

Abstract
Viral myocarditis is remarkably common, being detected in approximately 1% of unselected asymptomatic individuals. Many cases are attributable to enteroviral infection, and in particular to coxsackievirus B3. The underlying pathogenesis is controversial, but most studies admit the important immunopathological role of infiltrating CD8+ (cytotoxic) T lymphocytes (CTLs). We have previously shown that CTLs play conflicting roles in coxsackievirus B (CVB) myocarditis; they assist in controlling virus replication, but also are instrumental in causing the extensive inflammatory disease, which often results in severe myocardial scarring. A role for perforin, the major CTL cytolytic protein, in CVB myocarditis has been suggested, but never proven. In the present study we use perforin knockout (PKO) mice to show that perforin plays a major role in CVB infection; in broad terms, perforin is important in immunopathology, but not in CVB clearance. For example, PKO mice are better able to withstand a normally lethal dose of CVB (100% survival of PKO mice compared with 90% death in +/+ littermates). In addition, PKO mice given a nonlethal dose of CVB develop only a mild myocarditis, whereas their perforin+ littermates have extensive myocardial lesions. The myocarditis in PKO mice resolves more quickly, and these mice show minimal histological sequelae; in contrast, late in disease the perforin+ mice develop severe myocardial fibrosis. PKO mice, despite lacking this major CTL effector function, can control the infection and eradicate the virus; growth kinetics and peak CVB titers are indistinguishable in PKO and perforin+ mice. Therefore, the immunopathological and antiviral effects of CTLs can be uncoupled by ablation of perforin; this offers a promising target for therapy of myocarditis. Furthermore, we evaluate the possible roles of apoptosis, and of chemokine expression, in CVB infection. In perforin+ mice, apoptotic cells are detected within the inflammatory infiltrate, whereas in their PKO counterparts, apoptotic myocyte nuclei are seen. Chemokine expression in both PKO and perforin+ mice precedes and parallels the course of myocarditis. Several chemokines are detectable earlier in PKO mice than in perforin+ mice, but PKO mice show reduced peak levels, and chemokine expression decays sooner. In particular, MIP-1α expression is barely detectable at any time point in PKO mice, but it is readily identified in perforin+ animals, peaking just before the time of maximal myocarditis; this is particularly interesting, given that MIP-1α knockout mice are resistant to CVB myocarditis, but remain able to control viral infection. Thus, the chemokine pathway offers a second route of intervention to diminish myocarditis and its sequelae, while permitting the host to eradicate the virus.
Epidemiological and other studies suggest that 70% of the general public will be exposed to cardiotropic viruses, and half of them will have an episode of acute viral myocarditis. 1 Necropsy studies of victims of violent or accidental deaths show a remarkably high prevalence of myocarditis, approximately 1%, 2 although only a subset of these individuals, probably around 10%, have clinical disease, with symptoms such as chest pains, palpitations, or signs of heart failure. Although the majority of symptomatic patients recover well from acute myocarditis, the disease can have serious long-term sequelae; some 10 to 20% of symptomatic patients will develop chronic disease, which may progress over time to dilated cardiomyopathy (DCM), 1,3 in which one or both ventricles dilate and decompensate, with resulting cardiac failure. Dilated cardiomyopathy affects 20,000 to 40,000 Americans annually and has a 50% mortality in the 2 years after diagnosis, 4 and the most effective treatment is cardiac transplantation. 5
Many viruses can cause myocarditis, but enteroviruses in general, and coxsackieviruses in particular, are important human pathogens. Coxsackieviruses are members of the Picornaviridae family and fall into groups A and B, defined by their pathogenicity in newborn mice. 6 The proportion of cases of acute myocarditis, and of dilated cardiomyopathy, with suspected coxsackievirus etiology varies among studies, but is usually 25 to 50% and most commonly involves B group viruses (CVB). CVB has been isolated from stool or pharyngeal specimens of many patients with acute myocarditis. 7,8 Isolation of infectious virus particles directly from the human myocardium is less common, because myocardial biopsy remains unusual, but specimens obtained at necropsy after a lethal infection have yielded infectious CVB, 7,9,10 which causes myocarditis in mice. 9 Although five of the six CVB types can cause myocarditis, the majority of animal studies have focused on CVB3 and have shown unequivocally that this virus can cause cardiac disease. Studies have been carried out in many species, including primates, 11 but the most popular animal model is the mouse. The murine model systems show early death, acute myocarditis, and longer-term disease, thus paralleling the human situation. CVB3 infection of mice can result in death at around 4 days postinfection (p.i.), at which time histologically apparent myocarditis is rarely detectable. 12 In surviving mice of several strains, a biphasic myocarditis occurs; an acute stage, at which infiltration is first seen 4 to 7 days p.i., resolving by day 14 or so, and a chronic stage, in which low-grade inflammation is maintained. CVB can be isolated from myocardium up to day 9 to 10 p.i., but no infectious virus is detected beyond day 14 (at least in immunocompetent mice).
Although there is no doubt that CVB3 can cause myocarditis and myocyte destruction in mice, the precise mechanisms underlying these processes remain uncertain. The role of cytotoxic T lymphocytes (CTLs) in CVB-induced myocarditis was first shown more than 20 years ago 13 and has been confirmed in subsequent studies (see, eg, Refs. 12 and 14-16 ). Acute myocarditis becomes histologically evident 4 to 7 days p.i., and cellular infiltrate comprises CD8+ T cells, natural killer cells, and macrophages. 12,17-19 This infiltration usually resolves after viral clearance, leading to host recovery (sometimes with residual myocardial fibrosis). We have shown that CTLs play a major role in myocarditis; functional paralysis of this T-cell population (either by monoclonal antibody depletion, or by the use of mice deficient in class I recognition) greatly diminished the acute inflammatory response and prevented subsequent myocardial scarring. 12 However CD8+ T-cell depletion is a double-edged sword: in the absence of functional CTLs, viral titers were significantly increased, indicating that CTLs are both protective (diminishing virus titers) and immunopathological (causing extensive myocarditis).
The mechanisms by which CTLs exert their biological effects are varied. Major histocompatibility complex class I expression, required for CTL-mediated recognition and lysis, is low to undetectable on normal myocardiocytes in vivo, but can be readily detected in vivo after CVB infection, 20 perhaps being induced by interferon. CTLs have at least three methods whereby they may affect virus-infected target cells: 1) perforin-mediated lysis, 2) Fas-dependent lysis, and 3) cytokine release. Perforin is critical to the control of some virus infections, given that perforin knockout (PKO) mice are unable to clear infection by lymphocytic choriomeningitis virus. 21,22
In this article, we evaluate the role of perforin both in the etiology of CVB3 myocarditis and in CVB3 infection and clearance. Perforin-mediated lysis of CVB-infected cells is a plausible foundation for myocarditis and for control of myocardial virus titers, both of which we found to be CTL dependent. 12 Perforin-containing cells enter the myocardium soon after CVB infection, 18 and circumstantial evidence indicates that perforin may be required for CVB-induced myocarditis; 23 in addition, splenocytes from CVB-infected mice can lyse CVB-infected myocytes in vitro. 24 In this communication, we show that the absence of perforin is, broadly, beneficial to CVB-infected mice. PKO mice survive a CVB3 challenge that is lethal to 90% of perforin +/+ littermates, and although myocarditis occurs in the absence of perforin, it is much less extensive than in perforin+ mice, resolves more rapidly, and does not result in severe myocardial fibrosis. Despite the diminished myocardial infiltration in PKO mice, viral titers, myocardial distribution, and growth kinetics are indistinguishable in PKO and perforin+ mice, and virus is undetectable by 21 days p.i. in both mouse phenotypes. Therefore, in CVB myocarditis, the antiviral and immunopathological functions of CTLs can be uncoupled. We also evaluate apoptosis and chemokine expression in PKO and perforin+ mice. In both mouse genotypes, chemokine expression approximately parallels myocarditis. Expression of MIP-1α, a chemokine whose disruption renders mice resistant to myocarditis, 25 is barely detectable in PKO mice.
These findings have important therapeutic implications; the development of a specific inhibitor of perforin or of the chemokine pathways might permit us to prevent immunopathology without diminishing the antiviral efficacy of the immune response.
Materials and Methods
Cells and Virus
The cardiopathogenic H3 strain of CVB3 (Nancy) was utilized in all experiments. The titer of the virus was determined by plaque assay in HeLa cells according to previously published procedures.
Mice
PKO mice were generated by Drs. C. M. Walsh and W. R. Clark (University of California, Los Angeles, Los Angeles, CA), using AB-1 embryonal stem cells, which were introduced into the C57 background and bred on this background. 22 Heterozygous PKO mice were sent to Scripps Research Institute, where they were bred to generate homozygous knockout mice, as well as heterozygous and homozygous positive littermates (as controls). We determined the perforin gene status by polymerase chain reaction on tail DNA, for every mouse bred. All experiments utilized adult (at least 8 weeks old) male PKO mice and their transgenic wild-type (homozygous or heterozygous) littermates.
Infection Protocol and Organ Processing
Mice were infected (0.2 ml intraperitoneally) with CVB3 diluted in saline. For evaluation of susceptibility to early death, mice were given 100 plaque-forming units (pfu) CVB3 on day 0 and observed for 21 days. For evaluation of myocarditis, mice were given a sublethal dose (35 pfu) to ensure survival, and at the indicated time points, the organs were removed, dissected in equal parts, washed, and either snap frozen in liquid nitrogen or fixed in 10% zinc normal buffered formalin for histological analysis. Quantitation of virus from heart tissue was performed by homogenization of the tissue and centrifugal clarification of the supernatant, followed by virus plaque assay in HeLa cells as described.
Histology and Immunohistochemistry
For histological analysis, heart tissue was fixed in 10% neutral buffered formalin, sectioned into 5-μm slices, and stained either with hematoxylin and eosin (H&E) or with Masson’s trichrome as specified in the text. Inflammation was scored on a scale of 0 to 4 based on the number of lesions present in a section. A score of 1 represents 1 to 10 lesions; 2, 11 to 20; 3, 21 to 40; and 4, >40.
Multiprobe RNase Protection Assay
The RNase protection assay was performed as previously described. 26 Probes used were C10 (a murine member of the C-C chemokine family); MIP-1α and MIP-1β (macrophage inflammatory proteins 1α and 1β); MIP-2 (macrophage inflammatory protein 2); MCP-1 and MCP-3 (monocyte chemoattractant proteins 1 and 3); crg-2 (cytokine-regulated gene-2); and RANTES (regulated upon activation normal T cell expressed and secreted).
In Situ Hybridization
A previously published protocol 27 was followed on paraffin sections (5 μm) of heart tissue previously fixed in 10% normal buffered formalin. After preparation of the slides and prehybridization at 42°C, 1 × 106 cpm of a radiolabeled antisense RNA probe was applied and allowed to hybridize at 42°C overnight. After washing, slides were dipped in photographic emulsion and were held at 4°C for 3 to 14 days, when they were developed and fixed. To visualize the cells, the developed slides were stained in H&E for 2 to 3 minutes.
Identification of Apoptotic Cells
The Apoptag kit (Oncor, Gaithersburg, MD) was used according to the manufacturer’s instructions.
Results
Enhanced Survival in Absence of Perforin
Three groups of mice (10 mice per group) were infected with 100 pfu CVB3 and were observed for 21 days. As shown in Figure 1 ▶ , 90% of homozygous positive littermates (wild type, +/+) died, whereas 100% of PKO mice survived. Thus, CTL-mediated lysis appears to contribute to a lethal outcome of CVB infection. This is consistent with our findings of diminished susceptibility in β2mKO mice, which lack CD8+ T cells, and of protection against lethal outcome after CD8 depletion of CD4KO mice. 12 There may be a gene dosage effect, because 50% of heterozygous mice survived, compared with only 10% of homozygous positive mice. No animals died after day 10 p.i. Thus, perforin appears important to the immunopathological function of CD8+ T cells during lethal acute infection.
Figure 1.

Perforin contributes to lethal outcome of CVB3 infection. Three groups of male mice (10 mice per group) were infected with CVB3 (100 pfu intraperitoneally) and were observed daily for 21 days. No deaths occurred after day 10 p.i.
CVB3-Induced Myocarditis Is Present, But Much Reduced, in the Absence of Perforin
Results for +/+ and +/− mice were similar, and henceforth both genotypes will be referred to as “perforin+” mice. Figure 2 ▶ shows the development of myocarditis lesions over time in PKO and perforin+ mice. At each time point, in each group, a minimum of four mice was used (thus, perforin+ groups include at least eight mice, four +/+ and four +/−). At 4 days p.i., no infiltration was seen in PKO mice, and only very occasional small foci were detected in perforin+ mice. By 7 days p.i., mild myocarditis could be seen in both perforin+ and PKO mice. Myocarditis was much more marked by day 10 in perforin+ mice, but declined in PKO littermates. By day 14, myocarditis had almost resolved in PKO mice (four out of four mice), but remained severe in perforin+ animals. Note that this scoring system reflects only the number of individual lesions and not the total area of myocardium involved. In a particularly extensive myocarditis, the lesion score may even be reduced as adjacent lesions “fuse” to produce a single large lesion. Consequently, lesion scores must be complemented by representative histology (Figure 3) ▶ . The lesions in PKO mice were very much smaller, and more circumscribed, than those in perforin+ animals; the appearance in the PKO mice was similar to that seen after CD8 depletion of CD4KO mice and to that seen in β2mKO mice. 12
Figure 2.
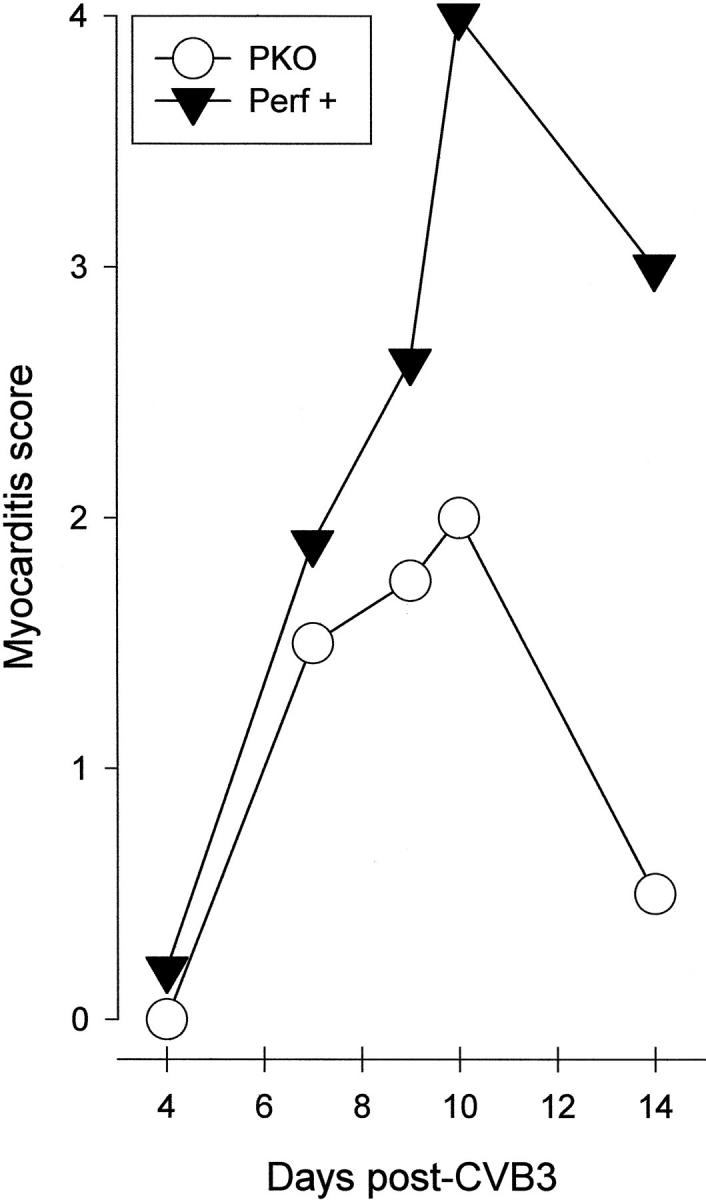
Myocarditis index in PKO and perforin+ mice. Mice were infected with 35 pfu of CVB3 and were sacrificed at the indicated time points (Days post-CVB3). Hearts were harvested, fixed, paraffin embedded and sectioned, and stained with H&E. Inflammatory lesions were counted. The scoring criteria are described in Materials and Methods and reflect only the number of lesions, rather than the total extent of myocardial involvement.
Figure 3.
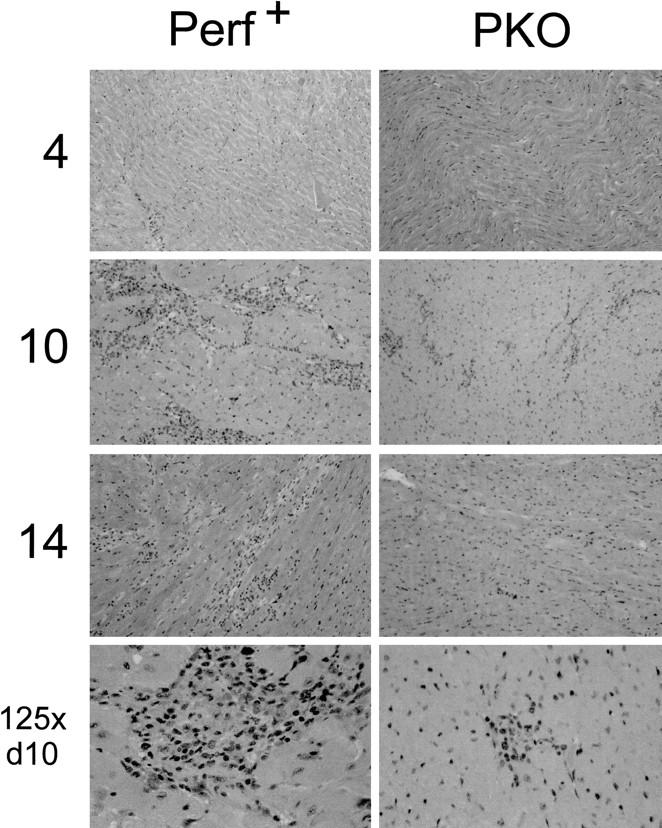
Myocarditis in perforin+ and PKO mice: perforin is required for extensive infiltration. Representative H&E-stained sections of hearts from CVB-infected perforin+ mice (left) and PKO mice (right) at days 4, 10, and 14 p.i. Original magnification, ×50. The final row shows enlargements (original magnification, ×25) of myocardial lesions at day 10 p.i.
Myocardial Fibrosis Is Reduced in the Hearts of PKO Mice
No infiltration was detectable in either genotype by day 21 p.i., but by >30 days p.i., marked myocardial fibrosis was present in perforin+ mice, but not in PKO animals (Figure 4) ▶ . Thus, perforin exacerbates myocarditis, and its ablation prevents not only acute disease, but also the subsequent scarring.
Figure 4.
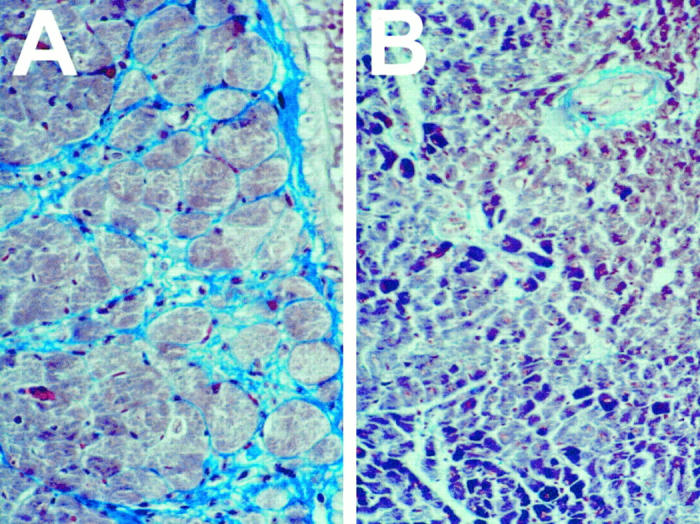
Reduced fibrosis in hearts of PKO mice. Representative sections from hearts of perforin+ mice (A) and PKO littermates (B) taken 30 to 40 days p.i., at which time no detectable infectious virus is present. In all cases, 35 pfu CVB was administered, and virus infection and replication were confirmed by serum titration during the first 10 days p.i. Sections are stained with Masson’s trichrome, which stains normal cardiac muscle cells red/purple and stains fibrous tissue light blue.
Deletion of Perforin Has No Apparent Effect on Virus Titers or Kinetics of Virus Replication in the Heart
CTLs are critical to the control of many virus infections, but, in combating picornavirus infections, antibodies appear to play a vital role. Nevertheless, we have shown that CD8+ T cells play some role in limiting CVB replication, because depletion of CD8+ T cells leads to increased CVB load in the heart. 12 To determine whether this antiviral effect was mediated by perforin, a time course of CVB3 replication in the heart was carried out, comparing PKO mice with perforin+ littermates. As shown in Figure 5 ▶ , there was no statistically significant difference between the mouse strains. Regardless of the perforin status, the growth curve peaked on day 4 p.i., falling rapidly thereafter, and no virus was detectable by 21 days p.i. (not shown). Furthermore, the absolute titers at each time point were not significantly different. Thus, perforin is not required for clearance of CVB3, and it appears to play little if any role in the limitation of viral replication in the heart.
Figure 5.

Perforin has no detectable effect on CVB3 titers or growth kinetics. PKO mice and perforin+ littermates were challenged with CVB3, and heart titers (pfu/g of tissue) were determined at the indicated times. For each strain, each time point represents the average titer of at least four mice; error bars are shown.
Virus Distribution in the Myocardium Is Not Altered by the Absence of Perforin
Although we show above that myocardial virus titers are similar regardless of the perforin status of the host, it remained possible that the infection would be more widely distributed in the myocardia of mice lacking perforin. Therefore, at the peak of virus titer, 4 days p.i., myocardial sections were evaluated by in situ hybridization with antisense probes for CVB3. For this positive-strand virus, the antisense probe detects the abundant viral genome and the almost identical single mRNA. Although the virus titer is very high (∼108 pfu/g), virus was detectable only in well separated and tightly circumscribed foci in both PKO and perforin+ hearts (shown by dark-field microscopy in Figure 6 ▶ ); the lesion numbers were similar in all mice, regardless of perforin status. Bright-field examination of the infected foci revealed no inflammation, consistent with our finding that myocarditis is absent on day 4 p.i., as shown in Figure 3 ▶ .
Figure 6.

Similar virus distribution 4 days post-CVB3 infection in PKO and +/− mice. PKO or perforin+ mice were infected with CVB3, and at 4 days p.i. (the peak of virus titer in the heart), mice were sacrificed. Paraffin sections of the myocardia were analyzed by in situ hybridization using an antisense probe for CVB3. After emulsification and development, slides were visualized by dark-field microscopy. Original magnification, ×80.
Apoptosis in CVB-Infected Myocardia in the Presence or Absence of Perforin
Apoptosis, or programmed cell death, is an important facet of the host defense against virus infection and also plays a critical part in several aspects of host development, including regulation of the immune response. Apoptosis can be induced by several mechanisms. We looked for apoptosis in +/+ and +/− littermates and found signal in the infiltrate, but not in noninflamed muscle (Figure 7) ▶ ; this is consistent with our previous observation in the CD4KO mouse strain (also perforin+). 12 The picture differed in PKO mice; little or no signal could be detected in the small inflammatory lesions, whereas many myocyte nuclei were positive. Therefore, in perforin+ mice, there was a marked inflammation, with apoptosis restricted to these areas whereas in PKO mice the myocarditis was reduced, but apoptosis was detected in myocyte nuclei.
Figure 7.
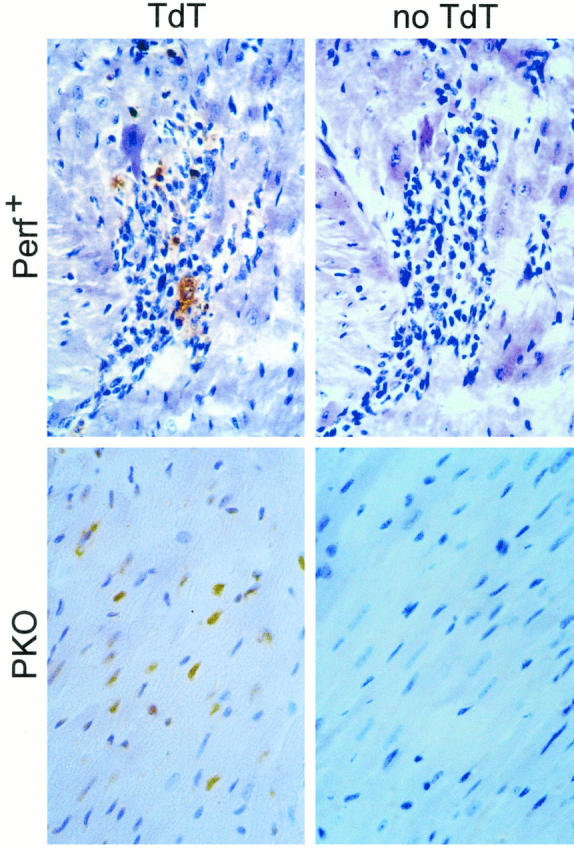
Apoptosis in CVB3-infected myocardium. The hearts harvested at 10 days p.i. and evaluated for myocarditis (shown in Figure 3 ▶ ) were tested for apoptosis using the terminal deoxynucleotidyl transferase (TdT)-mediated nick-end labeling assay. Reactions were carried out in the presence of the enzyme TdT (left; TdT) or, as a control, in its absence (right; no TdT). Signal in perforin+ mice was seen only within the inflammatory foci (top left), whereas in PKO mice signal was absent from the small foci, but was detected in myocyte nuclei (bottom left).
Kinetics of Chemokine Expression after CVB Infection and the Effects of Perforin Ablation
CVB-induced myocarditis does not occur in MIP-1α transgenic knockout mice. 25 This chemokine is secreted by macrophages, lymphocytes, and neutrophils and is chemoattractant for T cells. We considered it possible that perforin-mediated lysis of infected cells might increase neutrophil and macrophage infiltration, which would, through release of MIP-1α and other chemokines, increase the influx of CTLs. If so, the limited myocardial infiltration in PKO mice might result, in part, from lower chemokine levels in the infected myocardium, which in turn resulted from the reduced cytolysis. Normal and PKO mice were infected with a sublethal dose of CVB3 and chemokine mRNA expression in the hearts was examined at the indicated time points, using an RNase protection assay. As demonstrated in Figure 8 ▶ , many of the proinflammatory chemokines were up-regulated during the course of infection. Both the onset and decay of chemokine expression seemed advanced in PKO mice; crg-2 and MCP-1 were detectable at day 2 p.i. and peaked at day 4 p.i. In contrast, chemokine expression in perforin+ mice was undetectable on day 2, and barely detectable on day 4 p.i.; in these mice, expression peaked around day 7, when myocarditis developed. In both PKO and perforin+ mice, signal reached background levels by day 14. When considering specific chemokines, MIP1-α was barely detectable in PKO animals but was readily detectable in +/+ mice at 7 and 10 days p.i., whereas MCP-1, MCP-3, crg-2, and RANTES mRNA expression increased in response to infection in both PKO and wild-type mice. Thus, CVB3 infection resulted in an increase of proinflammatory chemokines that correlated with the development of myocarditis, and the expression kinetics differed between PKO and perforin+ mice.
Figure 8.

Chemokine mRNA levels in hearts of CVB3-infected PKO and perforin+ mice. RNAs were prepared from the hearts of perforin+ and PKO mice at the indicated time points p.i., and from uninfected mice (U). Chemokine mRNAs were detected by RNase protection, as described in Materials and Methods. Intact probes are shown in the right lane, and the positions of the protected bands are indicated (arrows) to the left of the figure, rp132 is a control probe, which detects an mRNA encoding a 32-kb ribosomal protein; this is included to confirm the assay execution and to ensure that all RNA samples were qualitatively and quantitatively comparable.
Localization of Chemokine Expression in CVB-Infected Myocardia
To confirm the above, and to localize the expression of the chemokines, in situ hybridization studies were carried out on the hearts of PKO and perforin+ mice, 7 days p.i. Antisense probes for MIP-1α, RANTES, and crg-2 were used, and specificity controls included sense probes (Figure 9 ▶ , B and D). In all cases, chemokine RNA expression was noted in infiltrating cells. Representative results for crg-2 are shown in Figure 9 ▶ for perforin+ mice (Figure 9 ▶ , A and B) and for PKO mice (Figure 9 ▶ , C and D).
Figure 9.
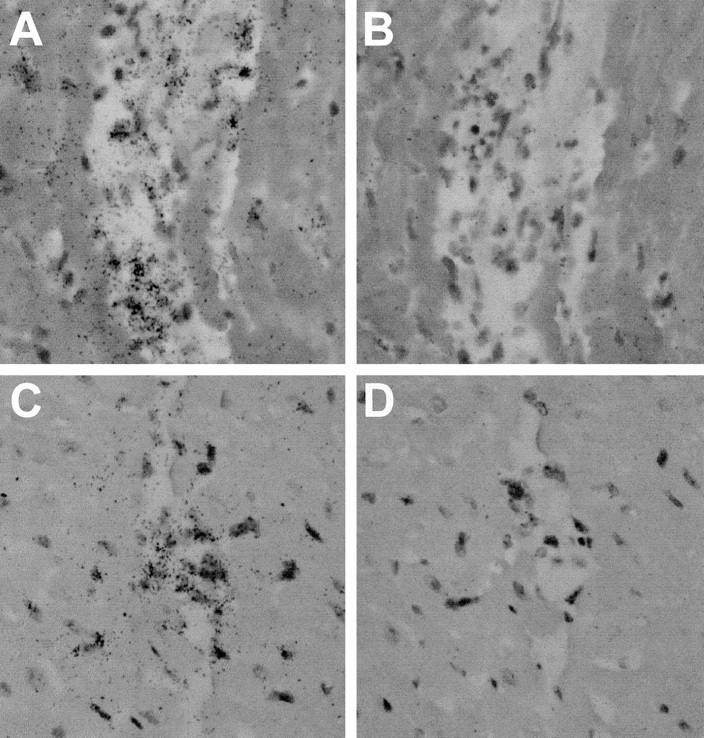
In situ hybridization for chemokine expression. Paraffin sections of hearts from CVB3-infected perforin+ mice (A and B) or PKO mice (C and D) were prepared 7 days p.i. and were analyzed by in situ hybridization using an antisense probe for crg-2 (A and C). As a control, adjacent sections of each heart were hybridized with a sense probe for crg-2 (B and D). After emulsification and development, slides were stained with H&E and were visualized by bright-field microscopy. Original magnification, ×160.
Discussion
CVB is a common associated factor in human subacute, acute, and chronic myocarditis. 8,28 Slot-blot hybridization studies have shown positive signal for coxsackievirus RNA in myocardial biopsy specimens of approximately 45% of patients with myocarditis or dilated cardiomyopathy, compared with none of the controls. 29 High levels of neutralizing antibodies are found in about 50% of patients, and serial antibody studies show a fourfold or greater change in paired sera in approximately half of patients. 29 The presence or absence of CVB infection appears to be important in determining the prognosis of individuals with acute myocarditis. In one 15-year follow-up study of myocarditis patients, 25% of patients with serological evidence of CVB infection died from subsequent chronic myocarditis or cardiomyopathy, whereas 0 of 26 patients with negative viral serology died. 30 The presence of enteroviral RNA in heart tissue also is associated with poorer prognosis. 31,32 Acute myocarditis and its long-term sequelae are, therefore, significant contributors to human morbidity and mortality, and the role of enteroviruses, and in particular of CVB, has been clearly demonstrated.
How does CVB cause myocarditis and long-term disease? Some proposed pathogenic mechanisms include 1) direct destruction of cardiac muscle cells by CVB3, 2) immunopathological destruction mediated by immune effector mechanisms directed against viral antigens, 3) immune-mediated destruction by effector mechanisms targeting novel host-encoded antigens expressed on the surface of cardiac cells after CVB3 infection, 4) immune-mediated destruction targeted against host antigens sharing determinants with CVB3 antigens (“molecular mimicry”), and 5) immune-mediated destruction targeted against host antigens normally expressed but made more immunogenic as a consequence of CVB3 infection. This array of possible mechanisms is further complicated by variability in outcome depending on virus genotype 33,34 and the strain, 35,36 sex, 37,38 age, 39 and immune status 8,40 of the murine host.
Our laboratory has chosen to focus on the first two mechanisms (1 and 2) described above, because they offer the most direct explanation for the pathology observed, and because there already is evidence in support of both. First, the virus itself can cause cell lysis. In the acute phase of infection, many organs have high viral titers (heart, pancreas, liver, spleen, lung, and kidney), and viral dissemination is largely hematogenous, as reflected by a high viremia. In most mouse strains, the virus titer in the heart peaks at 4 days p.i. and diminishes thereafter, with infectious virus being undetectable after 14 days. The virus appears to enter the heart by first infecting endothelial cells and then spreading to the myocardiocytes. 41 Myocardiocytes can be infected in vitro, 42,43 and infected cells are rapidly lysed. 44 The in vitro findings are corroborated by in vivo ultrastructural studies of myocardial tissue that show clear evidence of virus infection of cardiac muscle cells and cell death 45-47 and by studies showing that some pathology can occur even in immunocompromised mice unable to mount a normal T-cell response. 48,49 Thus, virus-mediated lysis of myocardiocytes in vivo almost certainly occurs and most probably contributes to myocarditis. Many laboratories have provided convincing evidence for the second mechanism, implicating T cells in myocarditis. 12,13,18,38,50-54 The relative contributions of CD4+ and CD8+ T cells appear to vary somewhat between different mouse strains, 38,54 and a role for γ-δ T cells has been suggested. 55,56
We previously showed that CD8+ T cells play two roles during CVB3 infection, one beneficial to the host, and the other harmful; CTLs limit viral titers in the myocardium, but also cause extensive myocarditis. 12 Here we establish the part played by perforin in these events and find that perforin function is, broadly, damaging to the host. Perforin expression results in a dramatically increased mortality upon CVB3 infection (Figure 1) ▶ ; some 90% of perforin+/+ mice succumb, in contrast to 100% survival in the PKO littermates, and there appears to be a gene dosage effect, as approximately half of the heterozygous littermates died. This parallels our previous finding that depletion of CD8+ T cells enhanced survival. 12 Furthermore, again consistent with our earlier study, we show that PKO mice have a much less extensive myocarditis than heterozygous or +/+ littermates. The lesions in PKO mice are less frequent, smaller, and better circumscribed, than those in perforin+ littermates (Figures 2 and 3) ▶ ▶ . Thus, perforin appears to play a major role in the CD8+ T cell-mediated immunopathology that accompanies CVB3 infection.
We have shown that CD8+ T cells limit viral titers in the heart, as depletion of these cells resulted in a 17-fold increase in CVB3 titer. 12 Here we demonstrate that this antiviral effect does not require perforin; virus growth kinetics, and viral titers, are indistinguishable in PKO and perforin+ mice (Figure 5) ▶ . It remained possible that, despite these findings, perforin played a role in restricting the myocardial distribution of CVB; however, in situ studies show that the number of infected foci at the peak of viral infection are similar in perforin+ and PKO mice (Figure 6) ▶ . Our demonstration that PKO mice clear virus in a manner indistinguishable from +/+ mice strengthens the argument that, although perforin plays a vital role in control of relatively “nonlytic” viruses (such as lymphocytic choriomeningitis virus), it may play less of a role in control of highly lytic viruses such as vaccinia, Semliki Forest, and vesicular stomatitis viruses 57 and CVB (our data). Thus, we show that the antiviral and immunopathological results of CTL activity can be uncoupled by removing perforin; perforin blockade therefore represents a promising therapeutic target in the treatment of CVB myocarditis.
How might CD8+ CTLs contribute to CVB clearance, given that our studies show that perforin is not required? CTLs have a second method of delivering a contact-mediated lethal signal, the fas pathway. Fas is a membrane protein, a member of the tumor necrosis factor receptor family, 58 which is commonly expressed on hematopoietic cells and whose activation leads to apoptosis of the carrier cell. The in vivo ligand for the fas protein, FasL, is expressed on CTLs and activated splenocytes; these cells can interact with, and induce apoptosis of, Fas+ target cells. 59,60 Whereas FasL expression is very restricted, Fas is abundantly expressed on a few nonhematopoietic tissues, including heart and liver. 61 We therefore consider it possible that CD8+ CTLs might control CVB replication by inducing apoptosis in infected myocardiocytes. We show here (Figure 7) ▶ that apoptosis is readily detectable in the hearts of both PKO and perforin+ mice. In the perforin+ mice, in which myocarditis is extensive, apoptosis is seen primarily within the infiltrates; in contrast, in PKO mice, we note clusters of myocardiocytes with nuclear staining consistent with apoptosis. This apoptosis may be T cell mediated (given that perforin is not required for the function of the Fas pathway), or it may be virus induced. Although these results are consistent with the hypothesis that the fas pathway is important in regulating the outcome of this infection, further studies, perhaps using mice defective in the fas pathway, 62 will be required to investigate this question. Alternatively, CTLs may exert their effects not through lysis but rather, as argued by Ramshaw et al 63 and others, 64 by showering infected cells and the surrounding area with a variety of cytokines. Others have recently analyzed cytokine expression in the CVB3-infected myocardium of normal mice and have found high levels of interleukin-2, interferon-γ, and tumor necrosis factor-β. 65
In addition, we have evaluated the expression of chemokines in CVB-induced myocarditis. Cook et al 25 found that MIP-1αKO mice were resistant to myocarditis, and that CVB titers were indistinguishable from those in normal animals. 25 As shown in Figure 8 ▶ , the mRNA encoding MIP-1α, a chemokine whose ablation prevents CVB-induced myocarditis, 25 is detectable in perforin+ mice, but barely detectable in the PKO littermates. Furthermore, the kinetics of chemokine mRNA expression vary somewhat between PKO and perforin+ mice. In general, responses are accelerated in PKO mice, in which mRNA expression is detectable by day 2 p.i., has lower levels of maximum expression, and recede sooner. It is tempting to suggest that this accelerated chemokine production may reflect enhanced replication of the CVB inoculum in the periphery of PKO mice and thus the earlier delivery of CVB to the PKO myocardium. However, our data (Figure 5) ▶ show high viral titers at 2 days p.i. in both PKO and perforin+ mice, and the similarity in growth kinetics renders this explanation less likely. The levels of chemokine mRNA expression approximate the severity of myocarditis. We have previously shown that macrophages are the largest cellular subset in the infiltrate, and that their recruitment is diminished by depletion of CD8+ T cells. 12 We suggest that, in the normal host, perforin-mediated CTL lysis of infected myocytes results in necrosis and a corresponding influx of macrophages and neutrophils; the chemokines expressed by these infiltrating cells (Figure 9) ▶ induce further lymphocytic infiltration, which in turn causes further tissue damage. In contrast, in PKO mice, antiviral CD8+ T cells invade the myocardium, limiting CVB replication, but cell death is largely apoptotic, and therefore the major trigger for myocarditis is absent. Consequently, phagocytic infiltration is minimized, and expression of the relevant chemokines is reduced, as we show here. This may offer a second avenue for therapeutic intervention; blockade of chemokine gene expression, or of their effector functions, at an early stage p.i. might greatly limit myocarditis without interrupting virus clearance.
The interaction between CVB and its host is complex; some immune components protect against myocarditis (CD4+ cells in C57BL/6 mice 12 ), whereas others greatly exacerbate disease (perforin+ CD8+ cells, shown here). In our initial work, we showed that depletion of CD8+ T cells was effective in diminishing myocarditis, but this therapeutic benefit was achieved at a cost, as viral titers were significantly elevated. 12 It is, therefore, perhaps not surprising that the outcome of wholesale immunosuppressive treatment is unpredictable, varying among different mouse strains. 66 Inhibition of T cells using lobenzarit disodium exacerbates disease, 67 and treatment with nonsteroidal anti-inflammatory agents is deleterious, 68,69 whereas FK-506 treatment reduces myocarditis. 70 A recent human clinical trial concluded that immunosuppressive drugs should not be routinely used. 71 Here we present evidence that the costs and benefits of anti-CVB T cell responses can be dissociated. This has important therapeutic implications, suggesting that blockade of the perforin pathway, or of chemokine responses, might ameliorate disease without diminishing the host’s capacity to eradicate the virus.
Acknowledgments
We thank Annette Lord for excellent secretarial support. This is publication number 10494-NP from the Scripps Research Institute.
Footnotes
Address reprint requests to Dr. J. Lindsay Whitton, Department of Neuropharmacology, CVN-9, The Scripps Research Institute, 10550 N. Torrey Pines Road, La Jolla, CA 92037. E-mail: lwhitton@scripps.edu.
Supported by National Institutes of Health grants AI 42314 (to JLW), and MH 50426 and MH 47680 (to ILC). IM was supported by a fellowship from the Fundacion Ramon Areces.
John R. Gebhard’s present address is Pharmadigm, Inc., Salt Lake City, UT.
References
- 1.O’Connell JB: The role of myocarditis in end-stage dilated cardiomyopathy. Tex Heart Inst J 1987, 14:268-275 [PMC free article] [PubMed] [Google Scholar]
- 2.Gravanis MB, Sternby NH: Incidence of myocarditis: a 10-year autopsy study from Malmo, Sweden. Arch Pathol Lab Med 1991, 115:390-392 [PubMed] [Google Scholar]
- 3.Sole MJ, Liu P: Viral myocarditis: a paradigm for understanding the pathogenesis and treatment of dilated cardiomyopathy. J Am Coll Cardiol 1993, 22:99A-105A [DOI] [PubMed] [Google Scholar]
- 4.Goodwin JF: Cardiomyopathies and specific heart muscle diseases: definitions, terminology, classifications and new and old approaches. Postgrad Med J 1992, 68(suppl 1):S3-S6 [PubMed] [Google Scholar]
- 5.Heck CF, Shumway SJ, Kaye MP: The Registry of the International Society for Heart Transplantation: sixth official report, 1989. J Heart Transplant 1989, 8:271-276 [PubMed] [Google Scholar]
- 6.Hyypia T, Kallajoki M, Maaronen M, Stanway G, Kandolf R, Auvinen P, Kalimo H: Pathogenetic differences between coxsackie A and B virus infections in newborn mice. Virus Res 1993, 27:71-78 [DOI] [PubMed] [Google Scholar]
- 7.Gelfand HM: The occurrence in nature of the coxsackie and ECHO viruses. Prog Med Virol 1961, 3:193-244 [PubMed] [Google Scholar]
- 8.Woodruff JF: Viral myocarditis: a review. Am J Pathol 1980, 101:425-484 [PMC free article] [PubMed] [Google Scholar]
- 9.Sutton GC, Harding HB, Trueheart RP, Clark HP: Coxsackie B4 myocarditis in an adult: successful isolation of virus from ventricular myocardium. Clin Aviation Aerospace Med 1967, 38:66-69 [Google Scholar]
- 10.Sutton GC, Tobin JR, Fox RT, Freeark RJ, Driscoll JF: Study of the pericardium and ventricular myocardium: exploratory mediastinotomy and biopsy in unexplained heart disease. JAMA 1963, 185:786-788 [DOI] [PubMed] [Google Scholar]
- 11.Paque RE, Gauntt CJ, Nealon TJ: Assessment of cell-mediated immunity against coxsackievirus B3-induced myocarditis in a primate model (Papio papio). Infect Immun 1981, 31:470-479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Henke A, Huber SA, Stelzner A, Whitton JL: The role of CD8+ T lymphocytes in coxsackie virus B3-induced myocarditis. J Virol 1995, 69:6720-6728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Woodruff JF, Woodruff JJ: Involvement of T lymphocytes in the pathogenesis of coxsackie virus B3 heart disease. J Immunol 1974, 113:1726-1734 [PubMed] [Google Scholar]
- 14.Hashimoto I, Tatsumi M, Nakagawa M: The role of T lymphocytes in the pathogenesis of Coxsackie virus B3 heart disease. Br J Exp Pathol 1983, 64:497-504 [PMC free article] [PubMed] [Google Scholar]
- 15.Ytterberg SR, Mahowald ML, Messner RP: T cells are required for coxsackievirus B1 induced murine polymyositis. J Rheumatol 1988, 15:475-478 [PubMed] [Google Scholar]
- 16.Kishimoto C, Abelmann WH: In vivo significance of T cells in the development of Coxsackievirus B3 myocarditis in mice: immature but antigen-specific T cells aggravate cardiac injury. Circ Res 1990, 67:589-598 [DOI] [PubMed] [Google Scholar]
- 17.Godeny EK, Gauntt CJ: In situ immune autoradiographic identification of cells in heart tissues of mice with coxsackievirus B3-induced myocarditis. Am J Pathol 1987, 129:267-276 [PMC free article] [PubMed] [Google Scholar]
- 18.Seko Y, Shinkai Y, Kawasaki A, Yagita H, Okumura K, Takaku F, Yazaki Y: Expression of perforin in infiltrating cells in murine hearts with acute myocarditis caused by coxsackievirus B3. Circulation 1991, 84:788-795 [DOI] [PubMed] [Google Scholar]
- 19.Chen SX, Mei SW, Bao SH, Zheng XJ, Chang PL, Yao JS, Yao S, Zhang DQ: Immunological status and pathology of coxsackie B viral myocarditis and dilated cardiomyopathy. Chin Med J (Engl) 1993, 106:659-664 [PubMed] [Google Scholar]
- 20.Seko Y, Tsuchimochi H, Nakamura T, Okumura K, Naito S, Imataka K, Fujii J, Takaku F, Yazaki Y: Expression of major histocompatibility complex class I antigen in murine ventricular myocytes infected with Coxsackievirus B3. Circ Res 1990, 67:360-367 [DOI] [PubMed] [Google Scholar]
- 21.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Olsen KJ, Podack ER, Zinkernagel RM, Hengartner H: Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 1994, 369:31-37 [DOI] [PubMed] [Google Scholar]
- 22.Walsh CM, Matloubian M, Liu CC, Ueda R, Kurahara CG, Christensen JL, Huang MT, Young JD, Ahmed R, Clark WR: Immune function in mice lacking the perforin gene. Proc Natl Acad Sci USA 1994, 91:10854-10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seko Y, Shinkai Y, Kawasaki A, Yagita H, Okumura K, Yazaki Y: Evidence of perforin-mediated cardiac myocyte injury in acute murine myocarditis caused by Coxsackie virus B3. J Pathol 1993, 170:53-58 [DOI] [PubMed] [Google Scholar]
- 24.Huber SA, Job LP: Cellular immune mechanisms in Coxsackievirus group B, type 3 induced myocarditis in BALB/C mice. Adv Exp Med Biol 1983, 161:491-508 [DOI] [PubMed] [Google Scholar]
- 25.Cook DN, Beck MA, Coffman TM, Kirby SL, Sheridan JF, Pragnell IB, Smithies O: Requirement of MIP-1α for an inflammatory response to viral infection. Science 1995, 269:1583-1585 [DOI] [PubMed] [Google Scholar]
- 26.Asensio VC, Campbell IL: Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J Virol 1997, 71:7832-7840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lane TE, Paoletti AD, Buchmeier MJ: Disassociation between the in vitro and in vivo effects of nitric oxide on a neurotropic murine coronavirus. J Virol 1997, 71:2202-2210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reyes MP, Lerner AM: Coxsackievirus myocarditis—with special reference to acute and chronic effects. Prog Cardiovasc Dis 1985, 27:373-394 [DOI] [PubMed] [Google Scholar]
- 29.Martino TA, Liu P, Petric M, Sole MJ: Enteroviral myocarditis and dilated cardiomyopathy: a review of clinical and experimental studies. Rotbart HA eds. Human Enterovirus Infections. 1995, :pp 291-351 ASM Press, Washington, DC [Google Scholar]
- 30.Levi G, Scalvini S, Volterrani M, Marangoni S, Arosio G, Quadri A: Coxsackie virus heart disease: 15 years after. Eur Heart J 1988, 9:1303-1307 [DOI] [PubMed] [Google Scholar]
- 31.Archard LC, Bowles NE, Cunningham L, Freeke CA, Olsen EG, Rose ML, Meany B, Why HJ, Richardson PJ: Molecular probes for detection of persisting enterovirus infection of human heart and their prognostic value. Eur Heart J 1991, 12(suppl D):56-59 [DOI] [PubMed] [Google Scholar]
- 32.Bowles NE, Rose ML, Taylor P, Banner NR, Morgan-Capner P, Cunningham L, Archard LC, Yacoub MH: End-stage dilated cardiomyopathy: persistence of enterovirus RNA in myocardium at cardiac transplantation and lack of immune response. Circulation 1989, 80:1128-1136 [DOI] [PubMed] [Google Scholar]
- 33.Gauntt CJ, Gomez PT, Duffey PS, Grant JA, Trent DW, Witherspoon AM, Paque RE: Characterization and myocarditic capabilities of coxsackievirus B3 variants in selected mouse strains. J Virol 1984, 52:598-605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Loudon RP, Moraska AF, Huber SA, Schwimmbeck P, Schultheiss P: An attenuated variant of Coxsackievirus B3 preferentially induces immunoregulatory T cells in vivo. J Virol 1991, 65:5813-5819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolfgram LJ, Beisel KW, Herskowitz A, Rose NR: Variations in the susceptibility to Coxsackievirus B3-induced myocarditis among different strains of mice. J Immunol 1986, 136:1846-1852 [PubMed] [Google Scholar]
- 36.Chow LH, Gauntt CJ, McManus BM: Differential effects of myocarditic variants of Coxsackievirus B3 in inbred mice: a pathologic characterization of heart tissue damage. Lab Invest 1991, 64:55-64 [PubMed] [Google Scholar]
- 37.Huber SA, Job LP, Auld KR, Woodruff JF: Sex-related differences in the rapid production of cytotoxic spleen cells active against uninfected myofibers during Coxsackievirus B-3 infection. J Immunol 1981, 126:1336-1340 [PubMed] [Google Scholar]
- 38.Huber SA, Pfaeffle B: Differential Th1 and Th2 cell responses in male and female BALB/c mice infected with coxsackievirus group B type 3. J Virol 1994, 68:5126-5132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khatib R, Chason JL, Silberberg BK, Lerner AM: Age-dependent pathogenicity of group B coxsackieviruses in Swiss-Webster mice: infectivity for myocardium and pancreas. J Infect Dis 1980, 141:394-403 [DOI] [PubMed] [Google Scholar]
- 40.Woodruff JF, Kilbourne ED: The influence of quantitated post-weaning undernutrition on coxsackievirus B3 infection of adult mice. I. Viral persistence and increased severity of lesions. J Infect Dis 1970, 121:137-163 [DOI] [PubMed] [Google Scholar]
- 41.Huber SA, Job LP, Woodruff JF: In vitro culture of coxsackievirus group B, type 3 immune spleen cells on infected endothelial cells and biological activity of the cultured cells in vivo. Infect Immun 1984, 43:567-573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heim A, Canu A, Kirschner P, Simon T, Mall G, Hofschneider PH, Kandolf R: Synergistic interaction of interferon-beta and interferon-gamma in coxsackievirus B3-infected carrier cultures of human myocardial fibroblasts. J Infect Dis 1992, 166:958-965 [DOI] [PubMed] [Google Scholar]
- 43.Kandolf R, Canu A, Hofschneider PH: Coxsackie B3 virus can replicate in cultured human foetal heart cells and is inhibited by interferon. J Mol Cell Cardiol 1985, 17:167-181 [DOI] [PubMed] [Google Scholar]
- 44.Herzum M, Ruppert V, Kuytz B, Jomaa H, Nakamura I, Maisch B: Coxsackievirus B3 infection leads to cell death of cardiac myocytes. J Mol Cell Cardiol 1994, 26:907-913 [DOI] [PubMed] [Google Scholar]
- 45.Hofschneider PH, Klingel K, Kandolf R: Toward understanding the pathogenesis of enterovirus-induced cardiomyopathy: molecular and ultrastructural approaches. J Struct Biol 1990, 104:32-37 [DOI] [PubMed] [Google Scholar]
- 46.Klingel K, Kandolf R: The role of enterovirus replication in the development of acute and chronic heart muscle disease in different immunocompetent mouse strains. Scand J Infect Dis Suppl 1993, 88:79-85 [PubMed] [Google Scholar]
- 47.Gomez RM, Lopez Costa JJ, Pecci Saavedra G, Berria MI: Ultrastructural study of cell injury induced by coxsackievirus B3 in pancreatic and cardiac tissues. Medicina (B Aires) 1993, 53:300-306 [PubMed] [Google Scholar]
- 48.McManus BM, Chow LH, Wilson JE, Anderson DR, Gulizia JM, Gauntt CJ, Klingel KE, Beisel KW, Kandolf R: Direct myocardial injury by enterovirus: a central roe in the evolution of murine myocarditis. Clin Immunol Immunopathol 1993, 68:159-169 [DOI] [PubMed] [Google Scholar]
- 49.Chow LH, Beisel KW, McManus BM: Enteroviral infection of mice with severe combined immunodeficiency: evidence for direct viral pathogenesis of myocardial injury. Lab Invest 1992, 66:24-31 [PubMed] [Google Scholar]
- 50.Seko Y, Yoshifumi E, Yagita H, Okumura K, Yazaki Y: Restricted usage of T-cell receptor V alpha genes in infiltrating cells in murine hearts with acute myocarditis caused by coxsackie virus B3. J Pathol 1996, 178:330-334 [DOI] [PubMed] [Google Scholar]
- 51.Guthrie M, Lodge PA, Huber SA: Cardiac injury in myocarditis induced by Coxsackievirus group B, type 3 in Balb/c mice is mediated by Lyt 2 + cytolytic lymphocytes. Cell Immunol 1984, 88:558-567 [DOI] [PubMed] [Google Scholar]
- 52.Wong CY, Woodruff JJ, Woodruff JF: Generation of cytotoxic T lymphocytes during coxsackievirus B-3 infection. II. Characterization of effector cells and demonstration of cytotoxicity against viral-infected myofibers. J Immunol 1977, 118:1165-1169 [PubMed] [Google Scholar]
- 53.Wong CY, Woodruff JJ, Woodruff JF: Generation of cytotoxic T lymphocytes during coxsackievirus B-3 infection. III. Role of sex. J Immunol 1977, 119:591-597 [PubMed] [Google Scholar]
- 54.Huber SA: Coxsackievirus-induced myocarditis is dependent on distinct immunopathogenic responses in different strains of mice. Lab Invest 1997, 76:691-701 [PubMed] [Google Scholar]
- 55.Huber SA, Moraska A, Choate M: T cells expressing the gamma delta T-cell receptor potentiate coxsackievirus B3-induced myocarditis. J Virol 1992, 66:6541-6546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huber SA, Mortensen A, Moulton G: Modulation of cytokine expression by CD4+ T cells during coxsackievirus B 3 infections of BALB/c mice initiated by cells expressing the γδ+ T-cell receptor. J Virol 1996, 70:3039-3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kagi D, Seiler P, Pavlovic J, Ledermann B, Burki K, Zinkernagel RM, Hengartner H: The roles of perforin- and Fas-dependent cytotoxicity in protection against cytopathic and noncytopathic viruses. Eur J Immunol 1995, 25:3256-3262 [DOI] [PubMed] [Google Scholar]
- 58.Itoh N, Yonehara S, Ishii A, Yonehara M, Mizushima S, Sameshima M, Hase A, Seto Y, Nagata S: The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 1991, 66:233-243 [DOI] [PubMed] [Google Scholar]
- 59.Rouvier E, Luciani MF, Golstein P: Fas involvement in Ca(2+)-independent T cell-mediated cytotoxicity. J Exp Med 1993, 177:195-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suda T, Nagata S: Purification and characterization of the Fas-ligand that induces apoptosis. J Exp Med 1994, 179:873-879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Watanabe-Fukunaga R, Brannan CI, Itoh N, Yonehara S, Copeland NG, Jenkins NA, Nagata S: The cDNA structure, expression, and chromosomal assignment of the mouse Fas antigen. J Immunol 1992, 148:1274-1279 [PubMed] [Google Scholar]
- 62.Nagata S, Suda T: Fas and Fas ligand: lpr and gld mutations. Immunol Today 1995, 16:39-43 [DOI] [PubMed] [Google Scholar]
- 63.Ramsay AJ, Ruby J, Ramshaw IA: A case for cytokines as effector molecules in the resolution of virus infection. Immunol Today 1993, 14:155-157 [DOI] [PubMed] [Google Scholar]
- 64.Guidotti LG, Chisari FV: To kill or to cure: options in host defense against viral infection. Curr Opinion Immunol 1996, 8:478-483 [DOI] [PubMed] [Google Scholar]
- 65.Seko Y, Takahashi N, Yagita H, Okumura K, Yazaki Y: Expression of cytokine mRNAs in murine hearts with acute myocarditis caused by coxsackievirus B3. J Pathol 1997, 183:105-108 [DOI] [PubMed] [Google Scholar]
- 66.Herzum M, Huber SA, Weller R, Grebe R, Maisch B: Treatment of experimental murine Coxsackie B3 myocarditis. Eur Heart J 1991, 12(suppl D):200-202 [DOI] [PubMed] [Google Scholar]
- 67.Kishimoto C, Takada H, Kuroki Y, Matsushita I, Hiraoka Y, Kurokawa M, Ochiai H, Sasayama S: Enhancement of coxsackievirus B3 myocarditis in mice by lobenzarit disodium through inhibition of splenic pan T cells. Cardiovasc Res 1993, 27:243-248 [DOI] [PubMed] [Google Scholar]
- 68.Rezkalla S, Khatib G, Khatib R: Coxsackievirus B3 murine myocarditis: deleterious effects of nonsteroidal anti-inflammatory agents. J Lab Clin Med 1986, 107:393-395 [PubMed] [Google Scholar]
- 69.Costanzo-Nordin MR, Reap EA, O’Connell JB, Robinson JA, Scanlon PJ: A nonsteroid anti-inflammatory drug exacerbates Coxsackie B3 murine myocarditis. J Am Coll Cardiol 1985, 6:1078-1082 [DOI] [PubMed] [Google Scholar]
- 70.Hiraoka Y, Kishimoto C, Kurokawa M, Ochiai H, Sasayama S: The effects of FK-506, a novel and potent immunosuppressant, upon murine Coxsackievirus B3 myocarditis. J Pharmacol Exp Ther 1992, 260:1386-1391 [PubMed] [Google Scholar]
- 71.Mason JW, O’Connell JB, Herskowitz A, Rose NR, McManus BM, Billingham ME, Moon TE: A clinical trial of immunosuppressive therapy for myocarditis: The Myocarditis Treatment Trial Investigators. N Engl J Med 1995, 333:269-275 [DOI] [PubMed] [Google Scholar]