

Abstract
Abnormal oxidative processes including a reduction in thiamine-dependent enzymes accompany many neurodegenerative diseases. Thiamine deficiency (TD) models the cellular and molecular mechanisms by which chronic oxidative aberrations associated with thiamine-dependent enzyme deficits cause selective neurodegeneration. The mechanisms underlying selective cell death in TD are unknown. In rodent TD, the earliest region-specific pathological change is breakdown of the blood-brain barrier (BBB). The current studies tested whether nitric oxide and microglia are important in the initial events that couple BBB breakdown to selective neuronal loss. Enhanced expression of endothelial nitric oxide synthase and nicotinamide adenine dinucleotide phosphate diaphorase reactivity in microvessels, as well as the presence of numerous inducible nitric oxide synthase-immunoreactive microglia, accompanied the increases in BBB permeability. Nitric oxide synthase induction appears critical to TD pathology, because immunoreactivity for nitrotyrosine, a specific nitration product of peroxynitrite, also increased in axons of susceptible regions. In addition, TD elevated iron and the antioxidant protein ferritin in microvessels and in activated microglia, suggesting that these cells are responding to an oxidative challenge. All of these changes occurred in selectively vulnerable regions, preceding neuronal death. These findings are consistent with the hypothesis that the free radical-mediated BBB alterations permit entry of iron and extraneuronal proteins that set in motion a cascade of inflammatory responses culminating in selective neuronal loss. Thus, the TD model should help elucidate the relationship between oxidative deficits, BBB abnormalities, the inflammatory response, ferritin and iron elevation, and selective neurodegeneration.
Abnormalities in oxidative metabolism appear central to aging and many pathological conditions including Alzheimer’s disease, 1-6 Parkinson’s disease, 7 Huntington’s disease, 8 ischemia-reperfusion injury, 9 Down’s syndrome, 10 amyotrophic lateral sclerosis, 11 chronic alcoholism, 12 and Wernicke-Korsakoff syndrome. 13 Reductions in α-ketoglutarate dehydrogenase, a thiamine-dependent, key enzyme of the Krebs cycle, occur in many of these diseases, such as Alzheimer’s disease, 14-16 Parkinson’s disease, 17 Wernicke-Korsakoff syndrome, 13 and Friedreich’s and type 1 hereditary spinocerebellar ataxias. 18 Nevertheless, the role of thiamine-dependent enzyme deficits in neurodegeneration is still unclear. Whether these deficits are the initiating event, part of a critical cascade, or merely a secondary phenomenon remains to be elucidated. Experimental thiamine deficiency (TD) provides a test of how chronic low-grade interruption of these enzymes contributes to the brain pathology.
TD models the molecular and cellular mechanisms by which a chronic, generalized impairment of oxidative metabolism leads to selective neuronal loss. In rodents, TD causes a generalized reduction in the α-ketoglutarate dehydrogenase activity. Breakdown of the blood-brain barrier (BBB) is the earliest region-specific pathological change during TD. 19,20 This is followed by cell loss and accumulation of β protein precursor (βPP) immunoreactivity in perikarya and abnormal neurites that occur either around the lesions in rats 21 or in neuritic clusters within vulnerable regions in mice. 22
The mechanisms responsible for the region-selective BBB breakdown, cell loss, and abnormal βPP expression in TD are unknown. One possibility is that factors released from endothelial cells and microglia such as nitric oxide (NO) or free radical generators induce the damage. Recently, increased cerebral free radical production has been reported in TD. 23 Although NO possesses important physiological roles in the nervous system, it is also a potential mediator of neurotoxicity in a variety of disease states. Previous studies from other models of disease suggest that excessive NO production may be linked to or even cause abnormalities in BBB permeability. 24-26 Microglia are prominently involved in the pathogenesis of neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. 27 In vitro experiments demonstrate that activated microglia mediate neuronal cell injury via NO. 28 The current studies tested whether NO and microglia participate in a cascade of initial events that culminate in selective cell loss during TD.
Materials and Methods
Induction of TD
Adult male C57BL/6 mice (20 to 30 g; Charles River Breeding Laboratories, Wilmington, MA) or Fischer 344 Brown Norway F1 hybrid rats (250 to 300 g; Harlan Sprague Dawley, Indianapolis, IN) were housed individually in suspended wire mesh cages. As in our previous reports, 19,21,22 TD was induced by ad libitum feeding of a thiamine-deficient diet (ICN Nutrition Biomedicals, Cleveland, OH) and daily intraperitoneal injection of the thiamine antagonist pyrithiamine hydrobromide (5 μg in 0.1 ml of saline/10 g body weight; Sigma Chemical Co., St. Louis, MO). Control animals received a thiamine-supplemented diet ad libitum and intraperitoneal saline injections daily (0.1 ml/10 g body weight). All animal procedures were approved by the Institutional Animal Care and Use Committee of Cornell University Medical College.
Tissue Preparation
At different stages of TD, mice (days 9, 10, and 11) and rats (days 9, 10, 11, 12, and 13) were euthanized with a lethal dose of sodium pentobarbital (6 mg/100 g intraperitoneally; Fort Dodge Laboratories, Fort Dodge, IA) and perfused transcardially with 0.9% NaCl containing heparin (10 units/ml) followed by 4% paraformaldehyde in 0.1 mol/L sodium phosphate buffer (PB; pH 7.4). Brains were removed and 35-μm free-floating sections were cut. Sections were stored in 0.1 mol/L sodium phosphate-buffered saline (PBS; pH 7.4). The current studies focused mainly on the thalamus, dorsal lateral geniculate nucleus, and inferior colliculus, three of the major brain regions consistently damaged during TD.
Immunohistochemical Detection of BBB Disturbances
Breakdown of the BBB was assessed by employing a one-step immunohistochemical detection of immunoglobulin G (IgG). 29 Sections were incubated for 1 hour in horseradish peroxidase-conjugated rabbit anti-rat or anti-mouse IgG (1:500 in PBS; Sigma). Visualization of the peroxidase activity was performed by incubating the sections for 5 to 10 minutes in a substrate solution containing 0.05% 3,3′-diaminobenzidine tetrahydrochloride dihydrate (DAB) and 0.003% H2O2 in PB.
Nicotinamide Adenine Dinucleotide Phosphate Diaphorase (NADPH-d) Histochemistry
NADPH-d activity is a reasonable but not an absolute correlate of NO synthase (NOS) activity. 30,31 In paraformaldehyde-fixed tissues, virtually all NADPH-dependent oxidative enzymes except NOS are inactivated. 32 To detect NADPH-d activity histochemically, sections were incubated at 37°C for 1 hour in a freshly prepared solution containing 1 mg/ml β-NADPH, 0.2 mg/ml nitroblue tetrazolium, and 0.3% Triton X-100 (all purchased from Sigma) dissolved in PB. The reaction was terminated by three washes with PB.
Immunocytochemistry
Sections were stained using modified avidin-biotin-peroxidase immunocytochemistry. 33 Endogenous peroxidase was blocked by incubating the sections in 0.3% H2O2 in PBS for 30 minutes. This was followed by sequential incubation in 1) PBS containing 1% bovine serum albumin and 0.2% Triton X-100 for 30 minutes, 2) one of the primary antisera (Table 1) ▶ diluted in PBS/0.5% bovine serum albumin for 18 hours, 3) biotinylated anti-rabbit IgG, 1:200 in PBS/0.5% bovine serum albumin for 1 hour, and 4) avidin-biotin-peroxidase complex, 1:50 in PBS for 1 hour. Biotinylated secondary antibodies and the avidin-biotin-peroxidase complex were both purchased from Vector Laboratories (Burlingame, CA). The immunoreaction was developed in a substrate solution containing 0.05% DAB and 0.003% H2O2 in PB. Sections were rinsed in PB, mounted on glass slides, dehydrated in increasing concentrations of ethanol, cleared in xylene, and coverslipped. Specificity controls were carried out by replacing the primary antibody with PBS/0.5% bovine serum albumin or, in the case of nitrotyrosine, by preadsorption with 10 mmol/L 3-nitrotyrosine (Sigma).
Table 1.
Antisera Used in the Experiments
| Antibodies | Source | Dilution |
|---|---|---|
| Endothelial NOS | Transduction Labs (Lexington, KY) | 1:1000 |
| Inducible NOS | Transduction Labs | 1:1000 |
| Brain NOS | Transduction Labs | 1:1000 |
| Nitrotyrosine | Upstate Biotechnology (Lake Placid, NY) | 1:100 |
| Ferritin | DAKO (Carpinteria, CA) | 1:7000 |
| GFAP | DAKO | 1:1000 |
| Galactocerebroside | Chemicon (Temecula, CA) | 1:1000 |
| βPP | Dr. Samuel E. Gandy | 1:7000 |
GFAP, glial fibrillary acidic protein.
Lectin Histochemical Staining of Microglia
Free-floating sections adjacent to those used for ferritin immunocytochemistry were stained using a lectin histochemical technique. 34 Sections were treated with 0.1% Triton X-100 in PBS for 30 minutes, rinsed three times in PBS, and incubated in peroxidase-labeled Bandeiraea simplicifolia BS-I isolectin B4 (20 μg/ml in PBS/0.1% Triton X-100; Sigma) overnight at 4°C. Sections were rinsed three times in PBS, and the lectin binding sites were localized with 0.05% DAB and 0.003% H2O2.
Iron Histochemistry
Free-floating sections were mounted on gelatin-coated slides and air dried. Sections were rinsed three times in deionized water for 30 minutes and stained for iron. 35,36 Sections were incubated in a solution containing 1% HCl and 1% potassium ferrocyanide at room temperature for 30 minutes and rinsed three times in deionized water for 30 minutes. The reaction was intensified by incubation in 0.5% DAB in PB for 20 minutes, followed by 0.5% DAB in PB containing 0.005% H2O2 for 15 minutes. Three rinses with deionized water for 30 minutes stopped the reaction.
Results
Behavior and Neuropathology of Thiamine-Deficient Animals
TD produced behavioral and neuropathological changes consistent with those observed in our previous studies. 19,21,22 Animals exhibited motor deficits and began to lose weight after 5 to 6 days (mice) or 8 to 9 days (rats) of thiamine deprivation. Weight loss was followed by ataxia and loss of righting reflex.
Similar neuropathological changes occurred in both mice and rats, although in the latter, the time course is longer and inducible NOS immunoreactivity was not detected. The observations described below apply to both species. Macroscopic examination of late-stage thiamine-deficient mouse (day 11) or rat (day 13) brains revealed pinpoint hemorrhages in the thalamus, mammillary body, dorsal lateral and medial geniculate nuclei, inferior colliculus, superior and inferior olives, and some periventricular regions. Brain ventricles displayed edematous enlargement. Tissue cavitation of the thalamus occurred in severe cases. Other regions such as the cortex and hippocampus were unaffected. Table 2 ▶ summarizes the sequence of microscopic pathological alterations during TD in mice. No apparent abnormalities were detected on day 9 of TD. At late stages, the vulnerable regions exhibited neuronal loss (Figure 1) ▶ , pale neuropil, perivascular edema, and extravasated erythrocytes. 19,21,22
Table 2.
Sequence of Pathological Changes in the Mouse Thalamus after TD
| Alterations | Control (n = 4) | TD, 9 days (n = 4) | TD, 10 days (n = 4) | TD, 11 days (n = 4) |
|---|---|---|---|---|
| IgG extravasation* | −−−− | −−−− | ++++ | ++++ |
| NADPH-d staining of microvessels† | −−−− | −−−− | ++++ | ++++ |
| Ferritin-positive activated microglia | −−−− | −−−− | ++++ | ++++ |
| Iron deposition in activated microglia | −−−− | −−−− | ++++ | ++++ |
| eNOS staining of microvessels | −−−− | −−−− | ++−+ | ++++ |
| iNOS staining of macrophage | −−−− | −−−− | −+−+ | ++++ |
| Nitrotyrosine staining of axons | −−−− | −−−− | ++++ | ++++ |
| Cell loss‡ | −−−− | −−−− | −−−+ | ++++ |
eNOS, endothelial NOS; iNOS, inducible NOS. Data represent the presence (+) or absence (−) of the abnormality in each animal.
*Region-specific increase in IgG immunoreactivity.
†Intense NADPH-d reactivity of microvessel walls compared with controls.
‡Measured by the presence of areas of cell death in βPP-stained sections.
Figure 1.
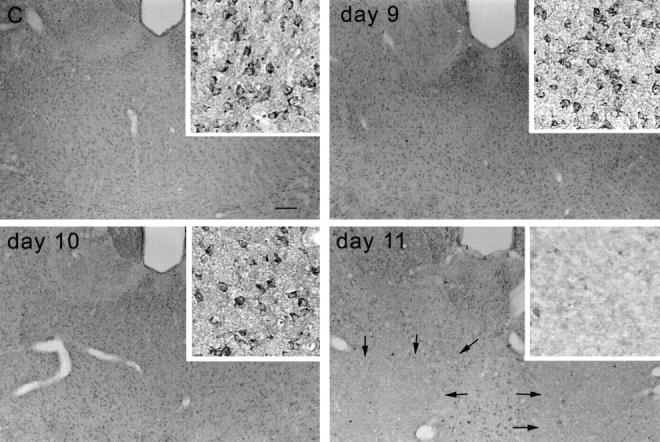
βPP immunoreactivity in the mouse thalamus of control (C) and days 9, 10, and 11 of TD, demonstrating areas of neuronal loss at day 11 (arrows). Insets: High magnification of the mediodorsal thalamic nuclei. Scale bar = 100 μm (15 μm for insets).
BBB Dysfunction
In thiamine-deficient mouse brain, small focal areas of increased IgG immunoreactivity appeared in susceptible regions as early as 10 days of TD before the onset of apparent cell loss and hemorrhage. These areas of IgG accumulation became larger on day 11 when hemorrhage and neuronal death were evident. On days 12 to 13, areas of IgG extravasation were very extensive.
NADPH-d Histochemistry
Under normal conditions, NADPH-d-labeled neurons occurred in specific nuclei throughout the mouse or rat brain, consistent with previous mapping studies in rats. 37,38 Intensely stained neurons were sparsely scattered in all layers of the cortex, striatum, and nucleus basalis. The thalamus, central nucleus of the inferior colliculus, and the dorsal lateral and medial geniculate nuclei were virtually devoid of NADPH-d-positive neurons, or they contained only a few weakly stained cells.
Striking changes occurred in vulnerable regions of thiamine-deficient brains. NADPH-d reactivity increased in microvessel walls within the thalamus, inferior colliculus (Figure 2) ▶ , mammillary body, and medial geniculate nucleus in TD as compared with controls. Elevated NADPH-d staining occurred as early as 10 days, which corresponds with the first appearance of IgG accumulation.
Figure 2.
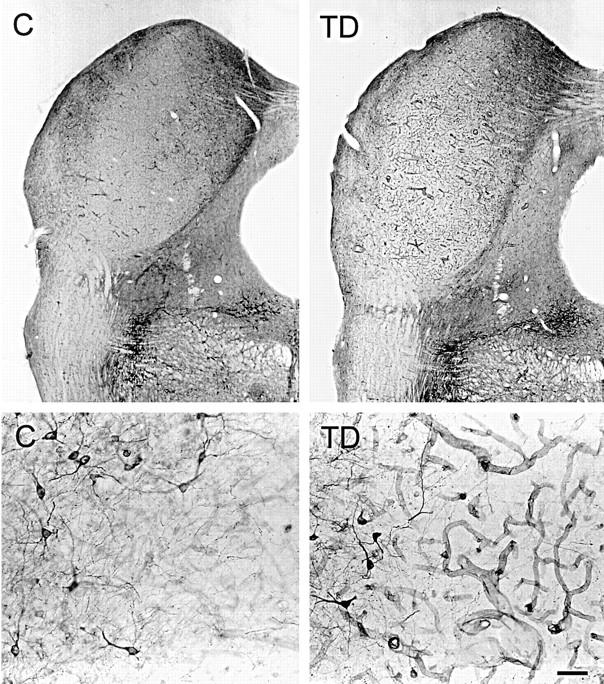
Low-magnification (top) and high-magnification (bottom) photomicrographs showing NADPH-d staining of the inferior colliculus of a 12-day thiamine-deficient (TD) rat compared with control (C). Note the enhanced NADPH-d reactivity of the microvessels after TD. NADPH-d-stained neurons occur along the periphery (bottom, left side of each photomicrograph) of the vulnerable central nucleus of the inferior colliculus that contains virtually no stained neurons (bottom, right side of each photomicrograph). Scale bar = 100 μm (top) and 10 μm (bottom).
NOS Induction
All isoforms of NOS possess NADPH-d activity. 31 The responses of the three distinct NOS types to thiamine deprivation were evaluated using antibodies that react selectively with peptide sequences unique to endothelial, inducible, and brain NOS. Ten days of TD enhanced endothelial NOS immunoreactivity in microvessel walls within the thalamus (Figure 3) ▶ . This alteration corresponded to increased NADPH-d reactivity in blood vessel walls. Numerous inducible NOS-immunoreactive macrophage-like cells occurred within the thalamus beginning at day 10 of TD in mice (Figure 3) ▶ but not in rats. Owing to the lack of a reliable marker to distinguish between microglia and macrophages, these cells were most likely macrophages on the basis of their morphology. Neither the control sections nor the nonvulnerable areas of thiamine-deficient brains exhibited these changes in NOS expression. Because of the restricted localization of endothelial and inducible NOS immunoreactivities, Western blot analysis was not performed.
Figure 3.
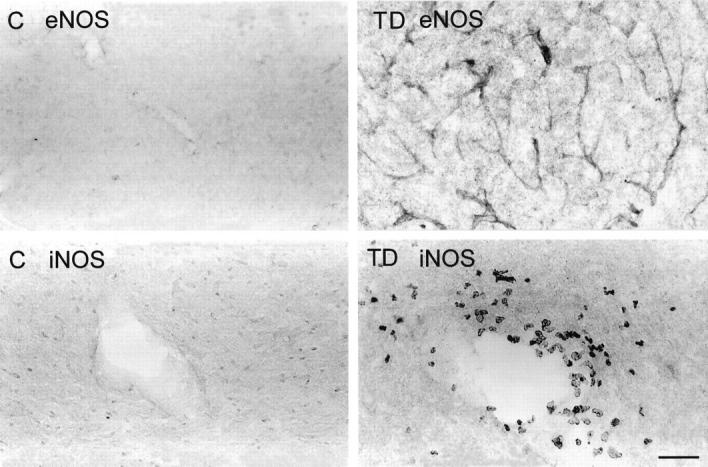
Endothelial NOS (eNOS) immunoreactivity (top) in microvessel walls within the thalamus of control (C) and thiamine-deficient (TD) mice depicting increased immunostaining after 10 days of TD. Inducible NOS (iNOS) immunodetection (bottom) in the thalamus of control (C) and thiamine-deficient (TD) mice shows immunoreactive macrophages around a blood vessel in the thalamus of TD. Scale bar = 100 μm (top) and 50 μm (bottom).
Nitrotyrosine Immunoreactivity
NO plays an important role in several physiological processes, including modulation of cerebral blood flow. 39,40 However, excessive NO produces oxidative damage. The reaction of superoxide with NO generates peroxynitrite, a potent biological oxidant that cannot be measured directly. One way to assess peroxynitrite-associated oxidative damage is by measuring nitrotyrosine, a specific nitration product of peroxynitrite. Localization of nitrotyrosine in axons and axon terminals of normal adult rats has been demonstrated ultrastructurally. 41 Thus, a rabbit polyclonal antibody was used to monitor nitrotyrosine-containing proteins. Intense nitrotyrosine immunoreactivity was not detected in any region of control brains. In 10- and 11-day thiamine-deficient animals, intensely immunolabeled axons and axon terminals were observed in the thalamus (Figure 4) ▶ . Moderate immunoreactivity also occurred in neuronal cytoplasm within the thalamus. Nitrotyrosine immunoreactive structures were not detected in the nonvulnerable areas of thiamine-deficient brains. Preadsorption of antinitrotyrosine with 10 mmol/L 3-nitrotyrosine abolished the intense immunoreactivity in the thalamus of TD brains (data not shown).
Figure 4.

Immunocytochemical detection of nitrotyrosine formation in the paraventricular thalamic nucleus of control (C) and thiamine-deficient (TD) mice. Prominent immunostaining occurs in axons after thiamine deprivation. Scale bar = 25 μm.
Ferritin, Iron, and Microglial Responses to TD
Ferritin immunoreactivity occurred in resting microglia throughout the gray matter of both control and thiamine-deficient animals. Lightly stained microglia with scanty cytoplasm and thin processes were scattered in many regions including the cerebral cortex, cerebellum, and hippocampus. Ferritin was not detectable in neurons in any region.
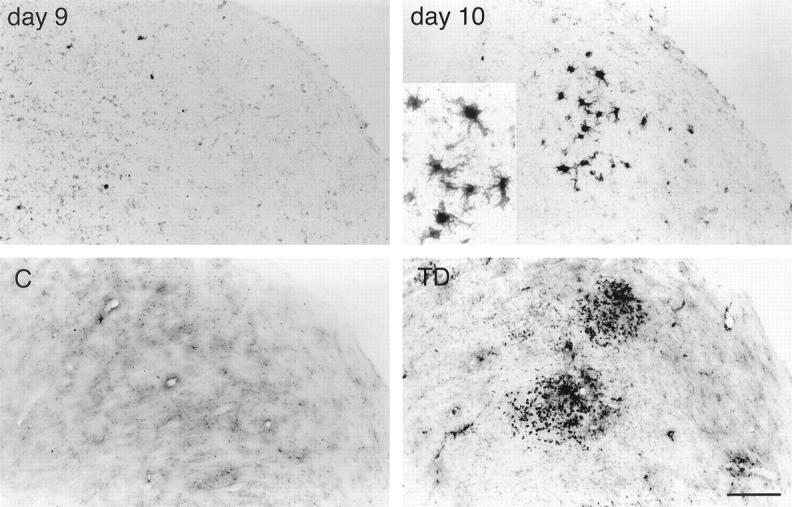
In thiamine-deficient brains, BBB breakdown was accompanied by the presence of many microglia with intense ferritin immunoreactivity in the thalamus, dorsal lateral, and medial geniculate nuclei (Figure 5) ▶ , mammillary body, and inferior colliculus. The immunolabeled cells possessed plump bodies with ramified processes (Figure 5 ▶ , inset), typical of the ferritin-positive activated microglia found in pathological brain tissues. 42,43 In adjacent sections, B. simplicifolia BS-I isolectin B4 stained cells with the same morphology and distribution as the ferritin-immunoreactive microglia (data not shown). These activated microglia were absent from unaffected TD brain regions and all areas of control brains. Light ferritin immunoreactivity occurred in microvessels within vulnerable regions. Intense ferritin immunolabeling was detected in microglia in the immediate vicinity of microvessels within the vulnerable regions (Figure 6) ▶ . Ferritin immunostaining was also prominent in the walls of large blood vessels and capillaries within susceptible areas, especially the thalamus of thiamine-deficient rats (Figure 6) ▶ . Blood vessels were also stained in the medial geniculate nucleus and inferior colliculus, although less frequently. Nonvulnerable regions did not exhibit strong immunoreactivity in microglia or microvessels.
Figure 5.

IgG (top) and ferritin (bottom) immunoreactivities in the dorsal lateral thalamic nucleus of 9-day and 10-day thiamine-deficient mice. Accumulation of IgG immunoreactivity on day 10 parallels ferritin-labeled microglial activation. Inset: Morphology of typical activated microglia. Scale bar = 100 μm (25 μm for inset).
Figure 6.
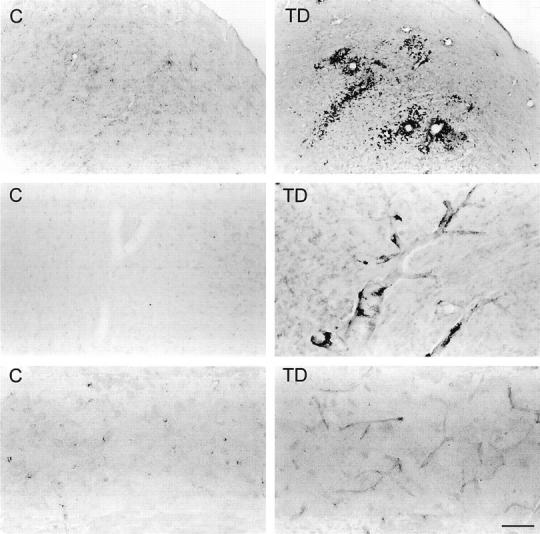
Top: Photomicrographs showing ferritin immunostaining in the inferior colliculus of control (C) and thiamine-deficient (TD) rats. Intensely labeled microglia occur prominently around blood vessels and are also scattered in the area of cell damage. Middle: Photomicrographs of ferritin in the thalamus of control and thiamine-deficient rats showing enhanced staining of large blood vessel walls in TD. Intensely labeled microglia occur along the walls of large vessels in TD. Bottom: Photomicrographs showing enhanced ferritin staining of capillaries in thiamine-deficient rat thalamus as compared with control. Scale bar = 250 μm (top) and 50 μm (middle and bottom).
In control mice and rats, light iron staining occurred mainly in microglia and oligodendrocytes, and to a lesser extent in the parenchyma throughout the brain. Parenchymal iron in control brains was more abundant in vulnerable regions such as the thalamus, mammillary body, and inferior colliculus as compared with the cortex, which is spared in TD. In thiamine-deficient mice, iron accumulated in microglia within the thalamus and dorsal lateral and medial geniculate nuclei (Figure 7) ▶ , beginning on day 10, coinciding with BBB breakdown and activation of ferritin-positive microglia. Iron deposition in glial cells was not detected in any nonvulnerable region. In thiamine-deficient rats, the iron-laden microglia formed clusters especially around blood vessels (Figure 7) ▶ . This pattern of excessive iron deposition in microglial cells paralleled the ferritin immunoreactivity. Glial fibrillary acidic protein immunostaining did not reveal any significant astrogliosis in thiamine-deficient brains compared with controls (data not shown).
Figure 7.
Iron histochemical staining of mouse control (C) and thiamine-deficient (TD) lateral dorsal geniculate nucleus (top) and rat control and thiamine-deficient inferior colliculus (bottom). In TD, iron accumulates in microglia with a similar distribution as ferritin (see Figures 5 ▶ , bottom, and 6 ▶ , top). Scale bar = 50 μm (25 μm for inset).
Discussion
The current experiments provide evidence that oxidative damage plays a role in the selective neuronal death induced by TD. The oxidative challenge was associated with a region-specific breakdown of the BBB before the onset of neuronal loss. Induction of endothelial and inducible NOS, as well as elevation of ferritin and iron, paralleled the BBB abnormalities within the vulnerable regions. NOS induction appeared critical to TD-induced cell loss because immunoreactivity for nitrotyrosine, a specific nitration product of peroxynitrite, increased in axons within the susceptible areas. Peroxynitrite is a powerful oxidant generated by reaction of superoxide with NO. In addition, elevation of iron paralleled increases in the antioxidant protein ferritin in blood vessels and in activated microglia. All of these changes occurred only in selectively vulnerable regions and preceded neuronal death. Taken together, these findings suggest that BBB breakdown and subsequent cerebral hemorrhage leads to or exacerbates the oxidative damage induced by the low-grade oxidative deficits during TD.
There are at least two possible mechanisms by which excessive vascular NO production contributes to neurodegeneration. First, NO can alter BBB permeability to damaging extraneuronal proteins, including IgG. For instance, inhibition of NOS by administration of NG-nitro-l-arginine methyl ester hydrochloride or aminoguanidine reverses the increased BBB permeability in focal ischemia, 44 meningitis, 26 and diabetes. 25 As has been suggested with amyotrophic lateral sclerosis patients, IgG can kill cells. 45 Internalization of specific plasma proteins such as immunoglobulins, transferrin, albumin, and α-2-macroglobulin has been reported to damage cerebellar Purkinje cells. 46 Indeed, blood proteins can inhibit oxidative metabolism, 47 which could be devastating to cells that are already metabolically compromised by TD. A second possibility is that vascular NO may react with superoxide, a free radical that is also increased in TD, 48 to generate peroxynitrite. Peroxynitrite can diffuse from its vascular and microglial origin and generate hydroxyl radicals that are deleterious to surrounding neurons. 49 The strong presence of ferritin in microvessels and in microglia around blood vessels suggests that these glial cells are responding to an oxidative challenge from the vessels. Earlier studies of hypoxia in cultured oligodendrocytes provide evidence that ferritin may serve as an antioxidant. 50,51 Even though NO and hemoglobin may serve a protective role, they failed to protect the microvessels from oxidative damage during TD. The current report is the first to document that the microvessels in vulnerable brain regions may be an important site of free radical production during TD. Inducible NOS expression in vascular cells has also been implicated in ischemic brain injury. 52
The enhancement of nitrotyrosine immunoreactivity in axons within vulnerable regions of thiamine-deficient brains provides strong evidence that oxidants derived from NO, such as peroxynitrite, are involved in TD pathology. Several mechanisms of peroxynitrite-mediated neurotoxicity have been reported. Peroxynitrite may cause neuronal energy deficiency by impairing the mitochondrial respiratory chain or the mitochondrial calcium metabolism, interfering with key enzymes of the tricarboxylic acid cycle, or DNA damage. 53 Nitrotyrosine immunoreactivity is present in neurons, including those containing neurofibrillary tangles in brain tissues from cases of Alzheimer’s disease but not in controls. 54,55 In TD, the nitration of tyrosine does not represent a global oxidative damage, but a local, selective change, as evidenced by the absence of intense nitrotyrosine staining in spared areas. This observation is consistent with the possible participation of peroxynitrite-associated oxidative damage in selective neurodegeneration in TD. However, the specificity of nitrotyrosine as a biomarker for peroxynitrite is still subject to question. 56
Ferritin induction has been reported as a measure of the brain’s ability to respond to damage such as hypoxia 50 and transient forebrain ischemia. 36 Ferritin is an intracellular iron-sequestering protein 57 found in resting microglia, 42 and to a lesser extent endothelial cells 58 and oligodendrocytes. 11 A polyclonal antiserum against ferritin was used in the current experiments to monitor ferritin synthesis in brain cells including microglia. Ferritin is a reliable marker for microglia. 42,43,59-61 In the current studies, demonstration of the specificity of ferritin antibody as a microglial marker consisted of 1) strong ferritin immunoreactivity of a known BV2 murine microglial cell line (unpublished results); 2) absence of glial fibrillary acidic protein or galactocerebroside immunoreactivity in cells similar to ferritin-positive, activated microglia within vulnerable regions in adjacent sections (data not shown) and 3) localization of the microglial marker B. simplicifolia BS-I isolectin B4 in cells with the same morphology and distribution as the ferritin-immunoreactive cells in adjacent sections (data not shown). Although ferritin is not restricted to microglia, 62 immunolabeling in the current experiments was more intense in microglia than in any other cell type. Altered ferritin expression in the vulnerable regions of thiamine-deprived brains further implicates oxidative damage in the pathogenesis of TD. Ferritin synthesis can be induced by oxidative damage after hypoxia, 50 by transient forebrain ischemia, 36 and by hemoglobin-derived heme. 51 NOS contains stoichiometric amounts of heme 63 that may also contribute to induction of ferritin synthesis.
Disruption of iron homeostasis is believed to contribute to oxidative damage in many neurodegenerative diseases. 5,59,64,65 Cellular iron is regulated by ferritin, which may serve as an antioxidant to protect cells from iron-catalyzed lipid peroxidation injury. 50,66 Enhanced ferritin immunoreactivity is a marker of excessive iron deposition. The parallel occurrence of increased ferritin with IgG accumulation in overlapping vulnerable regions during TD suggests that excess iron originates from peripheral blood and not from degenerating neurons. Hemin iron of disrupted erythrocytes that crosses the BBB by the same mechanism as IgG is a potential source of iron. The receptors for the iron-transport protein transferrin are localized only on the apical membrane of the BBB endothelial cells. 67 This highly polarized localization of transferrin receptors at the BBB supports a receptor-mediated endocytosis of iron-loaded transferrin at the BBB. 67 Iron can also be released from ferritin by the free radical form of toxins or superoxide. 66 Interestingly, the intrinsic iron in control animals was relatively abundant in vulnerable regions such as the thalamic nuclei, in agreement with a previous study, 36 and the inferior colliculus as compared with the nonsusceptible regions. It is possible that a relatively high intrinsic iron content predisposes the vulnerable regions to iron-induced oxidative stress. Iron can initiate free radical formation, leading to lipid peroxidation and subsequent neurodegeneration. 68-70 Ferritin elevation has been suggested to compensate for age-related increases in iron and to prevent accumulation of protein carbonyls, the principal product of protein oxidation. 71 In Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis, the localized elevation of iron identifies potential sites where iron could promote oxidative damage. 59,65 Recently, redox-active iron has been demonstrated in Alzheimer plaques and tangles of Alzheimer’s disease, suggesting that iron accumulation could be an important source of oxidative damage in Alzheimer’s disease. 72 These iron-containing neuritic plaques are surrounded by microglia and astrocytes that robustly immunostain for ferritin. 73 Thus, increases in ferritin immunoreactivity in blood vessels and microglia in vulnerable regions indicate a response to an increased iron load and therefore represent a neuroprotective and antioxidant response to TD.
The current results show that BBB damage and NOS induction are accompanied by microglial activation. Microglia are extremely sensitive to perturbations in the brain microenvironment. Glial activation usually has a protective and supportive function, but in disease, glial cells may produce excessive cytokines that compromise neuronal function. 74 Although overproduction of NO by endothelial cells, activated microglia, and macrophages may contribute to neuronal death, these cells may also be beneficial. 27,75 Factors secreted by activated macrophages have been shown to reduce Alzheimer β-amyloid protein accumulation in vascular smooth muscle cells by stimulating the nonamyloidogenic processing of βPP. 76 This finding, in combination with our current demonstration of region-specific microglial activation, may explain the failure to detect Alzheimer β-amyloid protein in plaque-like neuritic clusters that exhibit intense βPP immunoreactivity in affected regions from our previous mouse TD studies. 22 The early increase in BBB permeability, induction of NOS, elevation of ferritin and iron, and microglial activation at the same time course suggest that these responses are part of a cascade of events that contribute to TD pathogenesis. However, the temporal resolution of the current in vivo studies could not clarify the causal relationship among these abnormalities. Understanding this relationship will help elucidate the cellular basis for selective cell death. During late stages of TD, many events occur, including N-methyl-d-aspartate-mediated excitotoxicity, 77 seizures, and decreased NOS activities. 78 The decline in NOS activities in late stages of TD is attributable to the neuronal loss and is not a specific reduction.
The current observations support the hypothesis that a generalized metabolic injury from TD increases NO and other free radical generators in BBB endothelial cells, resulting in breach of the BBB. The free radical-mediated alteration of the BBB permits entry of immunoglobulins and iron into the brain. The extraneuronal proteins and iron, together with the metabolic impairment itself, activate microglia to initiate an inflammatory response including excessive NO production. NO reacts with superoxide to produce peroxynitrite, which diffuses from its cell origin to form an intermediate that nitrates proteins, thereby damaging the metabolically compromised neurons. Ferritin induction in microglia represents an important neuroprotective, antioxidant defense by storing and transporting iron in forms that will not generate reactive radicals. Taken together, the current observations suggest that oxidative damage may contribute to the selective neuronal death during TD. Thus, the TD model should be useful in elucidating the relationship between oxidative deficits, BBB abnormalities, inflammatory response, ferritin and iron elevation, and selective neurodegeneration. An understanding of these interactions may provide insight into the mechanism of cell demise in human diseases in which oxidative damage, NO, and microglial ferritin and iron responses have been implicated.
Acknowledgments
The authors are grateful to Charles Carver and Hui Zhang for helping with photography.
Footnotes
Address reprint requests to Dr. Gary E. Gibson, Cornell University Medical College at Burke Medical Research Institute, 785 Mamaroneck Avenue, White Plains, NY 10605. E-mail: ggibson@med.cornell.edu.
Supported by National Institutes of Health grants AG-14600 (to GEG); AG-11508, AG-09464, AG-10491, and AG-13780 (to SEG); and AG-09014-07 (to NYC).
References
- 1.Blass JP, Gibson GE: The role of oxidative abnormalities in the pathophysiology of Alzheimer’s disease. Rev Neurol (Paris) 1991, 147:513. [PubMed] [Google Scholar]
- 2.Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR: Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer’s disease. Proc Natl Acad Sci USA 1991, 88:10540-10543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA: A model for β-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer’s Disease. Proc Natl Acad Sci USA 1994, 91:3270-3274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nixon RA, Cataldo AM: Free radicals, proteolysis and degeneration of neurons in Alzheimer’s disease: how essential is the β-amyloid link? Neurobiol Aging 1994, 15:463-469 [DOI] [PubMed] [Google Scholar]
- 5.Smith MA, Perry G: Free radical damage, iron, and Alzheimer’s disease. J Neurol Sci 1995, 134:92-94 [DOI] [PubMed] [Google Scholar]
- 6.Smith MA, Sayre LM, Monnier VM, Perry G: Radical ageing in Alzheimer’s disease. Trends Neurosci 1995, 18:172-176 [DOI] [PubMed] [Google Scholar]
- 7.Dexter DT, Carter CJ, Wells FR, Javoy-Agid F, Jenner P, Marsden CD: Basal lipid peroxidation in substantia nigra is increased in Parkinson’s disease. J Neurochem 1987, 52:381-389 [DOI] [PubMed] [Google Scholar]
- 8.Beal MF: Aging, energy, and oxidative stress in neurodegenerative diseases. Ann Neurol 1995, 38:357-366 [DOI] [PubMed] [Google Scholar]
- 9.Cao W, Carney JM, Duchon A, Floyd RA, Chevion M: Oxygen free radical involvement in ischemia and reperfusion injury to brain. Neurosci Lett 1988, 88:233-238 [DOI] [PubMed] [Google Scholar]
- 10.Furuta A, Price DL, Pardo CA, Troncoso JC, Xu ZS, Taniguchi N, Martin LJ: Localization of superoxide dismutases in Alzheimer’s disease and Down’s syndrome neocortex and hippocampus. Am J Pathol 1995, 146:357-367 [PMC free article] [PubMed] [Google Scholar]
- 11.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, Donaldson D, Goto J, O’Regan JP, Deng H-X, Rahmani Z, Krizus A, McKenna-Yasek D, Cayabyab A, Gaston SM, Berger R, Tanzi R, Halpern JJ, Herzfeldt B, Van den Bergh R, Hung W-Y, Bird T, Deng G, Mulder DW, Smyth C, Laing NG, Soriano E, Pericak-Vance MA, Haines J, Rouleau GA, Gusella JS, Horvitz RS, Brown RH, Jr: Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362:20-21 [DOI] [PubMed] [Google Scholar]
- 12.Montoliu C, Vallés S, Renau-Piqueras J, Guerri C: Ethanol-induced oxygen radical formation and lipid peroxidation in rat brain: effect of chronic alcohol consumption. J Neurochem 1994, 63:1855-1862 [DOI] [PubMed] [Google Scholar]
- 13.Butterworth RF, Kril JJ, Harper CG: Thiamine-dependent enzyme changes in the brains of alcoholics: relationship to the Wernicke-Korsakoff syndrome. Alcohol Clin Exp Res 1993, 71:1084-1088 [DOI] [PubMed] [Google Scholar]
- 14.Gibson GE, Sheu K-FR, Baker AC, Carlson KC, Harding B, Perrino P: Reduced activities of thiamine-dependent enzymes in brains and peripheral tissues of Alzheimer patients. Arch Neurol 1988, 45:836-840 [DOI] [PubMed] [Google Scholar]
- 15.Mastrogiacomo F, Bergeron C, Kish SJ: Brain α-ketoglutarate dehydrogenase complex activity in Alzheimer’s disease. J Neurochem 1993, 61:2007-2014 [DOI] [PubMed] [Google Scholar]
- 16.Sheu K-FR, Cooper AJL, Koike K, Koike M, Lindsay JG, Blass JP: Abnormality of the α-ketoglutarate dehydrogenase complex in fibroblasts from familial Alzheimer’s disease. Ann Neurol 1994, 35:312-318 [DOI] [PubMed] [Google Scholar]
- 17.Mizuno Y, Matsuda S, Yoshino H, Mori H, Hattori N, Ikebe S-I: An immunohistochemical study on α-ketoglutarate dehydrogenase complex in Parkinson’s disease. Ann Neurol 1994, 35:204-210 [DOI] [PubMed] [Google Scholar]
- 18.Mastrogiacomo F, LaMarche J, Dozic S, Lindsay JG, Bettendorff L, Robitaille Y, Schut L, Kish SJ: Immunoreactive levels of α-ketoglutarate dehydrogenase subunits in Friedreich’s ataxia and spinocerebellar ataxia type 1. Neurodegeneration 1996, 5:27-33 [DOI] [PubMed] [Google Scholar]
- 19.Calingasan NY, Baker H, Sheu K-FR, Gibson GE: Blood-brain barrier abnormalities in vulnerable brain regions during thiamine deficiency. Exp Neurol 1995, 134:64-72 [DOI] [PubMed] [Google Scholar]
- 20.Harata N, Iwasaki Y: Evidence for early blood-brain barrier breakdown in experimental thiamine deficiency in the mouse. Metab Brain Dis 1995, 10:159-174 [DOI] [PubMed] [Google Scholar]
- 21.Calingasan NY, Gandy SE, Baker H, Sheu K-FR, Kim K-S, Wisniewski HM, Gibson GE: Accumulation of amyloid precursor protein-like immunoreactivity in rat brain in response to thiamine deficiency. Brain Res 1995, 677:50-60 [DOI] [PubMed] [Google Scholar]
- 22.Calingasan NY, Gandy SE, Baker H, Sheu K-FR, Smith JD, Lamb BT, Gearhart JD, Buxbaum JD, Harper C, Selkoe DJ, Price DL, Sisodia SS, Gibson GE: Novel neuritic clusters with accumulations of amyloid precursor protein and amyloid precursor-like protein 2 immunoreactivity in brain regions damaged by thiamine deficiency. Am J Pathol 1996, 149:1063-1071 [PMC free article] [PubMed] [Google Scholar]
- 23.Langlais PJ, Anderson G, Guo SX, Bondy SC: Increased cerebral free radical production during thiamine deficiency. Metab Brain Dis 1997, 12:137-143 [PubMed] [Google Scholar]
- 24.Mulligan MS, Hevel JM, Marletta MA, Ward PA: Tissue injury caused by deposition of immune complexes is l-arginine dependent. Proc Natl Acad Sci USA 1991, 88:6338-6342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Corbet JA, Tilton RG, Chang K, Hasan KS, Ido Y, Wang JL, Sweetland MA, Lancaster JR, Williamson JR, McDaniel ML: Aminoguanidine, a novel inhibitor of nitric oxide formation, prevents diabetic vascular dysfunction. Diabetes 1992, 41:552-556 [DOI] [PubMed] [Google Scholar]
- 26.Boje KM: An inflammatory role for nitric oxide during experimental meningitis in the rat. Fiskum G eds. Neurodegenerative Diseases. 1996, :pp 263-273 Plenum Press, New York [Google Scholar]
- 27.Barron KD: The microglial cell: a historical review. J Neurol Sci 1995, 134:57-68 [DOI] [PubMed] [Google Scholar]
- 28.Chao CC, Hu S, Molitor TW, Shaskan EG, Peterson PK: Activated microglia mediate neuronal cell injury via a nitric oxide mechanism. J Immunol 1992, 149:2736-2741 [PubMed] [Google Scholar]
- 29.Schmidt-Kastner R, Meller D, Bellander B-M, Stromberg I, Olson L, Ingvar M: A one-step immunohistochemical method for detection of blood-brain barrier disturbances for immunoglobulins in lesioned rat brain with special reference to false positive labelling in immunohistochemistry. J Neurosci Methods 1993, 46:121-132 [DOI] [PubMed] [Google Scholar]
- 30.Dawson TM, Bredy DS, Fotuhi M, Hwang PM, Snyder SH: Nitric oxide synthase and neuronal NADPH diaphorase are identical in brain and peripheral tissue. Proc Natl Acad Sci USA 1991, 88:7797-7801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tracey WR, Nakane M, Pollock JS, Forstermann U: Nitric oxide synthases in neuronal cells, macrophages and endothelium are NADPH diaphorases, but represent only a fraction of total cellular NADPH diaphorase activity. Biochem Biophys Res Commun 1993, 195:1035-1040 [DOI] [PubMed] [Google Scholar]
- 32.Matsumoto T, Nakane M, Pollock JS, Kuk JE, Forstermann U: A correlation between soluble brain nitric oxide synthase and NADPH-diaphorase activity is only seen after exposure of the tissue to fixative. Neurosci Lett 1993, 155:61-64 [DOI] [PubMed] [Google Scholar]
- 33.Hsu S, Raine L, Fanger H: Use of avidin-biotin-peroxidase techniques: a comparison between ABC and unlabeled antibody (PAP) procedures. J Histochem Cytochem 1981, 29:577-580 [DOI] [PubMed] [Google Scholar]
- 34.Streit WJ: An improved staining method for rat microglial cells using the lectin from Griffonia simplicifolia (GSA I-B4). J Histochem Cytochem 1990, 38:1683-1686 [DOI] [PubMed] [Google Scholar]
- 35.Hill JM, Switzer RC: The regional distribution and cellular localization of iron in the rat brain. Neuroscience 1984, 11:595-603 [DOI] [PubMed] [Google Scholar]
- 36.Kondo Y, Ogawa N, Asanuma M, Ota Z, Mori A: Regional differences in late-onset iron deposition, ferritin, transferrin, astrocyte proliferation, and microglial activation after transient forebrain ischemia in rat brain. J Cereb Blood Flow Metab 1995, 15:216-226 [DOI] [PubMed] [Google Scholar]
- 37.Leigh PN, Connick JH, Stone TW: Distribution of NADPH-diaphorase positive cells in the rat brain. Comp Biochem Physiol 1990, 97:259-264 [DOI] [PubMed] [Google Scholar]
- 38.Vincent SR, Kimura H: Histochemical mapping of nitric oxide synthase in the rat brain. Neuroscience 1992, 46:755-784 [DOI] [PubMed] [Google Scholar]
- 39.Moncada S, Palmer RMJ, Higgs EA: Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol Rev 1991, 43:109-136 [PubMed] [Google Scholar]
- 40.Prado R, Watson BD, Kuluz J, Dietrich WD: Endothelium-derived nitric oxide synthase inhibition: effects on cerebral blood flow, pial artery diameter and vascular morphology in rats. Stroke 1992, 23:1118-1124 [DOI] [PubMed] [Google Scholar]
- 41.Trifiletti RR, Bolan EA, Pickel VM: Nitrotyrosine immunolabeling is present in axons and glial processes in the globus pallidus of normal adult rat brain. Soc Neurosci Abstr 1996, 22:891 [Google Scholar]
- 42.Kaneko K, Chittamwood T, Takeshi J, Yamaguchi K: Ferritin immunohistochemistry as a marker for microglia. Acta Neuropathol 1989, 79:129-136 [DOI] [PubMed] [Google Scholar]
- 43.DiPatre PL, Gelman BB: Microglial cell activation in aging and Alzheimer Disease: partial linkage with neurofibrillary tangle burden in the hippocampus. J Neuropathol Exp Neurol 1997, 56:143-149 [DOI] [PubMed] [Google Scholar]
- 44.Chi OZ, Wei HM, Sinha AK, Weiss HR: Effects of inhibition of nitric oxide synthase on blood-brain barrier transport in focal cerebral ischemia. Pharmacology 1994, 48:367-373 [DOI] [PubMed] [Google Scholar]
- 45.Smith RG, Alexianu ME, Crawford G, Nyormoi O, Stefani E, Appel SH: Cytotoxicity of immunoglobulins from amyotrophic lateral sclerosis patients on a hybrid motoneuron cell line. Proc Natl Acad Sci USA 1994, 91:3393-3397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fishman PS, Farrand DA, Kristt DA: Internalization of plasma proteins by cerebellar Purkinje cells. J Neurol Sci 1990, 100:43-49 [DOI] [PubMed] [Google Scholar]
- 47.Tildon JT, McKenna MC, Stevenson J, Huang X: Modulation of central nervous system metabolism by macromolecules: effects of albumin and histones on glucose oxidation by synaptosomes. J Assoc Acad Minor Phys 1996, 7:47-52 [PubMed] [Google Scholar]
- 48.Todd KG, Butterworth RF: Evidence that oxidative stress plays a role in neuronal cell death due to thiamine deficiency. J Neurochem 1997, 69:S136A [Google Scholar]
- 49.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA: Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci USA 1990, 87:1620-1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Qi Y, Jamindar TM, Dawson G: Hypoxia alters iron homeostasis and induces ferritin synthesis in oligodendrocytes. J Neurochem 1995, 64:2458-2464 [DOI] [PubMed] [Google Scholar]
- 51.Vercellotti GM, Balla G, Balla J, Nath K, Eaton JW, Jacob HS: Heme and the vasculature: an oxidative hazard that induces antioxidant defenses in the endothelium. Artif Cells Blood Subst Immobil Biotech 1994, 22:207-213 [DOI] [PubMed] [Google Scholar]
- 52.Iadecola C, Zhang F, Casey R, Clark HB, Ross ME: Inducible nitric oxide synthase gene expression in vascular cells after transient focal cerebral ischemia. Stroke 1996, 27:1373-1380 [DOI] [PubMed] [Google Scholar]
- 53.Bolaños JP, Almeida A, Stewart V, Peuchen S, Land JM, Clark JB, Heales SJR: Nitric oxide-mediated mitochondrial damage in the brain: mechanisms and implications for neurodegenerative diseases. J Neurochem 1997, 68:2227-2240 [DOI] [PubMed] [Google Scholar]
- 54.Good PF, Werner P, Hsu A, Olanow CW, Perl DP: Evidence of neuronal oxidative damage in Alzheimer’s disease. Am J Pathol 1996, 149:21-28 [PMC free article] [PubMed] [Google Scholar]
- 55.Smith MA, Harris PLR, Sayre LM, Beckman JS, Perry G: Widespread peroxynitrite-mediated damage in Alzheimer’s disease. J Neurosci 1997, 17:2653-2657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Halliwell B: What nitrates tyrosine? Is nitrotyrosine specific as a biomarker of peroxynitrite formation in vivo? FEBS Lett 1997, 411:157-160 [DOI] [PubMed] [Google Scholar]
- 57.Rucker P, Torti FM, Torti SV: Role of H and L subunits in mouse ferritin. J Biol Chem 1996, 271:33352-33357 [DOI] [PubMed] [Google Scholar]
- 58.Juckett MB, Balla J, Balla G, Jessurun J, Jacob HS, Vercellotti GM: Ferritin protects endothelial cells from oxidized low density lipoprotein in vitro. Am J Pathol 1995, 147:782-789 [PMC free article] [PubMed] [Google Scholar]
- 59.Jellinger K, Paulus W, Grundke-Iqbal I, Riederer P, Youdim MB: Brain iron and ferritin in Parkinson’s and Alzheimer’s diseases. J Neural Transm Park Dis Dement Sect 1990, 2:237-240 [DOI] [PubMed] [Google Scholar]
- 60.Grundke-Iqbal I, Fleming JT, Tung YC, Lassmann H, Iqbal K, Joshi JG: Ferritin is a component of the neuritic (senile) plaque in Alzheimer’s dementia. Acta Neuropathol 1990, 81:105-110 [DOI] [PubMed] [Google Scholar]
- 61.Miyazono M, Iwaki T, Chittamwood T, Kaneko Y, Doh-ura K, Takeshi J: A comparative immunohistochemical study of kuru and senile plaques with a special reference to glial reactions at various stages of amyloid plaque formation. Am J Pathol 1991, 139:589-598 [PMC free article] [PubMed] [Google Scholar]
- 62.Benkovic SA, Connor JR: Ferritin, transferrin, and iron in selected regions of adult, and aged rat brain. J Comp Neurol 1993, 338:97-113 [DOI] [PubMed] [Google Scholar]
- 63.McMillan K, Bredt DS, Hirsch DJ, Snyder SH, Clark JE, Masters BS: Cloned, expressed rat cerebellar nitric oxide synthase contains stoichiometric amounts of heme, which binds carbon monoxide. Proc Natl Acad Sci USA 1992, 89:11141-11145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Connor JR, Menzies SL: Relationship of iron to oligodendrocytes and myelination. Glia 1996, 17:83-93 [DOI] [PubMed] [Google Scholar]
- 65.LeVine SM: Iron deposits in multiple sclerosis and Alzheimer disease brains. Brain Res 1997, 760:298-303 [DOI] [PubMed] [Google Scholar]
- 66.Aust SD: Ferritin as a source of iron and protection from iron-induced toxicities. Toxicol Lett 1995, 82–83:941-944 [DOI] [PubMed] [Google Scholar]
- 67.Roberts RL, Fine RE, Sandra A: Receptor-mediated endocytosis of transferrin at the blood-brain barrier. J Cell Sci 1993, 104:521-532 [DOI] [PubMed] [Google Scholar]
- 68.Watson BD, Busto R, Goldberg WJ: Lipid peroxidation in vivo induced by reversible global ischemia in rat brain. J Neurochem 1984, 42:268-274 [DOI] [PubMed] [Google Scholar]
- 69.Komara JS, Nayini NR, Bialick HA, Indrieri RJ, Evans AT, Garritano AM, Hoehner TJ, Jacobs WA, Huang RR, Krause GS, White BW, Aust SD: Brain iron delocalization and lipid peroxidation following cardiac arrest. Ann Emerg Med 1986, 15:384-389 [DOI] [PubMed] [Google Scholar]
- 70.Stadtman ER: Protein oxidation and aging. Science 1992, 257:1220-1224 [DOI] [PubMed] [Google Scholar]
- 71.Focht SJ, Snyder BS, Beard JL, Vangelder W, Williams LR, Connor JR: Regional distribution of iron, transferrin, ferritin, and oxidatively modified proteins in young and aged Fischer 344 rat brains. Neuroscience 1997, 79:255-261 [DOI] [PubMed] [Google Scholar]
- 72.Smith MA, Harris PLR, Sayre LM, Perry G: Iron accumulation in Alzheimer’s disease is a source of redox-generated free radicals. Proc Natl Acad Sci USA 1997, 94:9866-9868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Connor JR, Menzies SL: Cellular management of iron in the brain. J Neurol Sci 1995, 134:33-44 [DOI] [PubMed] [Google Scholar]
- 74.Griffin WST, Stanley LC: Glial activation as a common denominator in neurodegenerative disease: a hypothesis in neuropathophysiology. Fedoroff S Juurlink BHJ Doucette R Burkholder G eds. Biology and Pathology of Astrocyte-Neuron Interactions. 1993, :pp 359-381 Plenum Press, New York [Google Scholar]
- 75.Banati RB, Graeber MB: Surveillance, intervention, and cytotoxicity: is there a protective role of microglia? Dev Neurosci 1994, 16:114-127 [DOI] [PubMed] [Google Scholar]
- 76.MazurKolecka B, Frackowiak J, LeVine H, Haske T, Wisniewski HM: Factors produced by activated macrophages reduce accumulation of Alzheimer’s β-amyloid protein in vascular smooth muscle cells. Brain Res 1997, 760:255-260 [DOI] [PubMed] [Google Scholar]
- 77.Langlais PJ, Mair RG: Protective effects of the glutamate antagonist MK-801 on pyrithiamine-induced lesions and amino acid changes in rat brain. J Neurosci 1990, 10:1664-1674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rao VL, Mousseau DD, Butterworth RF: Nitric oxide synthase activities are selectively decreased in vulnerable brain regions in thiamine deficiency. Neurosci Lett 1996, 208:17-20 [DOI] [PubMed] [Google Scholar]