

Abstract
The mechanisms for neuronal apoptosis after axotomy and target deprivation in the adult central nervous system are poorly understood. We used a unilateral occipital cortex ablation model in the adult rat to test the hypothesis that apoptotic retrograde neurodegeneration in the dorsal lateral geniculate nucleus occurs in association with oxidative stress and mitochondrial abnormalities. Immunodetection of 8-hydroxy-2′-deoxyguanosine, a marker for oxidative injury to DNA, demonstrated that these apoptotic neurons undergo oxidative stress. Dual immunolabeling for the retrograde tracer Fluorogold to identify projection neurons and for 8-hydroxy-2′-deoxyguanosine demonstrated that apoptotic, oxidatively damaged neurons are geniculocortical projection neurons. By electron microscopy, degeneration of dorsal lateral geniculate nucleus neurons evolved in association with a transient increase in mitochondria within the perikaryon of dying neurons during the transition between chromatolysis and early apoptosis. The morphological integrity of mitochondria was preserved until late in the progression of apoptosis. The dorsal lateral geniculate nucleus ipsilateral to the cortical lesion had a transient increase in cytochrome c oxidase activity, and geniculocortical neurons at the transitional, early apoptotic stage accumulated cytochrome c oxidase activity. We conclude that axotomy-induced, retrograde neuronal apoptosis in the adult central nervous system occurs in association with the accumulation of functionally active mitochondria within the perikaryon and oxidative damage to nuclear DNA.
Free oxygen radicals have been implicated in the induction of neuronal apoptosis by oxidative injury in vitro 1 and in vivo during development. 2 Mitochondria may participate in the effector stage of apoptosis 3-5 by directly providing a rich source of reactive oxygen species (ROS) or by changes in mitochondrial membrane cell death proteins 6,7 that prevent apoptosis via an antioxidant pathway 8 by blocking release of cytochrome c, 9,10 a required molecule for apoptosis in cell-free systems, 11 or by regulating membrane potential and volume homeostasis of mitochondria. 7
Target deprivation produced by visual cortex ablation causes corticopetal projection neurons in the dorsal lateral geniculate nucleus (dLGN) to die by a process that closely resembles apoptosis. 12 The death of these axotomized, target-deprived thalamic neurons follows an ordered sequence of subcellular changes that begins as chromatolysis and evolves as apoptosis along a synchronous time course, whereby end-stage neuronal apoptosis is achieved at ∼7 days after the lesion occurs. Degenerative alterations in dLGN neurons are manifested as progressive cytoplasmic and nuclear condensation with chromatin compaction into uniformly large, round clumps. Cytoplasmic and then nuclear fragments bud into the surrounding neuropil and are engulfed by glial cells. 12 These changes are characteristic of apoptosis in nonneural 13 and neural tissues. 14,15
As in nonneuronal cells undergoing apoptosis, 13 the morphological integrity of mitochondria is preserved in apoptotic neurons until late in the progression of neurodegeneration. 12,14,15 Therefore, we have hypothesized that neuronal apoptosis evolves in association with perikaryal accumulation of mitochondria and oxidative stress. Our occipital cortex ablation model of neuronal apoptosis in the adult brain provides a unique opportunity to test the hypothesis in vivo that oxidative damage develops in central nervous system (CNS) neurons undergoing apoptosis and that this stress occurs along with changes in mitochondrial distribution and function in apoptotic neurons.
Materials and Methods
Lesion Paradigm to Study Retrograde Degeneration of dLGN Neurons
An occipital cortex aspiration served as the model for producing axotomy and target deprivation of dLGN neurons. 12 We used adult male Sprague-Dawley rats (Charles River, Wilmington, MA) (n = 52), weighing ∼150 to 200 g, that were housed in a colony room with a 12-hour light/dark cycle and ad libitum access to food and water. The animal protocol was approved by the Animal Care and Use Committee of The Johns Hopkins University School of Medicine. Rats were anesthetized with a mixture of enflurane:oxygen:nitrous oxide (1:33:66) and placed in a stereotaxic apparatus. After a midline scalp incision, a 2 × 2-mm craniotomy was made; the medial side of the craniotomy was located 1 mm lateral to the midline, and the caudal side was 1 mm rostral to λ. The dura was incised using a sharp 22-gauge needle. The cortex underlying the craniotomy was then aspirated using a blunt-tipped 22-gauge needle connected to a vacuum line, without damaging the surrounding venous sinuses and the underlying hippocampus. Postlesion survival times after occipital cortex ablation were: 1 day or 3, 4, 5, 6, and 7 days. In other rats (n = 4), a similar-sized aspiration lesion was made in the frontal cortex, which does not receive geniculocortical projections, to serve as a control for the effects of the surgical manipulation.
Retrograde Labeling of Corticopetal Projection Neurons in the dLGN
The retrograde tracer Fluorogold (FG; Fluorochrome, Inc., Englewood, CO) was injected into the visual cortex to prelabel corticopetal projection neurons in the dLGN. An occipital craniotomy was performed as described above, and, 0.7 mm below the cortical surface, 100 nl of 5% FG in deionized distilled H2O was injected slowly over 10 minutes using a 25-gauge 0.5-ml blunt-tip syringe (Hamilton, Reno, NV). Three days later, rats (n = 8) were anesthetized and underwent an occipital cortex ablation as described above. FG-injected rats with occipital cortex ablations recovered for 5 to 7 days before being perfused for light microscopic analysis. Identification of corticopetal projection neurons was done by direct fluorescence and by immunodetection of FG using a polyclonal antibody (diluted 1:10,000; Chemicon, Inc., Temecula, CA) in an immunoperoxidase procedure similar to that performed for the identification of oxidative damage to DNA in the dLGN (see below).
Light Microscopic Evaluation of Neurodegeneration in the dLGN
At 1 day or 3, 5, 6, and 7 days postlesion, rats (n ≥ 4 per time point) were anesthetized intraperitoneally with sodium pentobarbital (70 mg/kg) and perfused intra-aortically with 1% paraformaldehyde (PF) in ice-cold phosphate buffer (PB; 0.12 mol/L, pH 7.4) followed by 4% PF in ice-cold PB. Brains were removed from the skull, blocked in the coronal plane, postfixed in 4% PF for 1 to 2 hours at 4°C, cryoprotected in 30% sucrose in PB, and then frozen in isopentane chilled by dry ice. Frozen sections (40 μm) through the lateral geniculate nucleus were either stained with cresyl violet for light microscopy or were used for the immunocytochemical detection of oxidative injury marker or for the enzyme histochemical detection of cytochrome c oxidase (COX) activity.
Immunocytochemical Detection of Oxidative Damage to DNA in dLGN Neurons
The formation of 8-hydroxy-2′-deoxyguanosine (OH8dG) occurs as a product of oxidative injury to DNA by ROS. 16 Singlet oxygen, 17 as well as nitric oxide and superoxide, 18 produces OH8dG, the formation of which is inhibited by free radical scavengers. 18 Therefore, we immunocytochemically assayed for OH8dG in dLGN neurons in brain sections from rats at 5, 6, and 7 days postlesion (n = 2 or 3 per time point) using a monoclonal antibody (QED Bioscience, Inc., San Diego, CA). This antibody also reacts with 8-hydroxyguanine and 8-hydroxyguanosine according to the manufacturer’s specifications. Brain sections were first washed in Tris-buffered saline (TBS, pH 7.4) and then incubated in 2% normal goat serum and 0.35% Triton X-100 in TBS for 1 hour. Sections were then incubated at 4°C for ∼48 hours in anti-OH8dG antibody at a concentration of 2.9 μg/ml diluted in TBS with 2% normal goat serum and 0.18% Triton X-100. For competition controls, sections were reacted with antibody to OH8dG that was incubated at 4°C for 24 hours with 1000-fold concentrations of OH8dG and 8-hydroxyguanosine (Cayman Chemical, Ann Arbor, MI) and then centrifuged at 14,000 rpm for 2 minutes to pellet antigen-antibody complexes. The positive control for competition experiments was OH8dG antibody unexposed to antigen but subjected to the same procedure as preadsorbed antibody. As additional controls, sections were pretreated with DNase or RNase (1 mg/ml in magnesium sulfate buffer) or proteinase K (20 μg/ml in Tris buffer) before incubation with OH8dG antibody. After primary antibody incubation, sections were washed in TBS and incubated for 1 hour with biotinylated goat anti-mouse antibody (1:200 in 2% normal goat serum and 0.18% Triton X-100 at room temperature; Vector Laboratories, Burlingame, CA), washed again in TBS, and then incubated in an avidin and biotinylated horseradish peroxidase solution (1:70, Vector Laboratories). Sections were finally processed with a standard diaminobenzidine (Sigma Chemical Co., St. Louis, MO) chromagenic reaction (0.6 mg/ml diaminobenzidine and 0.01% H2O2 in TBS).
Assay for Mitochondrial Function in the dLGN during Neuronal Apoptosis
Mitochondrial function in the dLGN at 5 and 6 days after occipital cortex ablation was evaluated using the COX histochemical method of Wong-Riley, 19 as described previously. 20 The 5- and 6-day time points were chosen because these times correspond to the chromatolytic and early apoptotic stages of neurodegeneration in the dLGN. 12 The enzymatic reaction product in the dLGN ipsilateral and contralateral to the cortical lesion was quantified densitometrically as described. 20 Statistical analysis of the data was performed using the paired Student’s t-test.
Ultrastructural Analysis of Neuronal Injury and Death in the dLGN after Occipital Cortex Ablation
After 1 day and 3, 5, 6, or 7 days postlesion, rats (n = 2 per time point) were anesthetized with sodium pentobarbital and perfused intra-aortically with 1% PF/0.1% glutaraldehyde in ice-cold PB followed by 2% PF/2% glutaraldehyde in ice-cold PB. Brains were removed from the skull and postfixed overnight. Samples of lateral geniculate nucleus ipsilateral and contralateral to the cortical lesion were microdissected, rinsed in PB, placed in 2% osmium tetroxide for 2 hours, dehydrated, and embedded in plastic. Semithin sections (1 μm) stained with 1% toluidine blue were screened for regions of interest, and then thin sections (gold interference color) were cut on an ultramicrotome (Sorvall, Norwalk, CT), contrasted with uranyl acetate and lead citrate, and viewed with a JEOL 100S electron microscope.
Semiquantitative Analysis of Mitochondrial Accumulation within Neuronal Perikarya
The number of mitochondria per neuronal cross-section was analyzed using electron micrographs from cortically ablated rats at 1 day and 5, 6, and 7 days postlesion. Neurons were grouped as normal, 5-day chromatolytic, 6 to 7-day early apoptotic, and 6 to 7-day late apoptotic. In a random selection of five to eight micrographs per group, recognizable mitochondria were counted in complete cross-sectional profiles of neuronal perikarya. An average number of mitochondria per cross-section was calculated. Neuronal cross-sectional size was not taken into consideration to select as randomly as possible the micrographs. Statistical analysis of mitochondrial accumulation was performed using the paired Student’s t-test.
Results
Apoptotic dLGN Neurons Show Evidence for Oxidative Damage
This model of occipital cortex ablation causes apoptotic death of neurons within the dLGN over a time course of ∼1 week (Figure 1A) ▶ . 12 Immunoreactivity for OH8dG was detected in cells within the dLGN ipsilateral to the occipital cortex lesion at 5 to 7 days postlesion (Figures 1 ▶ , B and C, and Figure 2 ▶ , A and B). This time corresponds to the early to late stages of apoptosis in dLGN neurons. 12 OH8dG immunoreactivity was localized within the nucleus and perikaryon of neurons that had morphological features of apoptosis in cresyl violet-counterstained sections (Figure 1 ▶ , B and C). DNase and RNase abolished the nuclear and cytoplasmic immunolabeling, respectively, obtained with OH8dG antibodies, but proteinase K treatment neither altered the immunolabeling in the ipsilateral dLGN nor enhanced labeling of neurons in the contralateral dLGN (data not shown). In neurons at early stages of apoptosis (Figures 1B and 2A) ▶ ▶ , as indicated by the nascent condensation of chromatin into numerous small clumps and the absence of appreciable cell shrinkage, OH8dG immunoreactivity within and around the nucleus was faint and often punctate. In neurons at later stages of apoptosis (Figures 1C and 2 ▶ ▶ , A and B), as indicated by the advanced condensation of chromatin into a few large clumps and the presence of cell shrinkage, OH8dG immunoreactivity was dense and uniformly dark throughout the nucleus, except within regions of dense chromatin clumping that had lighter staining (Figure 2B) ▶ . This pattern of immunoreactivity was not seen outside the focus of neurodegeneration within the ipsilateral dLGN, nor was it observed in the contralateral dLGN at 5 to 7 days postlesion (Figure 2C) ▶ or in the dLGN ipsilateral to the frontal cortex lesion (data not shown). Preadsorption of OH8dG antibody with OH8dG and 8-hydroxyguanosine before the immunocytochemistry competed the signal in apoptotic neurons (Figure 2D) ▶ .
Figure 1.
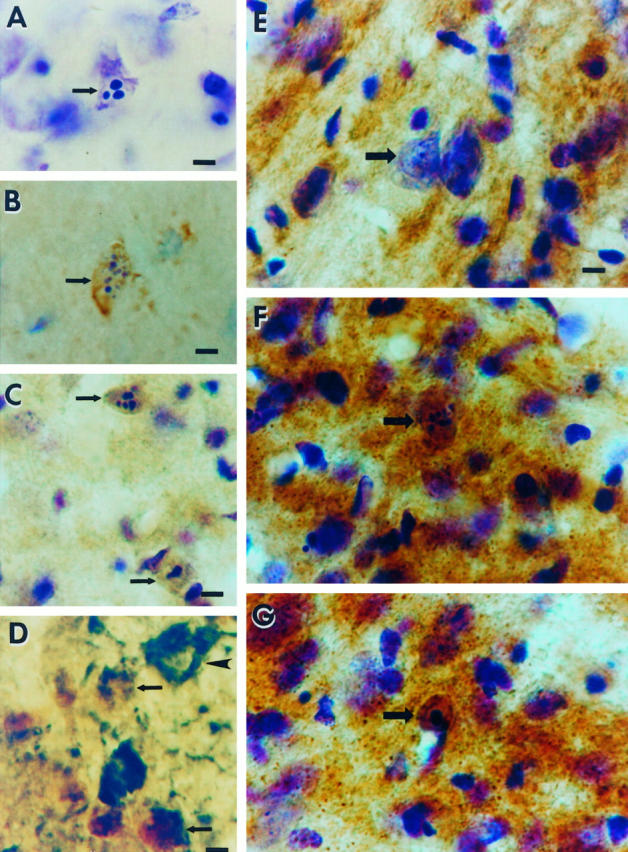
Occipital cortex ablation in the adult rat results in apoptosis of geniculocortical projection neurons within the dLGN that is associated with oxidative damage to the nucleus and increased mitochondrial COX activity within the perikaryon. A: Cresyl violet staining shows a representative apoptotic neuron (arrow) within the dLGN at 6 days postlesion. Note the three round clumps of chromatin within the nucleus, consistent with the morphology of apoptosis. The identification of these cells as neurons has been described. 12 Scale bar = 10 μm. B: Immunoreactivity (brown labeling) for the oxidative stress marker, OH8dG, is detected within apoptotic neurons (arrow) at 5 days postlesion. The cresyl violet counterstaining (purple) shows that this cell (arrow) is in the early stages of apoptotic chromatin condensation, as indicated by the formation of numerous, small aggregates of chromatin, and has not yet undergone shrinkage of the cell body (compare the size of the neuron in B with that of the apoptotic neurons in C). Scale bar = 10 μm. C: At 7 days postlesion, apoptotic neurons (arrows) show intense OH8dG immunoreactivity (brown) throughout the nucleus. The cresyl violet counterstaining (purple) reveals that these neurons are in the late stages of apoptosis, as indicated by the chromatin clumping and the cellular shrinkage. Scale bar = 10 μm. D: Double label immunodetection of OH8dG (brown) and the retrograde tracer FG (dark green) demonstrates that the cells that have evidence for oxidative stress within the nucleus are geniculocortical projection neurons (arrows), whereas a nearby undamaged geniculocortical projection neuron (arrowhead) has no OH8dG immunoreactivity within the nucleus and is not shrunken. Scale bar = 10 μm. E: In COX histochemical preparations counterstained with cresyl violet, COX activity (brown) in the control (contralateral to the occipital cortex ablation) dLGN is localized within the neuropil, and only faint activity appears within neuronal cell bodies (arrow). Scale bar = 10 μm (applies also to F and G). F and G: In the dLGN ipsilateral to the occipital cortex ablation, COX histochemistry/cresyl violet staining shows that axotomized neurons (arrows) at early stages of apoptosis at 5 days (F) and later stages of apoptosis at 6 days (G) postlesion are highly enriched in COX activity, indicating the accumulation of functional mitochondria within the perikaryon.
Figure 2.

Oxidative damage occurs within the ipsilateral dLGN after occipital cortex ablation, as determined by immunodetection of OH8dG. A and B: The detection of OH8dG immunoreactivity within cells (arrows) is widespread within the focus of axotomy-induced neurodegeneration in the ipsilateral dLGN. OH8dG immunoreactivity is primarily confined to the nucleus and is detected as small, granular immunoreactivity (arrowhead) or as large, condensed aggregates of immunoreactivity (arrows). B: At higher magnification, the nuclear OH8dG immunoreactivity is localized diffusely within the periphery of the nucleus as well as at the surfaces of round aggregates, corresponding to the round, condensed clumps of chromatin that are detected by cresyl violet staining (see Figure 1A ▶ ). Scale bar = 5.5 μm. C: The control dLGN (ie, contralateral of the occipital cortex ablation) has no OH8dG immunoreactivity. D: Preadsorption of the OH8dG monoclonal antibody with purified OH8dG abolishes the nuclear OH8dG immunolabeling within the ipsilateral dLGN. Scale bar = 25 μm (applies also to A and C).
Apoptotic Cells with Oxidative Damage Are Geniculocortical Projection Neurons
Injections of FG into the future center of the lesion site 3 days before the occipital cortex lesion labels the cytoplasm of neurons within the focus of neurodegeneration; therefore, the degenerating cells in the ipsilateral dLGN correspond to a subset of geniculocortical neurons. 12 Dual labeling immunocytochemistry revealed that OH8dG immunoreactivity was present in FG-labeled geniculocortical projection neurons (Figure 1D) ▶ . The cytoplasm of FG-labeled OH8dG-immunoreactive neurons was often incomplete, as the cytoplasmic fragmentation became more prominent during the progression of apoptosis. 12
Apoptotic dLGN Neurons Accumulate Mitochondria after Occipital Cortex Ablation
To provide insight into the possible mechanisms of oxidative injury in apoptotically dying neurons within the dLGN, electron microscopy (EM) was used to evaluate their ultrastructure (Figure 3) ▶ . Ultrastructural changes in injured dLGN neurons during the first 5 days postlesion included an early redistribution of the rough endoplasmic reticulum followed by its dispersion and fragmentation, release of free ribosomes, and dilation of Golgi cisterns (Figure 3 ▶ , B and C). These changes are in accord with our previous EM results 12 and are consistent with the axon reaction after axotomy. 21
Figure 3.
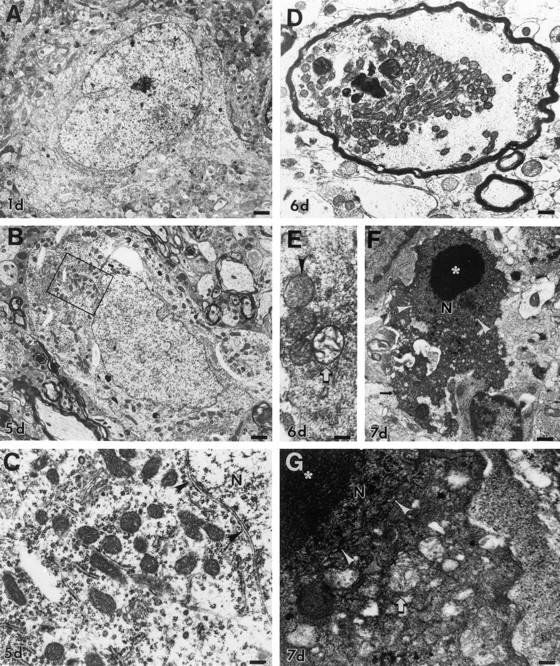
Degeneration of dLGN neurons resembles apoptosis ultrastructurally and is associated with accumulation of mitochondria and then mitochondrial swelling and dissolution. A: At 1 day postlesion, neurons in the ipsilateral dLGN appear healthy, with a normal nucleus and perinuclear distribution of organelles. Scale bar = 2 μm. B: By 5 days, the rough endoplasmic reticulum is redistributed to the perikaryal periphery and becomes more apparent. The Golgi cisterns become swollen. Mitochondria, which are all ultrastructurally intact at this time (box, shown enlarged in C), appear to be more numerous. The nucleus appears normal, although eccentrically placed. Scale bar = 1.75 μm. C: Chromatolytic neurons at 5 days postlesion appear to have accumulations of ultrastructurally normal mitochondria in some regions around the nucleus (N), which is still surrounded by an intact nuclear envelope (arrowheads). Scale bar = 0.5 μm. D: After axotomy, mitochondria also accumulate within large, swollen, myelinated axons within the dLGN. Scale bar = 0.8 μm. E: In the early stages of apoptosis at 6 days postlesion, the perikarya of dying neurons contain mitochondria that are pale and swollen or overtly showing cristaeolysis (open arrow). Scale bar = 0.3 μm. F: At 7 days postlesion, the apoptotic process is well advanced, and neurons are dark and shrunken. The condensed cytoplasm contains numerous small vacuoles, some of which are degenerating mitochondria (see G). The nucleus (N) is organized into large, round, compact chromatin clumps embedded within a homogeneously condensed nuclear matrix and is still surrounded partially by a nuclear membrane (arrowheads). These dying cells are identified as neurons by the presence of residual synapses (arrow). Scale bar = 1.5 μm. G: In apoptotic neurons at 7 days postlesion, the cytoplasm is fused and dark, containing many small vacuoles. Most mitochondria (arrow) are degenerating and appear as larger vacuoles containing cristae remnants. The nucleus (N), surrounded by a less conspicuous, undulated nuclear envelope (arrowheads), is condensed and dark, and it contains large, compact chromatin clumps (asterisk). Scale bar = 0.3 μm.
Chromatolytic neurons within the dLGN after occipital cortex ablation appeared to accumulate structurally intact mitochondria by 5 days postlesion (Figure 3 ▶ , B and C) as compared with uninjured neurons (Figure 3A) ▶ . In the surrounding neuropil, some axons were swollen and contained many mitochondria (Figure 3D) ▶ , possibly representing abnormal axonal trafficking of mitochondria in axotomized dLGN neurons. There was no ultrastructural evidence for mitochondrial proliferation (eg, partition membrane formation). At 6 days postlesion, as chromatolytic neurons progressed into the early stages of apoptosis, 12 some mitochondria that accumulated in the perikaryon underwent swelling and disintegration of the inner membrane, whereas other mitochondria remained intact (Figure 3E) ▶ . At 7 days postlesion, end-stage neuronal apoptosis was widespread (Figure 3F) ▶ . The organelles within the degenerated neurons were tightly packed within a dark cytoplasmic matrix of neurofilaments and microtubules, forming a lattice that was organized into condensed laminations (Figure 3 ▶ , F and G). In the late stages of apoptosis, the disintegrating mitochondria appeared as vacuoles within the condensed cytoplasm (Figure 3G) ▶ .
Mitochondria accumulated within the perikaryon before the early morphological stages of neuronal apoptosis. In neurons that were chromatolytic at 5 days postlesion, the number of mitochondria (per perikaryal cross-section) increased as compared with uninjured neurons (Figure 4) ▶ . As neurons entered the apoptotic stages of degeneration, their cell bodies underwent shrinkage and condensation, with perikaryal mitochondria number being similar to that of controls (Figure 4) ▶ . At the final stages of apoptosis, the number of recognizable mitochondria decreased significantly (Figure 4) ▶ , as mitochondria disintegrated (Figure 3 ▶ , F and G) and as components of the cytoplasm budded and dispersed into the surrounding neuropil. 12
Figure 4.

Histogram of the semiquantitative analysis of mitochondrial accumulation within neuronal perikarya in the ventromedial dLGN after occipital cortex ablation. Values are means ± SD. At 5 days postlesion, the number of recognizable mitochondria per neuronal cross-section was 176% of control, whereas by 6 to 7 days postlesion, the number of recognizable mitochondria was 41% of control in neurons at late stages of apoptosis. *Significant difference (P = 0.07) as compared with control; **significant difference (P = 0.02 and P = 0.007) as compared with control and 5 days postlesion, respectively.
Mitochondrial Function Is Increased Transiently in the dLGN after Occipital Cortex Ablation
The increased numbers of mitochondria within the perikaryon of apoptotically dying neurons within the dLGN, as determined by EM, were paralleled by an enhanced mitochondrial functional activity, as demonstrated by enzyme histochemical assay for COX (Figures 1 ▶ , E to G, and Figure 5 ▶ ). Histochemical preparations that were counterstained with cresyl violet revealed that COX activity was enriched within punctate structures in the neuropil and in neurons. These sections also demonstrated striking accumulations of COX activity within the somata of cells with morphological features consistent with apoptotic neurons (Figure 1 ▶ , F and G). Densitometric quantification of uncounterstained sections demonstrated that COX activity was increased in the ipsilateral dLGN as compared with the contralateral dLGN at 5 days after occipital cortex ablation (Figure 5) ▶ . This overall increase in mitochondrial function was transient, because, by 6 days postlesion, the ipsilateral and contralateral dLGN COX activity values were similar (Figure 5) ▶ .
Figure 5.

Histogram representation of the quantitative densitometric analysis of histochemical COX activity within the dLGN after occipital cortex ablation. Values are means ± SD of the optical density (OD) of the reaction product. At 5 days postlesion, COX activity was 130% of the contralateral control level but was unchanged at 6 days postlesion. *Significant difference (P = 0.03) as compared with contralateral, control dLGN.
Discussion
The retrograde degeneration of geniculocortical projection neurons that occurs after visual cortex ablation in the adult brain is apoptosis, 12 based on morphological criteria. 13-15 The experiments described here were conducted to explore the possible mechanisms for this apoptotic death of dLGN neurons after axotomy and target deprivation, by testing the hypothesis that this neuronal death occurs in association with oxidative injury and mitochondrial accumulation.
The present study is novel, because our data implicate the formation of ROS and oxidative damage to nucleic acids in an in vivo setting of retrograde neuronal apoptosis in the adult CNS. We found that OH8dG, a marker for oxidative damage in the form of DNA base lesions, 16,28,29 is detected immunocytochemically within the nucleus of apoptotic neurons; furthermore, cytoplasmic immunolabeling was observed with OH8dG antibodies. This immunolabeling was competed with both OH8dG and hydroxyguanosine, and the nuclear and cytoplasmic labeling was abolished, respectively, by DNase and RNase, demonstrating that this antibody detects both hydroxylated DNA and RNA. These observations suggest that free radical-mediated damage to DNA and RNA occur during the progression of neuronal apoptosis in the adult CNS. Alternatively, biochemical changes may occur within apoptotic cells that permit increased accessibility of OH8dG antibodies to nucleic acid antigens; nevertheless, this immunolabeling could be blocked by antibody preadsorption, and it was not observed in control dLGN.
By combining neuronal tract tracing with the immunodetection of OH8dG, we have demonstrated that the retrogradely dying geniculocortical projection neurons are the cells that are oxidatively damaged. The results also show that apoptosis of retrogradely degenerating neurons occurs in association with the accumulation of mitochondria within neurons. The relationships between cell volume and mitochondrial number during apoptosis were not measured in the present experiments. However, it appears that the transient accumulation of mitochondria occurs at a time when cell shrinkage is not yet prominent, 12 but as neurons shrink, once they have passed beyond the chromatolytic stage of apoptosis, mitochondrial number per cross-sectional area decreases (rather than increases), because mitochondria degenerate within the cytoplasm of apoptotic neurons instead of being maintained structurally and packaged into apoptotic bodies 12 as in classical apoptosis in nonneuronal cells. 13 Thus, the structural integrity of mitochondria is not preserved in degenerating neurons at late apoptosis. The finding that COX activity is increased at 5 days after axotomy suggests that some of these mitochondria are functionally active, although the efficiency of electron transfer in these mitochondria is uncertain. These mitochondrial changes provide a possible mechanism for the oxidative damage to macromolecules that occurs in apoptotic neurons. The morphological features of apoptosis and these histochemical observations on oxidative stress, as well as mitochondrial accumulation and activation, appear to be temporally correlated. Based on these data, we conclude that axotomy and target deprivation in this model cause alterations in the distribution of mitochondria within neurons, possibly because of abnormal trafficking of mitochondria, and subsequent perikaryal accumulation of functionally active mitochondria. These mitochondria within the perikaryon of neurons may provide a source of ROS that mediate oxidative damage to nucleic acids.
Oxidative stress can induce apoptosis in nonneuronal 8,22,23 and neuronal 1,24,25 in vitro models. In nonneuronal cells, ROS may contribute to the structural mechanisms of the apoptotic morphological phenotype. For example, the cytoplasmic budding and chromatin condensation during apoptosis are associated with cytoskeletal abnormalities after oxidation of sulfhydryl groups in actin filaments. 26,27 Among the ROS, hydroxyl radicals are highly reactive and are thought to be genotoxic by interacting with DNA and producing DNA strand breaks and base modifications. 28,29 Hydroxyl radicals are products of the transitional metal (eg, iron)-catalyzed, Haber-Weiss- and Fenton-type reactions that use superoxide and hydrogen peroxide as substrates, respectively. 30 Our experiments demonstrate, for the first time, the formation of hydroxyl radical-modified DNA during the progression of neuronal apoptosis in vivo.
The mitochondrial alterations that we have observed in dying dLGN neurons support our conclusion that oxidative stress participates in the induction of neuronal apoptosis in vivo. Three transient changes in mitochondria were observed during the progression of apoptotic degeneration of dLGN neurons: 1) mitochondria accumulated in the perikaryon of neurons at 5 days postlesion, a time corresponding to the chromatolytic, preapoptotic stage of degeneration that we have identified ultrastructurally; 12 2) mitochondrial function was increased at 5 days postlesion (as indicated by the elevated COX activity) in the focus of imminent apoptotic neurodegeneration within the dLGN and was enhanced within the cell body of dying neurons; and 3) mitochondria ultimately underwent swelling and then inner membrane disintegration in dLGN neurons during the early and late stages of apoptosis, respectively. These structural changes are consistent with the possible role for alterations in mitochondrial membrane potential, permeability, and volume homeostasis in apoptosis. 4-7
In the peripheral and central nervous systems, axotomy and axonal constriction result in the accumulation of mitochondria in proximal axons and in the perikaryon of neurons. 31-34 This accumulation may be due to an interruption of mitochondrial trafficking and to perturbations in cytoskeletal motors that may result from the axonal degeneration and dendritic attrition within dLGN neurons, 12 or it may reflect mitochondrial proliferation. This latter possibility is unlikely, because we did not observe the typical formation of a partition membrane that occurs during fission of elongated mitochondria. 35 Alternatively, apparent mitochondrial accumulation within dLGN neurons may be a consequence of cell body shrinkage during the process of apoptosis. The accumulation of mitochondria as an early event in apoptosis may be specific to apoptosis in retrogradely degenerating neurons (rather than a general feature of apoptosis in all cell types), because neurons are structurally distinct from nonneuronal cells because of their dendrites and axons. A possible consequence of distal axotomy of minimally collateralized neurons or proximal axotomy of collateralized neurons is that mitochondria are in closer proximity to each other and to the nucleus and other organelles within the perikaryon. This mitochondria-mitochondria or mitochondria-nucleus proximity hypothesis may be important mechanistically, because hydroxyl radicals are highly reactive and short-lived 30 and, therefore, need to be generated at sites near the target macromolecules (eg, nuclear or mitochondrial DNA or cytoplasmic RNA) for damage to occur.
We observed that mitochondrial function is enhanced transiently in the lateral geniculate nucleus after axotomy and target deprivation. ROS are products of oxidative metabolism. The mitochondrial electron-transfer chain is a primary generator of superoxide and peroxide, and damaged mitochondria are believed to produce even more superoxide ion. 30 Point mutations in mitochondrial DNA caused by oxidative damage may lead to protein conformational changes usually associated with an inefficient electron transfer to COX 36 and, hence, enhanced superoxide and peroxide formation. We found by EM that neurons in the early stages of apoptosis contain both morphologically intact mitochondria and damaged mitochondria. Consistent with our data, previous studies have also revealed that axotomy and axonal ligation in the peripheral nervous system cause an elevation of mitochondrial oxidative enzymes in the axon segment that is proximal to the site of injury. 37 We suggest that a result of mitochondrial accumulation and increased mitochondrial COX activity, regardless of whether electron transfer is efficient or inefficient, may be an overwhelming generation of ROS, depletion of mitochondrial and cytosolic antioxidant mechanisms, and subsequent oxidative damage to nucleic acids. During the stages of neuronal apoptosis at 6 to 7 days postlesion, the mitochondria become progressively damaged, as evidenced by the swelling and cristeolysis that appear concurrently with incipient cytoplasmic compaction, supporting the hypothesis that apoptosis-initiating factor(s) and cytochrome c are sequestered in the mitochondrial intermembrane space, 11,38 which on their release activate a cell death cascade. 11 In addition to their role as a source of ROS, mitochondria may also trigger changes in nuclear morphology. The opening of mitochondrial permeability transition pores and the consequent reduction of mitochondrial transmembrane potential has been shown to induce chromatin clumping in vitro. 5 However, in our in vivo experiments, it is still uncertain whether changes in mitochondria or oxidative stress are directly related to the activation of programmed cell death pathways or whether these changes are merely associations without causal implications for retrograde neuronal apoptosis. In our in vivo paradigm, we have provided several single-time-point data representations to reconstruct morphologically the process of neuronal death, 12 but we cannot track the evolution of oxidative stress and apoptosis in a single neuron in vivo. Thus, although we can associate mitochondrial accumulation, enhanced COX activity, and oxidative damage with neuronal apoptosis in the CNS after axotomy and target deprivation, this evidence is only indirect with regard to causality of neuronal death and may represent structural and biochemical end products of neurodegeneration.
Our findings showing that apoptosis of retrogradely degenerating neurons in the adult rat CNS occurs in association with the accumulation of mitochondria and oxidative damage may have important implications for the mechanisms of neuronal death in adult-onset, human neurodegenerative diseases such as amyotrophic lateral sclerosis and Alzheimer’s disease. Both diseases may have components of retrograde neuronal injury, which may contribute to the evolution of neurodegeneration, 14 and oxidative damage has been found in postmortem brains from individuals with amyotrophic lateral sclerosis 39 and Alzheimer’s disease. 40,41 In addition, in a small subset of subjects with familial amyotrophic lateral sclerosis, mutations have been identified in the superoxide dismutase 1 gene 42,43 that may result in a toxic gain of function in this enzyme and enhanced generation of ROS. 44 Although the contributions of neuronal apoptosis to the neurodegenerative processes in these human CNS diseases are still not clearly identified, 14 we found that the neurodegeneration in amyotrophic lateral sclerosis resembles apoptosis and may occur by a programmed cell death mechanism involving a subcellular redistribution of death proteins that are localized to mitochondria. 45 Therefore, it is possible that the CNS neurodegeneration in our animal model of axotomy/target deprivation and in human age-related diseases are mechanistically similar, although differing in the rate of progression of neuronal death.
Conclusions
An occipital cortex ablation model for producing neuronal apoptosis in the adult brain 12 was used to test the hypothesis that oxidative stress and abnormalities in mitochondria occur during the process of neuronal apoptosis in vivo. We conclude that oxidative damage to DNA occurs during the apoptotic retrograde degeneration of geniculocortical projection neurons induced by axotomy and target deprivation. This oxidative injury evolves in association with a transient accumulation of mitochondria and a functional increase in mitochondrial COX activity within the perikaryon of apoptotically dying neurons.
Acknowledgments
The authors thank Frank Barksdale and Ann Price, as well as the technical staff of the Department of Pathology EM core facility (Marilyn Miller, Barbara Plantholt, and Gerald Horne), for their assistance.
Footnotes
Address reprint requests to Dr. Lee J. Martin, Department of Pathology, The Johns Hopkins University School of Medicine, 720 Rutland Avenue, 558 Ross Research Building, Baltimore, MD 21205-2196. E-mail: lmartin@welchlink.welch.jhu.edu.
Supported by grants from the U.S. Public Health Service (NS34100) and the American Federation for Aging Research/Glenn Foundation.
References
- 1.Ratan RR, Murphy TH, Baraban JM: Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem 1994, 62:376-379 [DOI] [PubMed] [Google Scholar]
- 2.Wood KA, Youle RJ: The role of free radicals and p53 in neuron apoptosis in vivo. J Neurosci 1995, 15:5851-5857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newmeyer DD, Farschon DM, Reed JC: Cell-free apoptosis in Xenopus egg extracts: inhibition by bcl-2 and requirement for an organelle fraction enriched in mitochondria. Cell 1994, 79:353-364 [DOI] [PubMed] [Google Scholar]
- 4.Skulachev VP: Why are mitochondria involved in apoptosis? FEBS Lett 1996, 397:7-10 [DOI] [PubMed] [Google Scholar]
- 5.Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M, Kroemer G: Mitochondrial control of nuclear apoptosis. J Exp Med 1996, 183:1533-1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kroemer G, Petit P, Zamzami N, Vayssiere JL, Mignotte B: The biochemistry of programmed cell death. FASEB J 1995, 9:1277-1287 [DOI] [PubMed] [Google Scholar]
- 7.Vander Heiden MG, Chandel NS, Williamson EK, Schumacker PT, Thompson CB: Bcl-xL regulates the membrane potential and volume homeostasis of mitochondria. Cell 1997, 91:627-637 [DOI] [PubMed] [Google Scholar]
- 8.Hockenberry DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ: Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell 1993, 75:241-251 [DOI] [PubMed] [Google Scholar]
- 9.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X: Prevention of apoptosis by bcl-2: release of cytochrome c from mitochondria blocked. Science 1997, 275:1129-1132 [DOI] [PubMed] [Google Scholar]
- 10.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD: The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science 1997, 275:1132-1136 [DOI] [PubMed] [Google Scholar]
- 11.Liu X, Kim CN, Yang J, Jemmerson R, Wang X: Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell 1996, 86:147-157 [DOI] [PubMed] [Google Scholar]
- 12.Al-Abdulla NA, Portera-Cailliau C, Martin LJ: Occipital cortex ablation in adult rat causes retrograde neuronal death in the lateral geniculate nucleus that resembles apoptosis. Neuroscience 1998, 86:191-209 [DOI] [PubMed] [Google Scholar]
- 13.Kerr JFR, Gobé GC, Winterford CM, Harmon BV: Anatomical methods in cell death. Schwartz LM Osborne BA eds. Cell Death. 1995, :pp 1-27 Academic Press, New York [DOI] [PubMed] [Google Scholar]
- 14.Martin LJ, Al-Abdulla NA, Brambrink AM, Kirsch JR, Sieber FE, Portera-Cailliau C: Neurodegeneration in excitotoxicity, global cerebral ischemia, and target deprivation: a perspective on the contributions of apoptosis and necrosis. Brain Res Bull 1998, 46:281-309 [DOI] [PubMed] [Google Scholar]
- 15.Portera-Cailliau C, Price DL, Martin LJ: Excitotoxic neuronal death in the immature brain is an apoptosis-necrosis morphological continuum. J Comp Neurol 1997, 378:70-87 [PubMed] [Google Scholar]
- 16.Kasai H, Nishimura S: Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res 1984, 12:2137-2145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devasagayam TPA, Steenken S, Obendorf MSW, Schultz WA, Sies H: Formation of 8-hydroxy(deoxy)-guanosine and generation of strand breaks at guanine residues in DNA by singlet oxygen. Biochemistry 1991, 30:6283-6289 [DOI] [PubMed] [Google Scholar]
- 18.Inoue S, Kawanishi S: Oxidative DNA damage induced by simultaneous generation of nitric oxide, and superoxide. FEBS Lett 1995, 371:86-88 [DOI] [PubMed] [Google Scholar]
- 19.Wong-Riley MTT: Changes in the visual system of monocularly sutured or enucleated cats demonstrable with cytochrome oxidase histochemistry. Brain Res 1979, 171:11-28 [DOI] [PubMed] [Google Scholar]
- 20.Martin LJ, Brambrink A, Koehler RC, Traystman RJ: Primary sensory and forebrain motor systems in the newborn brain are preferentially damaged by hypoxia-ischemia. J Comp Neurol 1997, 377:262-285 [DOI] [PubMed] [Google Scholar]
- 21.Lieberman AR: The axon reaction: a review of the principal features of perikaryal responses to axon injury. Int Rev Neurobiol 1971, 14:49-124 [DOI] [PubMed] [Google Scholar]
- 22.Lin K-T, Xue J-Y, Nomen M, Spur B, Wong PYK: Peroxynitrite-induced apoptosis in HL60 cells. J Biol Chem 1995, 270:16487-16490 [DOI] [PubMed] [Google Scholar]
- 23.Verhaegen S, McGowan AJ, Brophy AR, Fernandes RS, Cotter TG: Inhibition of apoptosis by antioxidants in the human HL-60 leukemia cell line. Biochem Pharmacol 1995, 50:1021-1029 [DOI] [PubMed] [Google Scholar]
- 24.Ratan RR, Murphy TH, Baraban JM: Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci 1994, 14:4385-4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonfoco E, Krainc D, Ankarcrona M, Nicotera P, Lipton SA: Apoptosis and necrosis: two distinct events induced respectively by mild and intense insults with NMDA or nitric oxide/superoxide in cortical cell cultures. Proc Natl Acad Sci USA 1995, 92:72162-72166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mirabelli F, Salis A, Marinoni V, Finardi G, Bellomo G, Thor H, Orrenius S: Menadione-induced bleb formation in hepatocytes is associated with the oxidation of thiol groups in actin. Arch Biochem Biophys 1988, 264:261-269 [DOI] [PubMed] [Google Scholar]
- 27.Mirabelli F, Salis A, Perotti M, Taddei F, Bellomo G, Orrenius S: Alterations of surface morphology caused by metabolism of menadione in mammalian cells are associated with the oxidation of critical sulfhydryl groups in cytoskeletal proteins. Biochem Pharmacol 1998, 37:3423-3427 [DOI] [PubMed] [Google Scholar]
- 28.Kuchino Y, Mori F, Kasai H, Inoue H, Iwai S, Miura K, Ohtsuka E, Nishimura S: Misreading of DNA templates containing 8-hydroxydeoxyguanosine at the modified base and at adjacent residues. Nature 1987, 327:77-79 [DOI] [PubMed] [Google Scholar]
- 29.Fraga CG, Shigenaga MK, Park J-W, Degan P, Ames BN: Oxidative damage to DNA during aging: 8-hydroxy-2′-deoxyguanosine in rat organ DNA and urine. Proc Natl Acad Sci USA 1990, 87:4533-4537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boveris A, Cadenas E: Cellular sources and steady-state levels of reactive oxygen species. Biadasz Clerch L Massaro DJ eds. Oxygen, Gene Expression, and Cellular Function. 1997, :pp 1-25 Marcel Dekker, New York [Google Scholar]
- 31.Hartmann JF: Electron microscopy of motor nerve cells following section of axones. Anat Rec 1954, 118:19-33 [DOI] [PubMed] [Google Scholar]
- 32.Weiss P, Pillai A: Convection and fate of mitochondria in nerve fibers: axonal flow as vehicle. Proc Natl Acad Sci USA 1965, 54:48-56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blümcke S, Neidorf HR, Rode J: Axoplasmic alterations in the proximal and distal stumps of transsected nerves. Acta Neuropathol 1966, 7:44-61 [Google Scholar]
- 34.Hamori J, Lakos I: Ultrastructural alterations in the initial segments and in the recurrent collateral terminals of Purkinje cells following axotomy. Cell Tissue Res 1980, 212:415-427 [DOI] [PubMed] [Google Scholar]
- 35.Fawcett DW: Observations on the cytology and electron microscopy of hepatic cells. J Natl Cancer Inst 1955, 15:1475-1512 [PubMed] [Google Scholar]
- 36.Bandy B, Davison AJ: Mitochondrial mutations may increase oxidative stress: implications for carcinogenesis and aging? Free Rad Biol Med 1990, 8:523-539 [DOI] [PubMed] [Google Scholar]
- 37.Friede RL: Transport of oxidative enzymes in nerve fibers: a histochemical investigation of the regenerative cycle in neurons. Exp Neurol 1959, 1:441-466 [DOI] [PubMed] [Google Scholar]
- 38.Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G: Bcl-2 inhibits the mitochondrial release of an apoptogenic protease. J Exp Med 1996, 184:1331-1341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW, Brown RH: Increased 3-nitrotyrosine in both sporadic, and familial amyotrophic lateral sclerosis. Ann Neurol 1997, 42:646-654 [DOI] [PubMed] [Google Scholar]
- 40.Smith MA, Rudnicka-Nawrot M, Richey PL, Praprotnik D, Mulvihill P, Miller CA, Sayre LM, Perry G: Carbonyl-related posttranslational modification of neurofilament protein in the neurofibrillary pathology of Alzheimer’s disease. J Neurochem 1995, 64:2660-2666 [DOI] [PubMed] [Google Scholar]
- 41.Good PF, Werner P, Hsu A, Olanow CW, Perl DP: Evidence of neuronal oxidative damage in Alzheimer’s disease. Am J Pathol 1996, 149:21-28 [PMC free article] [PubMed] [Google Scholar]
- 42.Rosen DR, Siddique T, Patterson D, Figiewicz DA, Sapp P, Hentati A, Donalsson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston AM, Berger R, Tanzi RE, Halperin JJ, Harzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz HR, Brown RH: Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362:59-62 [DOI] [PubMed] [Google Scholar]
- 43.Deng H-X, Hentati A, Tainer JA, Iqbal Z, Cayabyab A, Hung W-Y, Getzoff ED, Hu P, Herzfeldt B, Roos RP, Warner C, Deng G, Soriano E, Smyth C, Parge HE, Ahmed A, Roses AD, Hallewell RA, Pericak-Vance MA, Siddique T: Amyotrophic lateral sclerosis and structural defects in Cu,Zn superoxide dismutase. Science 1993, 261:1047-1051 [DOI] [PubMed] [Google Scholar]
- 44.Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hantati A, Kwon YW, Deng H-X, Chen W, Zhai P, Sufit RL, Siddique T: Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264:1772-1775 [DOI] [PubMed] [Google Scholar]
- 45.Martin LJ: Neurodegeneration in amyotrophic lateral sclerosis (ALS) resembles apoptosis and may occur by a programmed cell death mechanism. Soc Neurosci Abstr 1998 (in press)