

Abstract
Spinal muscular atrophy is an autosomal recessive disorder characterized by the progressive loss or degeneration of the motor neurons. To investigate the expression of survival motor neuron (SMN), the spinal muscular atrophy-determining gene, and its relationship with the pathogenesis of the disease, we analyzed by means of in situ hybridization the location of SMN mRNA in fetal, newborn, infant, and adult human central nervous system tissues. The large motor neurons of the spinal cord are the main cells that express SMN together with the neurons of the medulla oblongata, the pyramidal cells of the cortex, and the Purkinje cells of the cerebellum. Some sensory neurons from the posterior horn and dorsal root ganglia express SMN to a lesser degree. Furthermore, strong SMN expression is detected in the ependymal cells of the central canal. The expression is present in the spinal cord at 8 weeks of fetal life throughout postnatal and adult life. The sharp expression of SMN in the motor neurons of the human spinal cord, the target cells in spinal muscular atrophy, suggests that this gene is implicated in neuronal development and in the pathogenesis of the disease. The location of the SMN gene expression in other neuronal structures not clearly or directly associated with clinical manifestations or pathological findings of spinal muscular atrophy may indicate a varying sensitivity to the absence or dysfunction of the SMN gene in motor neurons.
Spinal muscular atrophy (SMA) is an autosomal recessive neuromuscular disorder that results in the loss of motor neurons in the spinal cord. Although the pathological features of this disorder have been well characterized, the primary pathogenesis remains to be determined. The clinical expression of the disease is variable and classified by age of onset and maximal motor milestones achieved in type I (severe form), type II (intermediate form), and type III (mild/moderate form). The genomic region containing the defective gene(s) for SMA is localized in 5q13 and is particularly unstable and prone to large-scale deletions. Three genes and their homologous copies have been identified in this region: survival motor neuron (SMN), neuronal apoptosis inhibitor protein (NAIP), and p44. 1-3 SMN telomeric (SMNT) differs from its centromeric copy (SMNc) by only five nucleotides, none of which change the amino acid sequence of the putative protein. 1 A lack of SMNt is observed in more than 90% of the patients regardless of the phenotype, and this can be caused by gene deletion or gene conversion, the former being more common in severe cases and the latter more common in mild cases. 4 In the remaining patients, a frameshift mutation in exon 3 and other point mutations have been described. 1,4-9 The centromeric copy of the SMN gene is present in all patients and deleted in approximately 4% of healthy individuals. 1 None of the patients carried homozygous deletions of both SMNt and SMNc genes, suggesting that such a genotype may result in a lethal phenotype. All of these findings clearly indicate that SMNt is the determining gene for SMA.
SMN encodes a 38-kd protein that bears no resemblance to any other proteins in the data base. A number of observations indicate that SMN may be involved in RNA metabolism. First, a clustering of missense mutations in the SMN gene has been described in SMA patients in the region of amino acids 262 to 279, which contains a tyrosine/glycine-rich motif that is present in various RNA binding proteins. 6 Secondly, the SMN protein has been located by immunohistochemistry within novel structures named “gems,” which are more commonly associated with coiled bodies. Ramon y Cajal described the coiled bodies in 1903, and there is, at present, considerable evidence that both structures are involved in RNA metabolism. 10 Finally, SMN forms a complex with a novel protein named SIP1 (SMN interacting protein 1) and also interacts with small nuclear ribonucleoproteins U1 and U5 of the spliceosome, the catalytic core of the splicing reaction. 11,12
A detailed analysis of the expression of the SMN gene will assist in elucidating its role in the pathogenesis of the disease. The specific hypothesis regarding the function of the SMN gene can be tested with in vivo experimental systems, including the detection by in situ hybridization of SMN transcripts in the central nervous system tissues. This method allows specific and precise characterization of the temporal and spatial expression patterns of the gene. Studies using RNA in vitro methods and antibodies to the protein have shown that the SMN is expressed in a variety of human tissues, including muscle, heart, spinal cord, brain, liver, and pancreas. 1,13,14 However, a systematic description of the structural location and developmental patterns of SMN expression in human tissues has not been reported. We describe here the use of RNA in situ hybridization to define and characterize SMN expression in the central nervous system during human development.
Materials and Methods
Human Samples
Material from three fetuses was obtained from prostaglandin-induced, dilatation plus evacuation or spontaneous abortions at 8,12 and 17 weeks gestation. Postnatal tissues were obtained from one newborn (age 29 days), one infant (age 5 months), and three adults (ages 37, 42, and 60 years) from autopsy samples. None of them showed any histological abnormality in the tissues analyzed, which included spinal cord, medulla oblongata, cerebellum, and cerebral cortex. Tissues were fixed in 4% paraformaldehyde in phosphate-buffered saline and embedded in paraffin, and 10 to 12-μm sections were mounted onto silane-treated slides.
Probes
Human telomeric SMN cDNA inserts from nucleotide 1 to 1171 cloned in the Bluescript transcription vector (Stratagene, La Jolla, CA) and transcribed from either T3 or T7 promoters were used to generate single-stranded 35S-labeled antisense cRNA and the corresponding sense probes with an RNA transcription kit (Stratagene). Labeled cRNA was reduced in size to an average of 100 to 200 bp by limited alkali hydrolysis with 80 mmol/L NaHCO3 and 120 mmol/L Na2CO3 and separated from unincorporated nucleotides by fractionation on a Sephadex G-50 column. As a control, an R-domain probe of 480 bp of the CFTR gene 15 was used to hybridize some tissues of the individuals analyzed in this study.
In Situ Hybridization
The in situ protocol has been described elsewhere with slight modifications. 15 In brief, paraffin sections were dewaxed in xylene before being digested with 0.001% proteinase K at 37°C for 10 to 20 minutes, rinsed in 0.1 mol/L triethanolamine, acetylated in 0.25% acetic anhydride for 10 minutes, and dehydrated in a graded series of ethanol solutions. Hybridization was performed with 2 × 107 cpm/ml of probe in 50% formamide, 10% dextran sulfate, 0.3 mol/L NaCl, 10 mmol/L Tris, pH 8.0, 1× Denhardt’s solution, 0.5 mg/ml tRNA, and 10 mmol/L dithiothreitol at 48°C to 50°C overnight. Excess probe was removed by digestion with RNase A (10 μg/ml) and washed at a final stringency of 0.1× saline sodium citrate at 56°C. Slides were hand dipped in Kodak NTB2 emulsion, exposed for 3 to 4 weeks at 4°C, developed, and stained with hematoxylin and eosin and toluidine blue (TB). Photography was done with a Zeiss Axioscope microscope.
Interpretation
The relative levels of mRNA were quantified visually by grain count, comparing the positive antisense zones with the control sense zones. Background measurements were performed in areas without specific cells of the same slide, in an area similar to that of the serial section in which the sense probe had been used and in the total area of the sense hybridization.
Results
Spinal Cord
High levels of cell-specific SMN expression were detected at all of the developmental stages analyzed. The first stage of study was at 8 weeks, and the expression detected was diffuse in the epithelia of the central canal following the cell migration pattern to the ventral and posterior horn (Figure 1 ▶ , A to C). During the second trimester of fetal life, high expression was confined to neuroblasts of the anterior horn and ependymal cells and to a lesser degree in isolated cells of the posterior horn and dorsal root ganglia (Figure 1 ▶ , D to I). This pattern of expression was maintained throughout the postnatal life from neonatal to the adult period (Figure 2 ▶ , A to I). No significant expression was detected in cells of the neuroglia, neuropil, and white matter when compared with large motor neurons.
Figure 1.
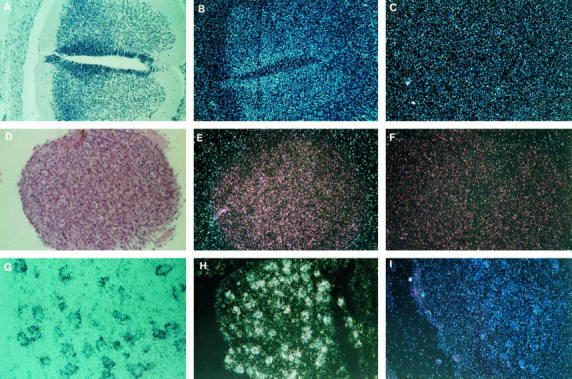
Expression of the SMN gene in fetal spinal cord. A to C: Spinal cord at 8 weeks of development. A: Untreated section, bright field. The cells from the neural epithelium migrate toward the posterior horn and, to a lesser degree, toward the anterior horn. B: Antisense probe, dark field, showing the diffuse expression in the neural epithelium following the pattern of migration of the neural cells into the two horns. C: Sense probe, dark field. Hematoxylin and eosin; (×100). D to F: Dorsal root ganglia at 12 weeks of development. D: Antisense probe, bright field. E: Antisense probe, dark field. F: Sense probe, dark field. Hematoxylin and eosin (×200). G to I: Anterior horn of the spinal cord at 17 weeks of development showing a large expression in neuroblasts. G: Antisense probe, bright field. TB (×400). H: Antisense probe, dark field. TB (×200). I: Sense probe. Hematoxylin and eosin (×200).
Figure 2.
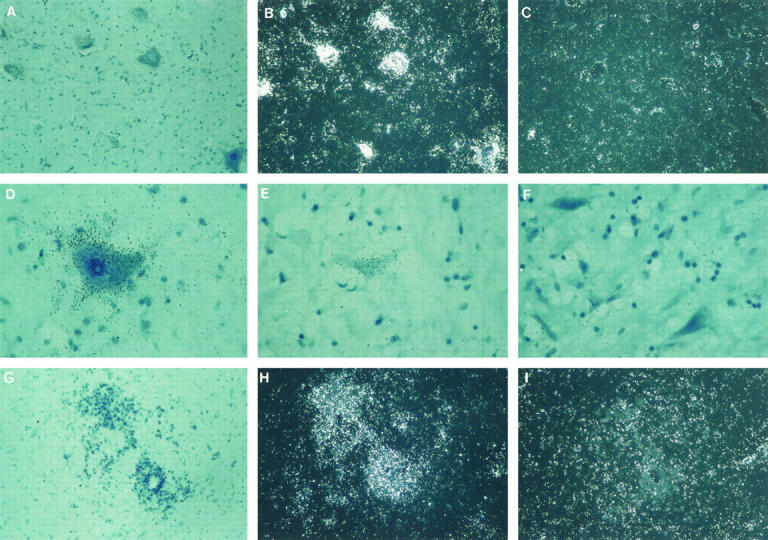
Expression of the SMN gene in adult spinal cord. A to C: Anterior horn. A: Antisense probe, bright field; strong expression in the motor neurons. B: Antisense probe, dark field. C: Sense probe. TB (×200). D: Higher magnification of one of the motor neurons from A. TB (×800). E and F: Expression in small neurons of the posterior horn. E: Antisense probe, bright field. F: Sense probe, bright field. TB (×800). G to I: Cells of the central canal expressing high level of SMN transcript. G: Antisense probe, bright field. H: Antisense probe, dark field. I: Sense probe, dark field. TB (×200).
Other Neuronal Tissues
Cerebral Cortex
During fetal life, the sample analyzed (17 weeks) demonstrated some expression in the precursor cells of the layers of the adult (data not shown). In adult life, a high level of expression was seen in the infragranular layers, particularly in pyramidal neurons (Figure 3 ▶ , A to C).
Figure 3.

Expression of the SMN gene in adult brain cortex, medulla oblongata, and cerebellum. A to C: Neurons of the parietal cerebral cortex expressing SMN transcript mainly in the pyramidal cells of the infragranular layer. A: Antisense probe, bright field. B: Antisense probe, dark field. C: Sense probe, dark field. TB (×200). D to F: Neurons of the medulla oblongata at the level of the olive showing high level of expression. D: Antisense probe, bright field. E: Antisense probe, dark field. F: Sense probe, dark field. TB (×200). G to I: Adult cerebellum with Purkinje cells being the main cell type that expresses SMN. G: Antisense probe, bright field. Arrow: Purkinje cell. TB (×400). H: Antisense probe, dark field. I: Sense probe, dark field. TB (×200).
Medulla Oblongata
A significant SMN expression was seen in the neurons of the medulla oblongata in adult stages. Further examination and level location of these cells indicated that these were neurons from the olive and the hypoglossal nuclei (Figure 3 ▶ , D to F).
Cerebellum
In the fetal sample analyzed (17 weeks), the expression was observed in the primordial cells of the external cortical layer and the internal granular layer (data not shown). No Purkinje cells were seen in the cerebellum at this point. In the adult samples, high levels of expression were detected mainly in the Purkinje cells and to a lesser degree in the cells of the granular layer (Figure 3 ▶ , G to I).
Discussion
The data presented here illustrate the expression pattern of the SMN gene during the human development of the central nervous system, indicating that specific neuronal populations express SMN.
The expression was detected in the spinal cord as early as 8 weeks of development, during which the neuroblasts are still not well distinguished, although migration of neural tube cells has already started. The expression was diffuse but predominant in the epithelium of the neural tube following the pattern of migration of the neural cells. During the second trimester, the neuroblasts are readily identified and specific, and high levels of expression were seen in these cells. Subsequently, the expression in motor neurons is detected throughout postnatal life, suggesting a prominent role of this gene in the development and maintenance of the integrity of motor neurons. This is in agreement with the neuropathological findings of the disease, given that the most important change observed in SMA is a pronounced loss of motor neurons, and those that remain show varying degrees of degeneration, 16 correlating with the course of neurogenic muscular atrophy. Our results at the mRNA level correlate well with the recent report of Lefebvre and colleagues, who detected SMN protein using monoclonal antibodies in the large cells of the anterior horns in a control human fetus. 13
A strong signal of the SMN transcript was also detected in the epithelial lining of the neural tube and subsequently in the cells of the central canal. These are less-differentiated cells derived from the primitive neuroepithelia, and their function is related to the exchange of water and ions in the cerebrospinal fluid. In SMA patients, there have been no descriptions concerning pathology in the ependymal cells or concerning alterations in the circulation, amount or composition of the cerebrospinal fluid. 17 Moreover, the central canal of the spinal cord generally becomes obliterated at the time of puberty, and in normal adults, the ependymal cells have completed their role as a generative epithelium. 18 In agreement with our results, SMN protein analysis in a control fetus has shown a positive signal around the central canal. 13 The exact role of SMN in these cells warrants further investigation.
Although there are no obvious clinical sensory disturbances in SMA, spinal ganglion cells undergo degenerative changes similar to those of the motor neurons in these patients. 16 Novelli et al 14 have noted SMN expression in dorsal root ganglia in a 10-week-old control fetus as well as SMN expression in the ventral and dorsal parts of the spinal cord, although no specific cell targets were reported. 14 In our study, two control fetal cases showed significant expression in dorsal root ganglia. Moreover, in the posterior horn, which has few small sensory neurons or neurons that participate in the trineuronal-reflex arc, we detected significant expression, although it was less intense when compared with neurons of the anterior horn. All of these findings suggest that SMN may play a role not only in motor neurons but also in the development and maintenance of sensory neurons.
Degeneration of cranial nerve nuclei IX to XII has been described in SMA type I and II. The consequences of hypoglossal nerve involvement (XII) are tongue fasciculation and atrophy. Involvement of nerves IX to XI includes dysphagia, dysphonia, hyperdiaphoresis, and increased mucus secretion. 16 Significant levels of expression of the SMN transcript were detected in the adult medulla oblongata corresponding to neurons from some of these structures, lending support to the neuropathological findings and the manifestations of the disease already described.
In the cerebral cortex and the cerebellum, the SMN gene appears to be expressed in a particular population of neurons. In the parietal cerebral cortex, expression was seen in the infragranular layers, and the pyramidal cells are the main type that expresses more significant levels of SMN transcript. In agreement with our results, SMN protein has been detected positively in the pyramidal cells of the human prefrontal cortex. 19 The consequences of SMN dysfunction in these cells are not clear. In type I cases, a diffuse increase in the number of astrocytes and microglia of the cerebral cortex has been reported. 16 Furthermore, few SMA patients have been described with SMN deletions and extended central nervous involvement with positive pyramidal signs. 20 In the cerebellum, a clear expression was detected prenatally in the fetal external layer and, to a lesser degree, in the internal layer. In postnatal life, this external layer disappears, and the expression was restricted mainly to the Purkinje cells. These cells were not observed in our fetal samples of the middle second trimester (17 weeks), because they probably appear later during development. Purkinje cells are efferent modulators of movement (extrapyramidal system), and the pathological findings described in the cerebellum of SMA patients include a slight loss of these cells, 17 in accordance with the expression that we noted pre- and postnatally in this organ.
The high level of expression of the SMN gene found in the spinal cord allows us to assume that the absence of SMN is clearly related to the pathological findings and the clinical manifestations of the disease. By contrast, the high levels of expression in the brain and the cerebellum in the absence of clinical symptoms make it difficult to hypothesize about a critical role for SMN in these structures. The study of SMA patients with cerebellar involvement could be a priori interesting to clarify this point. However, two recent reports indicate that the combination of motor neuronal degeneration and olivopontocerebellar hypoplasia are not likely to be the consequence of SMN deletions. 20,21
Previous Northern blot, reverse transcription-polymerase chain reaction, and Western studies of the SMN gene and protein have indicated a widespread SMN expression in a variety of human tissues, including brain and spinal cord. 1,13,14,22 Our in situ analysis confirmed the presence of expression in the latter two organs as well as in medulla oblongata and cerebellum. Moreover, the method allowed us to identify the different cells from these structures that express SMN at a significant level. The expression of the SMN gene is mainly located in specific neuronal populations, starting early during the development of the central nervous system. This was particularly noted in the differentiation of the epithelium of the neural tube in neuroblasts and in cells of the central canal, which corroborates the view that pathological changes of the disease begin during fetal life. 23 This pattern of expression is maintained during the rest of life until adult age and suggests that the function of the SMN gene is crucial to the development and maintenance of neuron integrity. Moreover, the lack of a significant expression in the glial cells, neuropil, and white matter when compared with the neurons, lends strong support to the fact that the pathology of SMA, the only disorder to date related to SMN mutations, is restricted to the neurons of the central nervous system. However, the expression in structures that are not clearly related to SMA manifestations or pathology (ie, ependymal cells or olive, cortical, or cerebellar neurons) suggests that these cells may be less sensitive to the absence or dysfunction of the SMN protein because of a compensation mechanism that could involves SMNc. There is evidence that SMNc expresses protein in the absence of SMNt 13,22 and that an increase in the copy number of SMNc may correlate with a decrease in disease severity. 1,4 However, according to a recent report, patients with type I and II SMA can have an identical copy number of SMNc, but fibroblasts of type II patients produce more gems than type I patients, suggesting that not all SMNc genes are equivalent. 22 In mouse, in which the SMN gene exists as a unique copy, the complete loss of the SMN gene produced by homologous recombination results in a lethal phenotype. 24 The massive cell death detected in these SMN-deficient mouse embryos reinforces the previous hypothesis that the complete absence of SMNt and SMNc is lethal in humans 1 and lends support to an indispensable role of SMN. Nevertheless, our results suggest that SMN, expressed in different neuronal populations, may have a puzzling regulatory mechanism that leads to the loss or degeneration of only part of a specific neuronal group when this gene is interrupted. The recent description of a complex SMN-Sp1-associated protein 11,12 with a possible role assembling the various spliceosomal small nuclear ribonucleoproteins proteins indicates that regulation of SMN may involve another modulating gene(s) or interaction with other cellular specific components. In this context, large motor neurons may have a varying sensitivity to the absence or dysfunction of the SMN gene. The clearing up of such doubts will improve our understanding of the pathophysiology of the disease and will assist in the search for therapeutic alternatives to circumvent the absence of SMNt.
Acknowledgments
We thank J. Melki for providing the SMN cDNA and for helpful comments and a critical reading of the manuscript. We are indebted to I. Ferrer, R. Bordes, and X. Matías for helpful comments; L. Giardino and L. Calzà for advice with the in situ hybridization; V. Montal for technical assistance; C. Rodríguez, J. M. Alvarez, I. Espinosa, V. Fumanal, and J. Parra for assistance in obtaining and preparing human material; and P. Ayesta and T. Torras for help with the photographic material.
Footnotes
Address reprint requests to Dr. Eduardo F. Tizzano, Servei de Genética, Hospital de Sant Pau, Padre Claret 167, 08025, Barcelona, Spain. E-mail: etizzano@santpau.es.
Supported by grant 98-556 from the Fondo de Investigacion Sanitaria and in part by the Institut de Recerca, Hospital de Sant Pau.
References
- 1.Lefebvre S, Bürglen L, Beboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Milasseau P, Zeviani M, Le Paslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J: Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995, 80:155-165 [DOI] [PubMed] [Google Scholar]
- 2.Roy N, Mahadevan MS, Mclean M, Shutler G, Yaraghi Z, Farahani R, Baird S, Besner Johnston A, Lefebvre C, Kang X, Salih K, Aubry H, Tamai K, Guan X, Ioannou P, Crawford TO, de Jong PJ, Surth L, Ikeda JE, Korneluk RG, McKenzie A: The gene for neural apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995, 80:167-178 [DOI] [PubMed] [Google Scholar]
- 3.Bürglen L, Seroz T, Miniou P, Lefebvre S, Burlet P, Munnich A, Viegas Pequignot E, Egly JM, Melki J: The gene encoding p44, a subunit of the transcription factor TFIIH, is involved in large-scale deletions associated with Werdnig-Hoffmann disease. Am J Hum Genet 1997, 60:72-79 [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell L, Potter A, Ignatius J, Dubowitz V, Davies K: Genomic variation and gene conversion in spinal muscular atrophy: implications for disease process and clinical phenotype. Am J Hum Genet 1997, 61:40-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bussaglia E, Clermont O, Tizzano E, Lefebvre S, Burglen L, Cruaud C, Urtizberea JA, Colomer J, Munnich A, Baiget M, Melki J: A frame-shift deletion in the survival motor neuron gene in Spanish spinal muscular atrophy patients. Nat Genet 1995, 11:335-337 [DOI] [PubMed] [Google Scholar]
- 6.Talbot K, Ponting C, Theodosiu AM, Rodrigues NR, Surtees R, Mountford R, Davies KD: Missense mutation clustering in the survival motor neuron gene: a role for conserved tyrosine and glycine rich region of the protein in RNA metabolism? Hum Mol Genet 1997, 6:497-500 [DOI] [PubMed] [Google Scholar]
- 7.Parsons DW, McAndrew PE, Monani UR, Mendell JR, Burghes AH: An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: further evidence for SMN as the primary SMA-determining gene. Hum Mol Genet 1996, 5:1727-1732 [DOI] [PubMed] [Google Scholar]
- 8.Brahe C, Clermont O, Zappata S, Tiziano F, Melki J, Neri G: Frameshift mutation in the survival motor neuron gene in a severe case of SMA type I. Hum Mol Genet 1996, 5:1971-1976 [DOI] [PubMed] [Google Scholar]
- 9.Hahnen E, Schönling J, Rudnik-Schöneborn S, Raschke H, Zerres K, Wirth B: Missense mutation in exon 6 of the survival motor neuron gene in patients with spinal muscular atrophy (SMA). Hum Mol Genet 1997, 6:821-825 [DOI] [PubMed] [Google Scholar]
- 10.Liu Q, Dreyfuss G: A novel nuclear structure containing the survival of motor neurons protein. EMBO J 1996, 15:3555-3565 [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Q, Fischer U, Wang F, Dreyfuss G: The spinal muscular atrophy disease gene product, SMN, and its associated protein SIP1 are in complex with spliceosomal snRNP proteins. Cell 1997, 90:1013-1021 [DOI] [PubMed] [Google Scholar]
- 12.Fischer U, Liu Q, Dreyfuss G: The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 1997, 90:1023-1029 [DOI] [PubMed] [Google Scholar]
- 13.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J: Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet 1997, 16:265-269 [DOI] [PubMed] [Google Scholar]
- 14.Novelli G, Calzà L, Amicucci P, Giardino L, Pozza M, SIlani V, Pizzuti A, Gennarelli M, Piombo G, Capon F, Dallapiccola B: Expression study of survival motor neuron gene in human fetal tissues. Biochem Mol Med 1997, 61:102-106 [DOI] [PubMed] [Google Scholar]
- 15.Tizzano EF, Silver M, Chitayat D, Benichou JC, Buchwald M: Differential cellular expression of cystic fibrosis transmembrane regulator in human reproductive tissues. Am J Pathol 1994, 144:906-914 [PMC free article] [PubMed] [Google Scholar]
- 16.Osawa M, Shishikura K: Werdnig-Hoffmann disease and variants. Vinken P Bruyn G Klawans H eds. Handbook of Clinical Neurology Diseases of the Motor System. 1991, vol 15.:pp 51-80 Elsevier Science Publishers, New York [Google Scholar]
- 17.Byers RK, Banker BQ: Infantile muscular atrophy. Arch Neurol 1961, 5:140-164 [DOI] [PubMed] [Google Scholar]
- 18.Fuller G, Burger P: Central nervous system. Sternberg S eds. Histology for Pathologists. 1992, :pp 145-159 Raven Press, New York [Google Scholar]
- 19.Battaglia G, Princivalle A, Forti F, Lizier C, Zeviani M: Expression of the SMN gene, the spinal muscular atrophy determining gene, in the mammalian central nervous system. Hum Mol Genet 1997, 6:1961-1971 [DOI] [PubMed] [Google Scholar]
- 20.Rudnik-Schöneborn S, Forket R, Hahnen E, Wirth B, Zerres K: Clinical spectrum and diagnostic criteria of infantile spinal muscular atrophy: Further delineation on the basis of SMN gene deletion findings. Neuropediatrics 1996, 27:8-15 [DOI] [PubMed] [Google Scholar]
- 21.Dubowitz V, Daniels RJ, Davies KE: Olivopontocerebellar hypoplasia with anterior horn cell involvement (SMA) does not localize to chromosome 5q. Neuromusc Disord 1995, 5:25-29 [DOI] [PubMed] [Google Scholar]
- 22.Coovert DD, Le TT, McAndrew P, Strasswimmer J, Crawford TO, Mendell JR, Coulson SE, Androphy Ej, Prior TW, Burghes AHM: The survival motor neuron protein in spinal muscular atrophy. Hum Mol Genet 1997, 6:1205-1214 [DOI] [PubMed] [Google Scholar]
- 23.Hausemanowa-Petrusewicz I: A research strategy for the resolution of childhood spinal muscular atrophy (SMA). Merlini L Granata C Dubowitz V eds. Current Concepts in Childhood Spinal Muscular Atrophy. 1989, :pp 21-32 Springer Verlag, Vienna [Google Scholar]
- 24.Schrank B, Götz R, Gunnersen J, Ure JM, Toyka KV, Smith AG, Sendtner M: Inactivation of the survival motor neuron gene, a candidate gene for human spinal muscular atrophy, leads to massive cell death in early mouse embryos. Proc Natl Acad Sci USA 1997, 94:9920-9925 [DOI] [PMC free article] [PubMed] [Google Scholar]