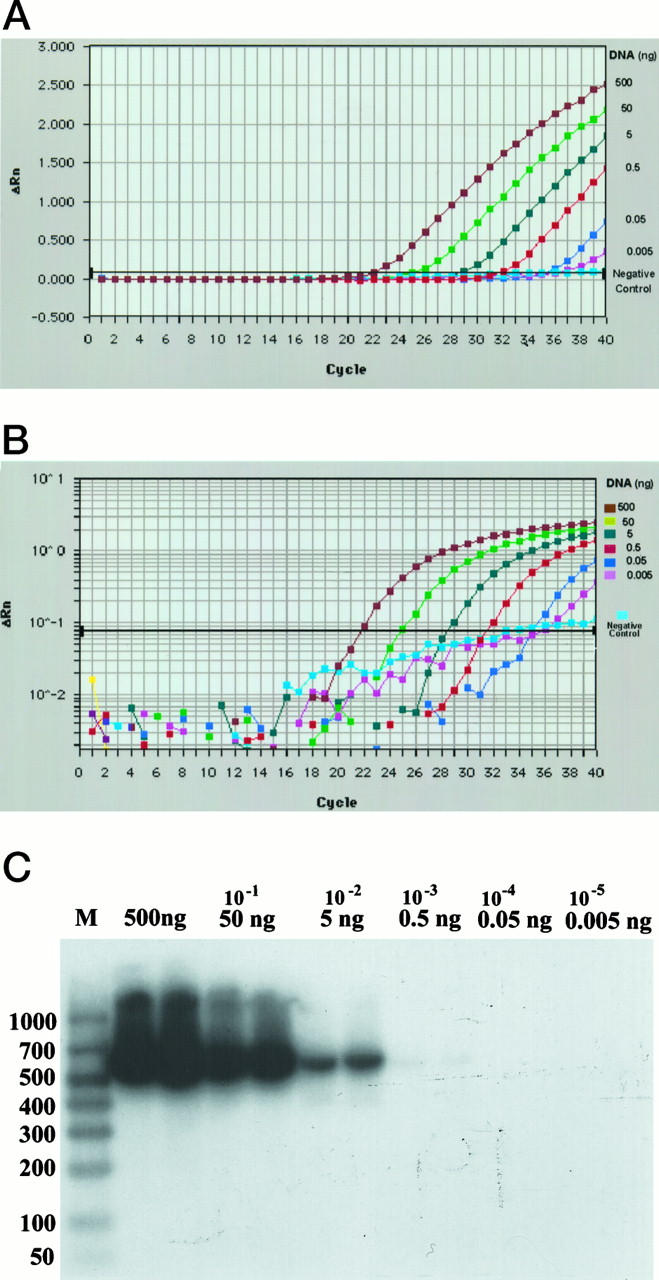
A comparison of detection sensitivities of exonuclease-based and conventional PCR assays for t(14;18)(q32;q21). Genomic DNA (500 ng) derived from a lymphoma cell line with t(14;18)(q32;q21) was diluted with DNA from nonneoplastic tissue to achieve the final concentration indicated and then subjected to either exonuclease-based or conventional PCR as described in Materials and Methods. DNA (500 ng) obtained from nonneoplastic tissue served as negative control. The fluorescence emission data collected during the extension phase of each cycle of exonuclease-based PCR were analyzed and plotted on a linear scale (A) or log scale (B). Although triplicate PCR reactions were performed for each DNA concentration, the data for only one are shown. C: Southern blot results of the conventional PCR assay performed in duplicate. Lane M: Molecular size marker. Molecular size in base pairs is indicated on the left. The final DNA concentration in the PCR assay is indicated on the top of the lanes.
Discussion
The PCR-mediated detection of t(14;18)(q32;q21) is frequently used to confirm the diagnosis of follicular lymphoma and to evaluate the course of the disease and the effect of therapy. Traditionally, amplified bcl-2/JH fusion sequences have been detected on ethidium bromide gels after electrophoresis. When high levels of sensitivity and specificity were required, the gels were blotted, hybridized with bcl-2-specific probes, and subjected to autoradiography. 1-3,8 Alternately, an aliquot of the amplification product was subjected to a second round of PCR (nested PCR), gel electrophoresis, Southern blotting, and autoradiography. 6,7,18 The 5′ exonuclease-based PCR assay combined with the laser-based detection system enables amplification and direct detection of specific target DNA sequences without the previously required post-PCR manipulations. We assessed the sensitivity and specificity of this novel assay in the detection of t(14;18)(q32;q21) associated with follicular lymphoma.
Exonuclease-based PCR detected bcl-2/JH fusion sequences in 24 (96%) of 25 cases that were positive by conventional PCR. The overall concordance between the two assays was 98% (52 of 53 cases concordant positive or negative). Serial dilution study demonstrated that exonuclease PCR was 100-fold more sensitive than conventional PCR. Failure to detect amplicons at 104-fold and higher dilutions by conventional PCR may be attributable to the fact that only 18 μl of the 50-μl PCR reaction mix was used for gel electrophoresis, in contrast to 50 μl used for signal detection by the 7700 Sequence Detector. Even after taking this factor into consideration, exonuclease-based fluorogenic PCR assay, however, was 35 times more sensitive than conventional PCR assay.
Because the amount of amplicon produced in any given cycle within the exponential phase of PCR is proportional to the initial number of template copies, a standard curve was generated using the fluorescence data from the serial dilution study (Figure 4) ▶ . The threshold cycle (y axis) is the cycle at which the fluorescence signal of the reporter dye rises above the baseline signal of the dye. Such standard curves can be utilized to determine the relative number of cells carrying bcl-2/JH fusion sequences (tumor burden) in test samples before and after treatment. However, the mere detection of PCR-amplifiable bcl-2/JH fusion sequences does not necessarily indicate whether translocation-bearing cells in the remission or follow-up samples represent the original clone or the emergence of a new or a secondary clone. Hence, it is important to confirm the clonal relatedness by sequence analysis, when analyzing sequential samples from a patient. It is also known that bcl-2/JH fusion sequences can be detected by PCR in peripheral blood of healthy individuals 5,6,18 and in normal lymphoid tissue in the setting of hyperplasia. 4,19 bcl-2/JH fusion sequences, however, were not detected in nonneoplastic tissues in our study. It should be noted that only 35 ng of DNA per PCR reaction was used in the present study, in contrast to 1000 to 7000 ng of DNA per PCR reaction that was used in the above cited studies. As the sensitivity of the PCR assay ultimately depends on the maximal number of cells tested, the amount of DNA used in the exonuclease-based PCR assay may have contributed to our inability to detect t(14;18) in nonneoplastic proliferations.
Figure 4.
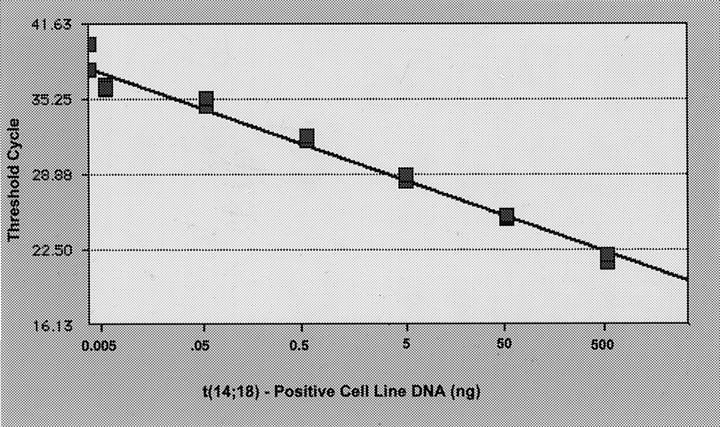
Standard curve showing the input DNA concentration versus threshold cycle. The standard curve was generated from the fluorescence data shown in Figure 3 ▶ . Each point represents the mean of the triplicate PCR amplifications.
In conclusion, this study demonstrated that the novel exonuclease-based PCR assay for the detection of bcl-2/JH fusion sequences is both specific and highly sensitive. The simplicity and the ability to acquire data as soon as PCR is completed ie, within 2 hours instead of the 5 days generally required for conventional PCR, make this assay ideal for the detection and quantitation of t(14;18)(q31;q21)-carrying cells in patients with follicular lymphoma. In addition, the ability of the instrument to detect multiple fluorescent dyes in a single reaction allows co-amplification and detection of a normalizer gene (positive internal control) that can be used to obtain accurate quantitative measurements and to confirm the presence of amplifiable DNA in a test sample. Preliminary results indicate that the exonuclease-based PCR assay can also successfully detect other lymphoma-associated chromosomal translocations.
Acknowledgments
We thank John Pfeifer of PE Applied Biosystems for his assistance in designing the fluorogenic probes, and we thank Bernadina Waasdorp and Cara Sutcliffe for performing some of the PCR assays. We also acknowledge Jeanne Knight for her assistance in preparing the illustrations and the manuscript.
Footnotes
Address reprint requests to Rajyalakshmi Luthra, Ph.D., Department of Pathology, The University of Texas M.D. Anderson Cancer Center, 1515 Holcombe Boulevard, Box 085, Houston, TX 77030.
References
- 1.Crescenzi M, Seto M, Herzig G, Weiss P, Griffith R, Korsmeyer S: Thermostable DNA polymerase chain amplification of t(14;18) chromosome breakpoints and detection of minimal residual disease. Proc Natl Acad Sci USA 1988, 85:4869-4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee M-S, Chang K-S, Cabanillas F, Freireich E, Trujillo J, Stass S: Detection of minimal residual cells carrying t(14;18) by DNA sequence amplification. Science 1987, 237:175-178 [DOI] [PubMed] [Google Scholar]
- 3.Stetler-Stevenson M, Raffeld M, Cohen P, Cossman J: Detection of occult follicular lymphoma by specific DNA amplification. Blood 1988, 72:1822-1825 [PubMed] [Google Scholar]
- 4.Limpens J, de Jong D, van Krieken J, Price C, Young B, van Ommen G-J, Kluin P: Bcl-2/JH rearrangements in benign lymphoid tissues with follicular hyperplasia. Oncogene 1991, 6:2271-2276 [PubMed] [Google Scholar]
- 5.Limpens J, Stad R, Vos C: Lymphoma-associated translocation t(14;18) in blood cells of normal individuals. Blood 1995, 9:2528-2536 [PubMed] [Google Scholar]
- 6.Ji W, Qu G, Ye P, Zhang X-Y: Frequent detection of bcl-2/JH translocations in human blood and organ sample by a quantitative polymerase chain reaction assay. Cancer Res 1995, 55:2876-2882 [PubMed] [Google Scholar]
- 7.Gribben J, Freedman A, Woo S: All advanced stage non-Hodgkin’s lymphomas with a polymerase chain reaction amplifiable breakpoint of bcl-2 have residual cells containing the bcl-2 rearrangement at evaluation and after treatment. Blood 1991, 78:3275-3280 [PubMed] [Google Scholar]
- 8.Ngan B-Y, Nourse J, Cleary ML: Detection of chromosomal translocation t(14;18) within the minor cluster region of bcl-2 by polymerase chain reaction and direct genomic sequencing of the enzymatically amplified DNA in follicular lymphomas. Blood 1989, 73:1759-1762 [PubMed] [Google Scholar]
- 9.Holland PM, Abramson RD, Watson R, Gelfand DH: Detection of specific polymerase chain reaction product by utilizing the 5′–3′ exonuclease activity of Thermus aquaticus DNA polymerase. Proc Natl Acad Sci USA 1991, 88:7276-7280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Livak KJ, Flood SJA, Marmaro J, Giusti W, Deetz K: Oligonucleotides with fluorescent dyes at opposite ends provide a quenched probe system useful for detecting PCR product and nucleic acid hybridization. PCR Methods Appl 1995, 4:357-362 [DOI] [PubMed] [Google Scholar]
- 11.Higuchi R, Dollinger G, Walsh P, Griffith R: Simultaneous amplification and detection of specific DNA sequences. Biotechnology 1992, 10:413-417 [DOI] [PubMed] [Google Scholar]
- 12.Higuchi R, Fockler C, GD, Watson R: Kinetic PCR: real time monitoring of DNA amplification reactions. Biotechnology 1993, 11:1026-1030 [DOI] [PubMed] [Google Scholar]
- 13.Heid CA, Stevens J, Livak KJ, Williams PM: Real time quantitative PCR. Genome Res 1996, 6:986-994 [DOI] [PubMed] [Google Scholar]
- 14.Gibson UEM, Heid CA, Williams PM: A novel method for real time quantitative RT-PCR. Genome Res 1996, 6:995-1001 [DOI] [PubMed] [Google Scholar]
- 15.Faas SJ, Menon R, Braun ER, Rudert WA, Trucco M: Sequence-specific priming and exonuclease-released fluorescence detection of HLA-DQB1 alleles. Tissue Ant 1996, 48:97-112 [DOI] [PubMed] [Google Scholar]
- 16.Lakowitz J: Energy transfer. Principles of Fluorescent Spectroscopy. 1983, :pp 303-309 Plenum Press, New York [Google Scholar]
- 17.Luthra R, McBride J, Hai S, Cabanillas F, Pugh W: The application of fluorescence-based PCR and PCR-SSCP to monitor the clonal relationship of cells bearing the t(14;18)(q32;q21) in sequential biopsy specimens from patients with follicle center cell lymphoma. Diagn Mol Pathol 1997, 6:71-77 [DOI] [PubMed] [Google Scholar]
- 18.Liu Y, Hernandez AM, Shibata D, Cortopassi GA: BCL2 translocation frequency rises with age in humans. Proc Natl Acad Sci USA 1994, 91:8910-8914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aster JC, Kobayashi Y, Shiota M, Mori S, Sklar J: Detection of the t(14;18) at similar frequencies in hyperplastic lymphoid tissues from American and Japanese patients. Am J Pathol 1992, 141:291-299 [PMC free article] [PubMed] [Google Scholar]