

Abstract
Biopsies routinely performed for the histopathological diagnosis of oral epithelial lesions before treatment were screened for chromosomal imbalances by comparative genomic hybridization. Comparative genomic hybridization was performed on 12 oral premalignant lesions (OPLs; dysplasias and carcinomas in situ) and 14 oral squamous cell carcinomas (OSCCs). Eight biopsies displayed areas of different histopathological appearance, so that OPLs and OSCCs from the same patient were analyzed. To avoid contamination with nonneoplastic cells, defined cell populations were isolated by micromanipulation with a glass needle. Before comparative genomic hybridization analysis, universal DNA amplification was performed using the DOP-polymerase chain reaction protocol. In the 14 OSCCs examined, the average number of chromosomal imbalances was significantly higher than in the 12 OPLs (mean ± SEM: 11.9 ± 1.9 versus 3.2 ± 1.2; P = 0.003). The DNA copy number changes identified in more than one OPL were gains on 8q (3 of 12) and 16p (2 of 12), as well as losses on 3p (5 of 12); 5q (4 of 12); 13q (3 of 12); and 4q, 8p, and 9p (2 of 12 each). In more than 30% of OSCCs, gains of chromosomal material were identified on 20q (8 of 14); 8q, 11q, 22q (7 of 14 each); 3q, 15q, and 17p (6 of 14 each); and 14q, 17q, and 20p (5 of 14 each), and losses were identified on 3p and 4q (9 of 14 each), 5q (7 of 14), 13q (6 of 14), and 2q and 9p (5 of 14 each). These results were validated by positive and negative control comparative genomic hybridization experiments and microsatellite analysis for the detection of allelic loss. The vast majority of genomic alterations found in OPLs were again identified in OSCCs from the same biopsy, supporting the hypothesis that multiple lesions in the same patient are clonally related. In summary, we show that comprehensive information on the genomic alterations in oral epithelial lesions can be obtained from small biopsies. Such data may identify prognostic indicators that could eventually assist in designing therapeutic strategies.
Oral and oropharyngeal squamous cell carcinoma is the sixth most frequently occurring cancer worldwide, with approximately 400,000 new cases diagnosed each year. 1 A high incidence of second primary lesions, both malignant and premalignant, was observed in patients with oral squamous cell carcinoma (OSCC), leading to the concept of field cancerization. 2 This phenomenon is partly attributed to the fact that the entire oral mucosa is exposed to exogenous cancer-promoting substances such as alcohol and tobacco. Some OSCCs are preceded by oral premalignant lesions (OPLs), which include dysplasias and carcinomas in situ (CIS) of the oral mucosa. However, 64% of OPLs do not progress to malignancy.3
In most studies on the genetic alterations in cancer of the upper aerodigestive tract, OSCCs are included in the more heterogeneous group of squamous cell carcinomas (SCCs) of the head and neck (HNSCCs). In HNSCCs, losses of genomic material were identified on 3p, 5q, 7q, 8p, 9p, 11q, 13q, 17p, and 18q by chromosome banding and/or allelotyping. 4-8 Cytogenetic gains were found on 1q, 3q, 8q, and 15q. 4,5 By comparative genomic hybridization (CGH), 9-13 gains in HNSCCs were most frequently detected on 1q, 3q, 5p, 7q, and 8q, whereas losses were most commonly found on 1p, 3p, 5q, 11q, 13q, and 19. 14-16 Less is known about genetic changes in OPLs. Molecular genetic analyses revealed loss of heterozygosity (LOH) at 3p, 9p, 17p, and 18q in dysplasias and additionally at 11q, 13q, and 14q in CIS. 17-20
Most of the above-mentioned studies were performed on resection specimens obtained at surgery when therapy decisions had already been made. It was the aim of our study to make use of the little material obtained on biopsy of oral epithelial lesions of unknown malignancy that were made for diagnostic reasons before the patients were started on any therapy regimen. Biopsies were histopathologically classified as dysplasia (n = 8), CIS (n = 4), or OSCC (n = 14) and screened for chromosomal imbalances by CGH. To exclusively analyze cells representative of the diagnosis, precise tissue areas were microdissected and their DNA was universally amplified before CGH analysis. As this approach provides comprehensive information on the genomic alterations of an oral epithelial lesion before treatment, it could supplement histopathological findings and assist in identifying prognostic parameters as a basis for therapy decisions.
Materials and Methods
Biopsy Samples and Clinical Data
Material was obtained from oral biopsies routinely performed for diagnostic purposes in the Clinic for Oral and Maxillofacial Surgery, University of Heidelberg, Germany. The biopsy material was fixed in 4% PBS-buffered formalin for no longer than 4 hours, paraffin-embedded, and sectioned. The hematoxylin and eosin (H&E)-stained sections were classified by an oral pathologist (IAB) according to the World Health Organization classification. 21 Lesions consisted of 8 dysplasias (1 mild, 5 moderate, and 2 severe dysplasias), 4 CIS, and 14 carcinomas (3 carcinomas of grade 1, 9 of grade 2, and 2 of grade 3). The clinical course of the patients whose biopsies were examined was monitored from January 1996 to December 1997. Of the patients with OSCC, one had metastases at diagnosis (patient 4) and two developed metastases during the follow-up period (patients 5 and 14). None of the patients diagnosed with OPL developed an OSCC during the follow-up period of 23 months. Consumption of alcohol and tobacco was assessed in all patients. Patients with a regular alcohol intake of greater than 50 g per day were given the symbol A, and patients who regularly smoked more than 10 cigarettes per day were given the symbol T in Figure 1 ▶ .
Figure 1.
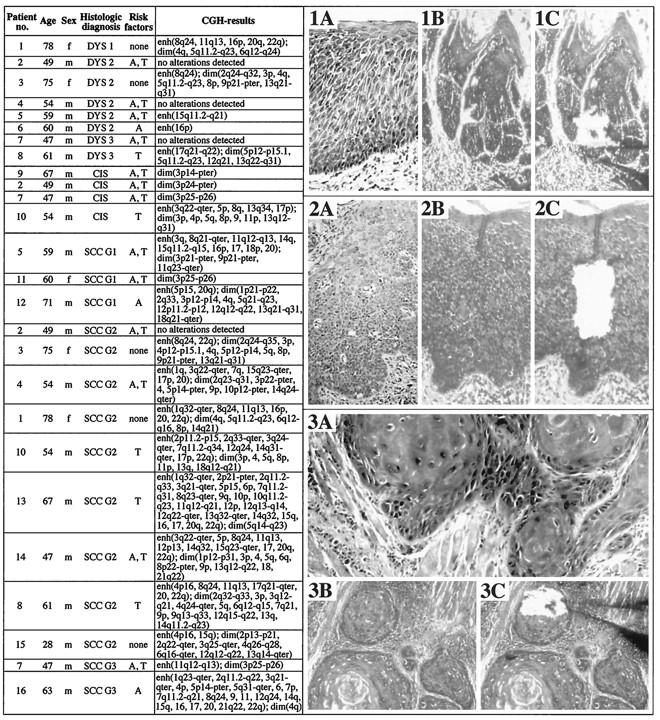
Left: Summary of selected clinical data, histopathological characteristics, and CGH results in dysplasias, CIS, and OSCCs investigated. m, male; f, female; DYS 1, mild dysplasia; DYS 2, moderate dysplasia; DYS 3, severe dysplasia; G1, G2, and G3, grades 1, 2, and 3, respectively; A, alcohol consumption (as defined in Materials and Methods); T, tobacco consumption (as defined in Materials and Methods); enh, enhanced number of chromosomes or chromosome segments; dim, reduced number of chromosomes or chromosome segments. Right: Covered H&E-stained tissue sections of oral biopsies representative of a dysplasia (1A), a CIS (2A), and a SCC (3A) viewed through a brightfield microscope used for diagnosing the lesions (magnification, ×100). Noncovered H&E-stained tissue sections before (1B, dysplasia; 2B, CIS; 3B, SCC) and after (1C, dysplasia; 2C, CIS; 3C, SCC) microdissection viewed through an inverted microscope used for microdissection (×100). The tip of a glass needle can be seen in images (1C) and (3C).
DNA Extraction, Microdissection and Proteinase K Digestion
The preparation of high molecular weight DNA from peripheral blood leukocytes as control DNA was carried out as described. 22 The test DNA was obtained by microdissection of 50–100 cells of a defined histopathological entity from an H&E-stained, formalin-fixed, paraffin-embedded biopsy section. For microdissection, a glass needle was produced from a glass capillary using a microforge. Microdissection was then performed using an inverted microscope with the glass needle attached to a de Fonbrune micromanipulator. The microdissected tissue was placed into a tube containing 20 μl 100 mmol/L Tris-HCl buffer (pH 7.5) and digested with 1 μl proteinase K (10 mg/ml) for 3 h at 55°C. The enzyme was then inactivated by incubation at 90°C for 10 minutes.
Degenerate Oligonucleotide Primed-Polymerase Chain Reaction
Degenerate oligonucleotide primed-polymerase chain reaction (DOP-PCR) was used to universally amplify both the test and the control DNA in a thermal cycler. The protocol used was modified from Telenius et al. 23 PCR was done in a final volume of 50 μl (4 μl 25 mmol/L MgCl2, 5 μl 10× PCR buffer (500 mmol/L KCl, 100 mmol/L Tris-HCl, pH 8.3) with 2 μl 5 mmol/L deoxynucleotide triphosphate, 5 μl 17 μM primer 6MW (5′-CCG ACT CGA GNN NNN NAT GTG G-3′), 0.5 μl (2.5 U) Taq polymerase, and test or control DNA in 20 μl 100 mmol/L Tris-HCl, pH 7.5). The PCR conditions used were: 10 minutes at 93°C, followed by 10 cycles of 1 minute at 94°C, 1.3 minutes at 30°C, 3-minute transition at 30–72°C, and 3-minute extension at 72°C, followed by 35 cycles of 1 minute at 94°C, 1 minute at 62°C, 3 minutes at 72°C with an addition of 1 second/cycle to the extension step, and a final extension of 10 minutes.
CGH
Following standard procedures, metaphase spreads were prepared from stimulated peripheral blood lymphocytes obtained from a healthy male subject (46,XY). CGH was performed with test and control DNA amplified by DOP-PCR as described in detail elsewhere. 24 For image acquisition and processing, an epifluorescence microscope (Zeiss Axiophot, Jena, Germany) was used that was equipped with a cooled charge-coupled device camera (Photometrics, Tucson, AZ). Digital images were processed with a program developed by du Manoir et al 25 on the basis of the software package TCL-Image (TNO Institute of Applied Physics, Delft, The Netherlands). Mean ratio profiles were determined from the analysis of 10 metaphase spreads. The diagnostic threshold values used to score losses and gains were 0.75 (lower threshold) and 1.25 (upper threshold), respectively, in accordance with previously reported CGH analysis protocols. 25 Due to the suppression with Cot1 DNA, the fluorescence intensities were not representative at chromosome regions with tandem repetitive DNA clusters. These areas were excluded from evaluation. Banding assignment of losses and gains was based on the comparison of CGH average ratio profiles and chromosome ideograms.
Microsatellite Analysis
Microsatellite locus D4S426 (4q35) was studied for LOH in the dysplasia and the carcinoma from patient 1. PCR amplification, denaturing polyacrylamide electrophoresis of PCR products, silver staining of the gels, and assessment for allelic loss was performed as described. 26
Control Experiments
To validate our approach combining the microdissection of H&E-stained biopsy sections followed by DOP-PCR and CGH, various control experiments were performed.
Negative control experiments were carried out to specifically validate the DOP-PCR protocol used by hybridizing differently labeled, DOP-PCR-amplified control DNAs to reference chromosomes. As a further control experiment, DNA obtained from connective tissue microdissected from biopsy sections and amplified by DOP-PCR was hybridized together with differently labeled DOP-PCR-amplified control DNA to reference chromosomes. In these control experiments, no imbalances were detected on any chromosome except on 19, X and Y once each (four experiments). As artifactual results had been occasionally observed in our and other laboratories on chromosome 19 and chromosomal bands 1p34–p36, 27-29 these regions as well as the sex chromosomes were excluded from the analysis.
Positive control experiments were performed with DNA extracted conventionally from OSCC tissue. Results from CGH experiments performed with 1) OSCC DNA versus control DNA and 2) the same OSCC DNA versus control DNA, but both amplified by DOP-PCR, were compared. The chromosomal imbalances identified by CGH in the OSCC DNAs that had not been subjected to DOP-PCR were again detected in the experiments with the same OSCC DNAs that had been universally amplified by DOP-PCR. To validate CGH results by an independent method, microsatellite analysis was performed at D4S426 on DOP-PCR-amplified DNA from a dysplasia and a carcinoma of patient 1. LOH at this microsatellite marker was identified in both the dysplasia and the carcinoma of this patient. This result was concordant with the CGH findings that loss on chromosome arm 4q was present in both lesions.
Statistics
Given are means ± SEM. After assessment for normal distribution by the Shapiro-Wilks test, comparisons between groups were performed by Mann-Whitney U test. P < 0.05 was considered statistically significant.
Results
Figure 1 ▶ summarizes the histopathological diagnoses, selected clinical data, and all CGH results from the biopsies tested. In addition, H&E-stained tissue sections from a representative dysplasia, CIS, and OSCC are shown.
Oral Dysplasias and CIS (Premalignant Lesions)
An average of 2.9 ± 1.3 genomic alterations was detected in the 8 dysplasias. In the 4 CIS studied, the average number of chromosomal imbalances was slightly higher (3.8 ± 2.8). When combining all 12 premalignant lesions, an average of 3.2 ± 1.2 genomic changes per case was found (Figure 2) ▶ .
Figure 2.

Accumulation of genetic alterations during progression from OPL to OSCC. The number of CGH alterations detected in an individual lesion are shown in the respective group. The OPL group comprises dysplasias (○) and CIS (•). CIS. Next to the column of data points, mean ± SEM are given. *, significant difference compared with values for OPLs (P < 0.05).
The imbalances identified in more than one premalignant lesion were gains on 8q (3 of 12) and 16p (2 of 12), as well as losses on 3p (5 of 12); 5q (4 of 12); 13q (3 of 12); and 4q, 8p, and 9p (2 of 12 each) (Figures 3 and 4) ▶ ▶ .
Figure 3.
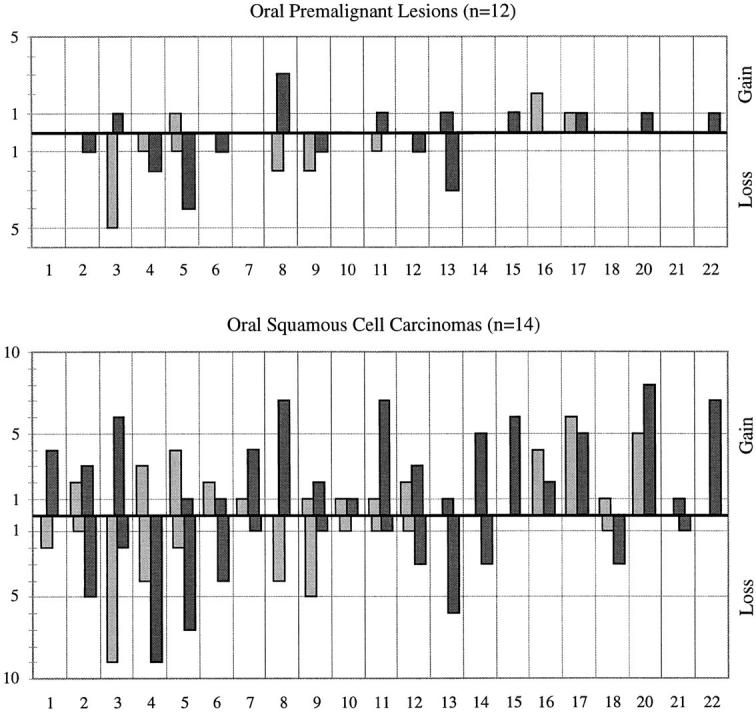
Frequency of all CGH results obtained in OPLs (top) and OSCCs (bottom). x axis, chromosomes 1–22; y axis, number of cases. Ascending bars represent gains on the short arms (light bars) or long arms (dark bars), whereas descending bars represent losses on the short arms (light bars) or long arms (dark bars) of the respective chromosomes.
Figure 4.

Location of chromosomal imbalances detected by CGH in OPLs (discontinuous lines) and OSCCs (continuous lines). Vertical lines on the right side of a chromosome ideogram represent gains of genetic material, whereas vertical lines on the left side correspond to losses. Chromosomal region 1p34–p36 and chromosome 19 are shaded gray; they were excluded from the evaluation (see Materials and Methods).
OSCCs
In the 14 carcinomas examined, the average number of chromosomal imbalances was significantly higher (11.9 ± 1.9) than in the OPLs (P = 0.003) (Figure 2) ▶ .
The following chromosomal alterations were identified in more than three carcinomas: gains on 20q (8 of 14, 57%); 8q, 11q, and 22q (7 of 14, 50% each); 3q, 15q, and 17p (6 of 14, 43% each); 14q, 17q, and 20p (5 of 14, 36% each); 1q, 5p, 7q, and 16p (4 of 14, 29% each) and losses on 3p and 4q (9 of 14, 64% each); 5q (7 of 14, 50%); 13q (6 of 14, 43%); 2q and 9p (5 of 14, 36% each); and 4p, 6q, and 8p (4 of 14, 29% each) (Figures 3 and 4) ▶ ▶ .
Of the 14 OSCCs investigated, 3 were metastasizing. The mean number of genomic alterations was higher in these 3 cases than in the 11 nonmetastasizing tumors (15 ± 2.5 versus 11.1 ± 2.4). The DNA copy number changes common to all 3 metastasizing OSCCs were gains on 3q, 15q, 17p, and 20q, as well as losses on 3p and 9p.
Comparison of Lesions of Different Histopathology within One Biopsy
In eight biopsies with the histopathological diagnosis of carcinoma, premalignant lesions were also found in other areas of the biopsy material. In these cases, cells from dysplasia, CIS, and carcinoma from the same patient were microdissected separately and analyzed.
In 4 biopsies (patients no. 2, 4, 5, and 7), no or only one genomic alteration was found in the premalignant lesions and 0, 2, 12, and 13 abnormalities in the respective carcinomas. Figure 5A ▶ shows CGH results obtained for patient 5 comparing average ratio profiles detected in the dysplasia and the carcinoma of this patient. In the other 4 biopsies (patients no. 1, 3, 8, and 10), 5 or more changes were identified in the premalignant lesions and 11 or more anomalies in the respective carcinomas (5, 8, 8, and 12 (changes in OPLs) versus 18, 11, 11, and 15 (changes in OSCCs)). Chromosomal imbalances found in the dysplasia and the carcinoma of patient 1 are shown in Figure 5B ▶ . Of the 36 CGH abnormalities detected in premalignant lesions, 30 (83%) were again found in the respective carcinomas (Figure 6) ▶ .
Figure 5.

Comparison of chromosomal imbalances identified in the dysplasia (DYS, top) and the carcinoma (SCC, bottom) of patient 5 (A) and patient 1 (B). Selected average ratio profiles are shown. The three vertical lines to the right of each chromosome ideogram represent the balanced state (center) and the lower (left) and upper (right) thresholds used as diagnostic cutoff values for losses and gains, respectively. The areas of tandem repetitive DNA clusters are gray shaded. Because no representative fluorescence intensities can be measured in these regions due to suppression with Cot1 DNA, they were excluded from the evaluation. Whereas only one imbalance was found in the dysplasia of patient 5 (A), almost all alterations identified in the SCC of patient 1 (B) were already present in the respective dysplasia. The losses on 4q detected in the dysplasia and SCC of patient 1 by CGH (B) were confirmed by LOH analysis at the microsatellite locus D4S426 (4q35) (data not shown).
Figure 6.

Number of distinct and shared genomic alterations identified in lesions of different histopathology within one biopsy. Each box represents one chromosomal imbalance detected only in the OPL (dark gray), in both OPL and OSCC from the same patient (light gray), or in the OSCC only (white). Patients are divided into those with zero to one copy number change (left) and those with more than five copy number changes (right) in the OPL of their biopsy. Pt. no., patient number; no. chr. imb. in OPLs, number of chromosomal imbalances detected in OPLs.
Discussion
Experimental Approach
In this study, we used CGH to identify chromosomal imbalances in 26 oral lesions from 16 patients diagnosed as dysplasia, CIS, or invasive SCC. The stringent diagnostic thresholds that we use for CGH analysis 25 require a high percentage of cells to be tested within the tissue used for DNA extraction and amplification. As CIS and particularly dysplasias represent alterations of a few defined cells within the oral epithelium, the isolation of a precise tissue area is a prerequisite for CGH analysis. Similarly, SCCs may contain a significant component of nonneoplastic cells, such as inflammatory cells, as well as connective tissue. In our study, we therefore performed microscopic dissection of areas of 50 to 100 histopathologically defined cells with the aid of a glass needle and a micromanipulator. To achieve a precise cellular identification, H&E-stained tissue sections were used for microdissection. This approach allowed a more reliable microdissection of defined cells as compared with using an unstained section consecutive to a H&E-stained section.
The DNA from the microdissected tissue was then universally amplified using a modified DOP-PCR protocol 23 (see Materials and Methods). Negative and positive control CGH experiments confirmed the validity of using this DOP-PCR protocol for the generation of genomic DNA as probes for CGH (see Materials and Methods). The reliability of this approach was further corroborated by the detection of LOH on 4q in a dysplastic lesion and an OSCC from the same patient that had both been found to have losses on 4q by CGH. Thus, tissue microdissection followed by universal DNA amplification and CGH represent a strategy that allows the detection of chromosomal imbalances even in small precursor lesions representing an early stage of the tumor progression pathway. Previously, a combination of these techniques was performed on a few tumors of different types. These included one human testicular tumor versus adjacent normal testis tissue, 30 three primary human cutaneous malignant melanomas in different growth phases, 31 and seven ductal CIS that represent precursor lesions of invasive ductal breast cancer. 32
Genomic Alterations in OPLs and OSCCs
In 8 of the 12 OPLs analyzed here, no or only one genomic alteration was detected, whereas 5 or more genetic changes were found in the other 4 lesions. The most frequent losses identified included those on 3p, 9p, and 13q, which had previously been reported in OPLs by LOH analyses. 7,17-19,33 Additionally, losses on 4q, 5q, and 8p were detected in our study. The gains identified by CGH in more than one case were located on 8q and 16p.
One of the most frequent alterations detected in OSCCs was loss on 3p. On this chromosome arm, three distinct regions of deletion were previously identified, of which the most proximal (3p13–p21.1) and the most distal (3p25) were found to occur in oral dysplastic lesions at the same frequency as in OSCCs. 19 In our study, a commonly deleted region in OPLs and OSCCs encompassed 3p25–p26. This region contains the VHL (von Hippel-Lindau) tumor suppressor gene. 34 However, in 26 upper aerodigestive tract SCCs that were known to have lost one allele at 3p, neither mutations nor hypermethylation was found in the remaining allele of the VHL gene, 35 making VHL an unlikely “target” for the distal 3p deletion. Losses detected on 9p in OPLs and OSCCs in this report included the locus of tumor suppressor gene CDKN2A (p16/MTS1/INK4a). Loss of p16 expression was found by immunohistochemistry in 38% of oral dysplastic lesions 36 and in 50% of HNSCC cell lines. 37 In our study, loss on 13q was detected in 25% of OPLs, a percentage similar to that identified by LOH studies. 17 The smallest commonly deleted region found comprised 13q22–q31.
Losses on 4q, 8p, and 5q, which were identified in OPLs and in higher percentages of OSCCs in our study, had previously only been identified in HNSCCs: LOH on 4q was seen in the majority of HNSCCs with a common region of deletion at 4q25 containing the EGF locus; 38 on 8p, multiple distinct regions of allelic loss (8p21, 8p22–p23, and 8p23) were detected in SCC of the supraglottic larynx; 39 LOH on 5q was shown to involve the APC gene in OSCC, 40 which is located within the commonly deleted region identified in OPLs (5q11.2–q23) and OSCCs (5q21–q23) here. According to our results, particularly the loss on 5q that was found in one-third of OPLs may in fact be a previously unidentified genetic alteration that occurs in the progression pathway before the invasive carcinoma stage.
In our study, 3 of 12 OPLs and 7 of 14 OSCCs revealed gain on 8q with a smallest region of common gain including the MYC proto-oncogene locus at 8q24. 41 In oral SCCs, MYC was previously shown to be amplified42,43 and overexpressed, 44 making this gene the likely “target” for the gain on 8q in OPLs and OSCCs described here.
Clonality of Different Lesions within the Oral Mucosa
The observation that histopathological abnormalities are common in the vicinity of invasive carcinomas from the oral cavity was first made by Slaughter et al. and termed field cancerization. 2 In eight patients with OSCC studied here, areas of dysplasia and/or CIS were identified within the same biopsy and examined additionally. Six OSCCs displayed the same alterations as the respective OPLs and additional DNA copy number changes. In only three cases, a small fraction of chromosomal imbalances identified in the OPLs were not found in the respective OSCCs. These data support the hypothesis that field cancerization is caused by the migration and expansion of clonally related cells in a high proportion of cases. Additional evidence that multiple lesions within the mucosa of the head and neck arise from a single progenitor clone was provided by studies of allelic loss and X chromosome inactivation in lesions with different histopathological appearance and multiple primary tumors from the same patients. 17,45
Genomic Alterations as Prognostic Parameters
The chromosomal imbalances identified in biopsy material diagnosed as OSCC in this study are similar to those detected in a CGH study on 30 primary tumor specimens of HNSCC obtained at surgical resection. 16 This is particularly interesting, as it demonstrates that CGH can be reliably performed on biopsy material, which is obtained from the patient when the oral lesion is first diagnosed, before a therapeutic decision has been made. Together with the histopathological classification, the genomic alterations of an oral lesion determined at such an early time point could eventually be used as prognostic indicators that could assist in the choice of the best therapeutic strategy.
As a first step in this direction, it is of interest to determine the most likely sequence in which genomic alterations occur during the progression from premalignant lesions to invasive carcinomas. Such genetic models have been proposed for the development from normal mucosa to hyperplasia (9p loss), dysplasia (3p, 17p loss), CIS (11q, 13q and 14q loss), and invasive HNSCC (6p, 8, and 4q loss) based on microsatellite analyses for allelic loss at 10 chromosomal loci. 17 Other models are based on compilations of the literature. 20,46 A CGH study postulated that distinct patterns of gains and losses were associated with HNSCC of different histological grades (grade 1, losses on 3p, 9p, and gain on 3q; grade 3, additional losses on 4q, 8p, 11q, 13q, 18q, and 21q, and gains on 1pter, 11q13, 19, and 22q). 16 In our study, a genome-wide screening for chromosomal imbalances was possible not only in pure tumor cell populations from OSCCs but also in oral dysplasias and CIS. Compared with the above-mentioned reports, the additional alterations identified here in more than one-fourth of OPLs were loss on 5q and gain on 8q and in more than one-third of OSCCs gains on 14q and 15q and losses on 2q.
To be able to base therapy decisions on the genetic abnormalities identified, it is necessary to correlate the respective chromosomal changes with the clinical course of the patient, ie, with the development of invasive carcinoma from OPLs or of metastases from SCCs, or with patient survival. Mao et al 33 found that detection of allelic loss at 9p21 and 3p14 in OPLs may aid in assessing the risk for progression to carcinoma. They reported that more than one-third of patients with LOH at one of these loci developed OSCCs, whereas only 6% of patients without allelic loss did so. In our study, the mean number of genomic alterations was higher in the 3 metastasizing carcinomas than in the 11 nonmetastasizing tumors. This is in line with the findings by Soder et al 47 that diploidy was significantly more common in nonmetastasizing carcinomas and that aneuploidy predominated in the group of metastasizing HNSCCs. The chromosomal imbalances attributed to metastasizing versus nonmetastasizing HNSCCs by Bockmühl et al 48 differ from our results for metastasizing OSCCs except for overrepresentation of 20q, which was frequently found in both studies. In some reports, genetic changes were correlated with patient survival. In HNSCCs, the fractional allele loss calculated for 52 tumors, 49 LOH at 3p, 50 and rearrangements at 11q13 51 were associated with a reduction in survival time.
By our combined approach, comprehensive information on the chromosomal imbalances in small biopsies of oral epithelial lesions could be obtained. Such data may assist in identifying prognostic parameters that could in future be used as a basis for patient counseling and therapeutic decisions.
Acknowledgments
We are indebted to Thomas Cremer (München, Germany) for generous support and fruitful scientific discussions and to Dieter Haffner (Heidelberg, Germany) for statistical advice and compilation of graphs. Heidi Holtgreve-Grez, Anna Jauch, Susanne Popp, and Brigitte Schoell (all of Heidelberg, Germany); Sabine Irmer and Jürgen Kraus (both of München, Germany); and Jeff Craig (Edinburgh, United Kingdom) are gratefully acknowledged for technical advice, as is Anja I. Soder (Heidelberg, Germany) for reading the manuscript.
Footnotes
Address reprint requests to Dr. Ruthild G. Weber, Deutsches Krebsforschungszentrum, Abt. H0700, INF 280, 69120 Heidelberg, Germany. E-mail: r.weber@dkfz-heidelberg.de.
Supported by the Deutsche Krebshilfe (grants 10-1124-Li1 and 10-0976-Re1).
References
- 1.Pisani P, Parkin DM, Ferlay J: Estimates of the worldwide mortality from eighteen major cancers in 1985: implications for prevention and projections of future burden. Int J Cancer 1993, 55:891-903 [DOI] [PubMed] [Google Scholar]
- 2.Slaughter DP, Southwick HW, Smejkal W: “Field cancerization” in oral stratified squamous epithelium: clinical implications of multicentric origin. Cancer 1953, 6:963-968 [DOI] [PubMed] [Google Scholar]
- 3.Papadimitrakopoulou VA, Hong WK, Lee JS, Martin JW, Lee JJ, Batsakis JG, Lippman SM: Low-dose isotretinoin versus β-carotene to prevent oral carcinogenesis: long-term follow up. J Natl Cancer Inst 1997, 89:257-258 [DOI] [PubMed] [Google Scholar]
- 4.Jin Y, Mertens F, Mandahl N, Heim S, Olegard C, Wennerberg J, Biörklund A, Mitelman F: Chromosome abnormalities in eighty-three head and neck squamous cell carcinomas: influence of culture conditions on karyotypic pattern. Cancer Res 1993, 53:2140-2146 [PubMed] [Google Scholar]
- 5.Rao PH, Sreekantaiah C, Schantz SP, Chaganti RSK: Cytogenetic analysis of 11 squamous cell carcinomas of the head and neck. Cancer Genet Cytogenet 1994, 77:60-64 [DOI] [PubMed] [Google Scholar]
- 6.Nawroz H, van der Riet P, Hruban RH, Koch W, Ruppert JM, Sidransky D: Allelotype of head and neck squamous cell carcinoma. Cancer Res 1994, 54:1152-1155 [PubMed] [Google Scholar]
- 7.El-Naggar AD, Hurr K, Batsakis JG, Luna MA, Goepfert H, Huff V: Sequential loss of heterozygosity at microsatellite motifs in preinvasive and invasive head and neck squamous carcinoma. Cancer Res 1995, 55:2656-2659 [PubMed] [Google Scholar]
- 8.Jones JW, Raval JR, Beals TF, Worsham MJ, Van Dyke DL, Esclamado RM, Wolf GT, Bradford CR, Miller T, Carey TE: Frequent loss of heterozygosity on chromosome arm 18q in squamous cell carcinomas. Arch Otolaryngol Head Neck Surg 1997, 123:610-614 [DOI] [PubMed] [Google Scholar]
- 9.Kallioniemi A, Kallioniemi O-P, Sudar D, Rutovitz D, Gray JW, Waldman F, Pinkel D: Comparative genomic hybridization for molecular cytogenetic analysis of solid tumors. Science 1992, 258:818-821 [DOI] [PubMed] [Google Scholar]
- 10.Du Manoir S, Speicher MR, Joos S, Schröck E, Popp S, Döhner H, Kovacs G, Robert-Nicoud M, Lichter P, Cremer T: Detection of complete and partial chromosome gains and losses by comparative genomic in situ hybridization. Hum Genet 1993, 90:590-610 [DOI] [PubMed] [Google Scholar]
- 11.Forozan F, Karhu R, Kononen J, Kallioniemi A, Kallioniemi O-P: Genome screening by comparative genomic hybridization. Trends Genet 1997, 13:405-409 [DOI] [PubMed] [Google Scholar]
- 12.Zitzelsberger H, Lehmann L, Werner M, Bauchinger M: Comparative genomic hybridisation for the analysis of chromosomal imbalances in solid tumours and haematological malignancies. Histochem Cell Biol 1997, 108:403-417 [DOI] [PubMed] [Google Scholar]
- 13.van Dekken H, Rosenberg C, Krijtenburg PJ, Alers JC: Interphase cytogenetics and comparative genomic hybridization of human epithelial cancers and precursor lesions. Histochem Cell Biol 1997, 108:419-430 [DOI] [PubMed] [Google Scholar]
- 14.Speicher MR, Howe C, Crotty P, du Manoir S, Costa J, Ward DC: Comparative genomic hybridization detects novel deletions and amplifications in head and neck squamous cell carcinomas. Cancer Res 1995, 55:1010-1013 [PubMed] [Google Scholar]
- 15.Brzoska PM, Levin NA, Fu KK, Kaplan MJ, Singer MI, Gray JW, Christman MF: Frequent novel DNA copy number increase in squamous cell head and neck tumors. Cancer Res 1995, 55:3055-3059 [PubMed] [Google Scholar]
- 16.Bockmühl U, Schwendel A, Dietel M, Petersen I: Distinct pattern of chromosomal alterations in high- and low-grade head and neck squamous cell carcinomas. Cancer Res 1996, 56:5325-5329 [PubMed] [Google Scholar]
- 17.Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W, Sidransky D: Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res 1996, 56:2488-2492 [PubMed] [Google Scholar]
- 18.Emilion G, Langdon JD, Speight P, Partridge M: Frequent gene deletions in potentially malignant oral lesions. Br J Cancer 1996, 73:809-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roz L, Wu CL, Porter S, Scully C, Speight P, Read A, Sloan P, Thakker N: Allelic imbalances on chromosome 3p in oral dysplastic lesions: an early event in oral carcinogenesis. Cancer Res 1996, 56:1228-1231 [PubMed] [Google Scholar]
- 20.Mao L: Leukoplakia: molecular understanding of pre-malignant lesions and implications for clinical management. Mol Med Today 1997, 3:442-448 [DOI] [PubMed] [Google Scholar]
- 21.Pindborg JJ, Reichart PA, Smith CJ, van der Waal I: Histological Typing of Cancer and Precancer of the Oral Mucosa, ed 2. Berlin, Springer-Verlag, 1997
- 22.Sambrook J, Fritsch EF, Maniatis T: Molecular Cloning: A Laboratory Manual. 1989. New York, Cold Spring Harbor Laboratory Press, Cold Spring Harbor
- 23.Telenius H, Carter NP, Bebb CE, Nordenskjöld M, Ponder BAJ, Tunnacliffe A: Degenerate oligonucleotide-primed PCR: general amplification of target DNA by a single degenerate primer. Genomics 1992, 13:718-725 [DOI] [PubMed] [Google Scholar]
- 24.Lichter P, Bentz M, Joos S: Detection of chromosomal aberrations by means of molecular cytogenetics: painting of chromosomes and chromosomal subregions and comparative genomic hybridization. Methods Enzymol 1995, 254:334-359 [DOI] [PubMed] [Google Scholar]
- 25.Du Manoir S, Schröck E, Bentz M, Speicher MR, Joos S, Ried T, Lichter P, Cremer T: Quantitative analysis of comparative genomic hybridization. Cytometry 1995, 19:27-41 [DOI] [PubMed] [Google Scholar]
- 26.Boström J, Mühlbauer A, Reifenberger G: Deletion mapping of the short arm of chromosome 1 identifies a common region of deletion distal to D1S496 in human meningiomas. Acta Neuropathol 1997, 94:479-485 [DOI] [PubMed] [Google Scholar]
- 27.Kallioniemi O-P, Kallioniemi A, Piper J, Isola J, Waldman FM, Gray JW, Pinkel D: Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes Chromosomes Cancer 1994, 10:231-243 [DOI] [PubMed] [Google Scholar]
- 28.Lichter P, Bentz M, du Manoir S, Joos S: Comparative genomic hybridization. Verma RS Babu A eds. Human Chromosomes. 1995, :pp 191-210 McGraw Hill, New York [Google Scholar]
- 29.Weber RG, Boström J, Wolter M, Baudis M, Collins VP, Reifenberger G, Lichter P: Analysis of genomic alterations in benign, atypical, and anaplastic meningiomas: toward a genetic model of meningioma progression. Proc Natl Acad Sci USA 1997, 94:14719-14724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Speicher MR, Jauch A, Walt H, du Manoir S, Ried T, Jochum W, Sulser T, Cremer T: Correlation of microscopic phenotype with genotype in a formalin-fixed, paraffin-embedded testicular germ cell tumor with universal DNA amplification, comparative genomic hybridization, and interphase cytogenetics. Am J Pathol 1995, 146:1332-1340 [PMC free article] [PubMed] [Google Scholar]
- 31.Wiltshire RN, Duray P, Bittner ML, Visakorpi T, Meltzer PS, Tuthill RJ, Liotta LA, Trent JM: Direct visualization of the clonal progression of primary cutaneous melanoma: application of tissue microdissection and comparative genomic hybridization. Cancer Res 1995, 55:3954-3957 [PubMed] [Google Scholar]
- 32.Kuukasjarvi T, Tanner M, Pennanen S, Karhu R, Kallioniemi O-P, Isola J: Genetic changes in intraductal breast cancer detected by comparative genomic hybridization. Am J Pathol 1997, 150:1465-1471 [PMC free article] [PubMed] [Google Scholar]
- 33.Mao L, Lee J, Fan YH, Ro JY, Batsakis JG, Lippman S, Hittelman W, Hong WK: Frequent microsatellite alterations at chromosomes 9p21 and 3p14 in oral premalignant lesions and their value in cancer risk assessment. Nat Med 1996, 2:682-685 [DOI] [PubMed] [Google Scholar]
- 34.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L: Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993, 260:1317-1320 [DOI] [PubMed] [Google Scholar]
- 35.Waber PG, Lee NK, Nisen PD: Frequent allelic loss at chromosome arm 3p is distinct from genetic alterations of the von Hippel-Lindau tumor suppressor gene in head and neck cancer. Oncogene 1996, 12:365-369 [PubMed] [Google Scholar]
- 36.Papadimitrakopoulou V, Izzo J, Lippman SM, Lee JS, Fan YH, Clayman G, Ro JY, Hittelman WN, Lotan R, Hong WK, Mao L: Frequent inactivation of p16INK4a in oral premalignant lesions. Oncogene 1997, 14:1799-1803 [DOI] [PubMed] [Google Scholar]
- 37.Lydiatt WM, Murty VVVS, Davidson BJ, Xu L, Dyomina K, Sacks PG, Schnatz SP, Chaganti RSK: Homozygous deletions and loss of expression of the CDKN2 gene occur frequently in head and neck squamous cell carcinoma cell lines but infrequently in primary tumors. Genes Chromosomes Cancer 1995, 13:94-98 [DOI] [PubMed] [Google Scholar]
- 38.Pershouse MA, El-Naggar AK, Hurr K, Lin H, Yung WK, Steck PA: Deletion mapping of chromosome 4 in head and neck squamous cell carcinoma. Oncogene 1997, 14:369-373 [DOI] [PubMed] [Google Scholar]
- 39.Sunwoo JB, Holt MS, Radford DM, Deeker C, Scholnick SB: Evidence for multiple tumor suppressor genes on chromosome arm 8p in supraglottic laryngeal cancer. Genes Chromosomes Cancer 1996, 16:164-169 [DOI] [PubMed] [Google Scholar]
- 40.Largey JS, Meltzer SJ, Sauk JJ, Hebert CA, Archibald DW: Loss of heterozygosity involving the APC gene in oral squamous cell carcinomas. Oral Surg Oral Med Oral Pathol 1994, 77:260-263 [DOI] [PubMed] [Google Scholar]
- 41.Neel BG, Jhanwar SC, Chaganti RSK, Hayward WS: Two human c-onc genes are located on the long arm of chromosome 8. Proc Natl Acad Sci USA 1982, 79:7842-7846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Saranath D, Panchal RG, Nair R, Metha AR, Des MG: Oncogene amplification in squamous cell carcinoma of the oral cavity. Jpn J Cancer Res 1989, 80:430-437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Leonard JH, Kearsley JH, Chenevix-Trench G, Hayward NK: Analysis of gene amplification in head-and-neck squamous-cell carcinoma. Int J Cancer 1991, 48:511-515 [DOI] [PubMed] [Google Scholar]
- 44.Spandidos DA, Lamothe A, Field JK: Multiple transcriptional activation of cellular oncogenes in human head and neck solid tumours. Anticancer Res 1985, 5:221-224 [PubMed] [Google Scholar]
- 45.Bedi GC, Westra WH, Gabrielson E, Koch W, Sidransky D: Multiple head and neck tumors: evidence for a common clonal origin. Cancer Res 1996, 56:2484-2487 [PubMed] [Google Scholar]
- 46.Todd R, Donoff RB, Wong DTW: The molecular biology of oral carcinogenesis: toward a tumor progression model. J Oral Maxillofac Surg 1997, 55:613-623 [DOI] [PubMed] [Google Scholar]
- 47.Soder AI, Hopman AHN, Ramaekers FCS, Conradt C, Bosch FX: Distinct nonrandom patterns of chromosomal aberrations in the progression of squamous cell carcinomas of the head and neck. Cancer Res 1995, 55:5030-5037 [PubMed] [Google Scholar]
- 48.Bockmühl U, Petersen S, Schmidt S, Wolf G, Jahnke V, Dietel M, Petersen I: Patterns of chromosomal alterations in metastasizing and nonmetastasizing primary head and neck carcinomas. Cancer Res 1997, 57:5213-5216 [PubMed] [Google Scholar]
- 49.Field JK, Kiaris H, Risk JM, Tsiriyotis C, Adamson R, Zoumopourlis V, Rowley H, Taylor K, Whittacker J, Howard P, Beirne JC, Gosney JR, Woolgar J, Vaughan ED, Spandidos DA, Jones AS: Allelotype of squamous cell carcinoma of the head and neck: fractional allele loss correlates with survival. Br J Cancer 1995, 72:1180-1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Partridge M, Emilion G, Langdon JD: LOH at 3p correlates with a poor survival in oral squamous cell carcinoma. Br J Cancer 1996, 73:366-371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Akervall JA, Yuesheng J, Wennerberg JP, Zätterström UK, Kjellén E, Mertens F, Willén R, Mandahl N, Heim S, Mitelman F: Chromosomal abnormalities involving 11q13 are associated with poor prognosis in patients with squamous cell carcinoma of the head and neck. Cancer 1995, 76:853-859 [DOI] [PubMed] [Google Scholar]