

Abstract
We have analyzed the mechanism of human endothelial injury in a human peripheral blood lymphocyte-severe combined immunodeficient (huPBL-SCID) mouse/human skin graft model of allograft injury and examined the effect of immunosuppressive drugs on this process. In this model, split-thickness human skin containing the superficial dermal microvessels was grafted onto immunodeficient C.B-17 SCID or SCID/beige mice and allowed to heal. Human peripheral blood mononuclear cells (PBMCs) allogeneic to the skin, when subsequently introduced by intraperitoneal injection, caused destruction of the human dermal microvasculature by day 16, evident as endothelial cell sloughing and thrombosis. In the same specimens, mouse microvessels that invaded the human skin graft were uninjured. Human microvascular cell injury was accompanied by a mononuclear cell infiltrate consisting of approximately equal numbers of human CD4+ and CD8+ T cells, some of which contained perforin-positive granules. We found no evidence of human natural killer cells and noted occasional human, but not mouse, macrophages at a frequency indistinguishable from that resident in skin on animals not receiving human PBMCs. These human T cell infiltrates did not extend into adjacent mouse skin. Human immunoglobulin G antibody was detected in the blood and was diffusely present throughout mouse and human tissues in SCID mice receiving PBMCs. Mouse C3 was detected on human dermal vessels in both unreconstituted control animals and those that received PBMCs. Blood and tissues from mice injected with PBMCs depleted of B cells showed no human immunoglobulin, but circulating CD3+ cells were detected by flow cytometry at levels comparable with those of animals receiving whole PBMCs. Significantly, skin graft infiltration by human T cells and human dermal microvascular injury were equivalent in the B cell-depleted and whole-PBMC-reconstituted mice. Mice inoculated with PBMCs depleted of CD8+ T cells developed microvascular injury and infiltrates containing perforin-expressing CD4+ T cells. These data suggested a cytolytic T cell-dependent mechanism of microvessel injury. We then tested the ability of T cell immunosuppressants, cyclosporine and rapamycin, to attenuate vessel damage. Neither cyclosporine nor rapamycin alone effectively reduced either mononuclear cell infiltration or vascular injury. However, a combination of the two agents reduced both parameters. We conclude that the huPBL-SCID/skin allograft model may be used both to study cytolytic T cell-mediated rejection and to test the effect of immunosuppressive drug strategies in vivo in a small-animal model of human immune responses.
Acute cell-mediated allograft rejection is a host anti-graft immune response. It is initiated when circulating recipient T cells recognize graft alloantigens expressed on passenger “professional” antigen-presenting cells, such as monocytes/macrophages, B cells, and dendritic cells, 1 or “semiprofessional” antigen-presenting cells, such as vascular endothelial cells. 2 Immune recognition of the graft as foreign is thought to induce T cell-mediated injury of the microvasculature and parenchymal elements, largely mediated by CD8+ cytolytic T lymphocytes (CTLs). 3,4 In addition to causing direct cytotoxicity, alloactivated T cells, especially CD4+ helper T lymphocytes, produce a variety of cytokines, such as interferon-γ and tumor necrosis factor-α, which in turn recruit and activate mononuclear phagocytes that participate in injury to the allograft, ie, through a delayed-type hypersensitivity mechanism. 5 The recruitment of T cells and mononuclear phagocytes to the site of the rejecting allograft depends on the inducible expression of vascular adhesion molecules, chemokines, and vasoactive substances by the graft microvascular endothelium. 6
Investigation of the mechanisms of allograft rejection have been explored largely in rodent systems and have generally not focused on the role of the graft microvasculature either in immune recognition, ie, alloantigen presentation by endothelial cells to the recipient T cells, or as targets of the rejection response. The vascular endothelium may play a somewhat different role in the immune system in rodents versus humans. For example, a striking difference in the phenotype of small-vessel endothelial cells between rodents and humans is the lack of constitutive expression of major histocompatibility complex (MHC) class II gene products on the rat endothelium in contrast to the abundant expression on human endothelial cells. 7,8 In vitro models suggest that human endothelial cells, stimulated to express MHC class II gene products by interferon-γ, activate antigen-specific CD4+ memory T cells to secrete cytokines and proliferate, 9,10 whereas rat endothelial cells are poor stimulators of T-cell proliferation even when class II MHC molecules have been induced. 8,11 Highly purified cultures of human endothelial cells also have the capacity to stimulate resting allogeneic T cells to proliferate in the absence of other accessory cells. 12-16 Mouse endothelial cells, which can be induced to express class II MHC molecules in vivo, 17 can, after interferon-γ treatment, also activate alloreactive CD4+ T cells in vitro. 18 However, mouse endothelial cells preferentially activate T-helper 2 memory clones, 19,20 whereas human endothelial cells preferentially stimulate interleukin 2 and interferon-γ secretion, the hallmarks of a T-helper-1-like response. 13,14,16
Another important difference between human allograft rejection and rodent models is that in allogeneic mouse skin graft models, the principal target of the cytotoxic T-cell response is the epithelium, most clearly shown using skin grafts from tetraparental mice. 3,4 In contrast, the initial injury of human allogeneic skin grafts occurs to the dermal microvascular endothelial cells. 21 Keratinocyte injury in humans is a late event, probably resulting from graft ischemia. 21 Similarly, dermal microvascular endothelial cells are injured early in the development of graft-versus-host disease in allogeneic bone marrow transplant patients. 22
We recently described a model of human allograft injury in immunocompromised mice. 23 This model uses severe combined immunodeficient (SCID) mice, which rearrange their T- and B-cell receptor genes at a very low frequency and therefore lack functional, mature T and B cells. 24,25 A human microvascular bed, in the form of a split-thickness skin graft, is placed on the animal. As the graft heals, human microvessels spontaneously inosculate with the mice microvessels of the graft bed, and by 2 to 3 weeks, the graft is largely perfused through retained microvessels lined by human endothelial cells. At this time, human lymphocytes are introduced intraperitoneally (i.p.) as a suspension of 1 to 3 × 108 human peripheral blood mononuclear cells (PBMCs) allogeneic to the skin donor. In more than 95% of the animals, human CD3+ T cells are detectable in the mouse circulation as a discrete population in 3 to 7 days. In essentially all of these animals, injury of the graft dermal microvascular bed occurs with kinetics and histological features that resemble first-set rejection in humans. 23
In this report, we present evidence that microvessel injury in this model appears to be mediated by perforin-expressing CTLs. Endothelial injury is independent of human B-cell engraftment and antibody production and also appears to be independent of mononuclear phagocytes. Skin grafts harvested from mice reconstituted with PBMCs depleted of CD8+ T cells show marked CD4+ T-cell infiltration and microvessel damage. In this setting, CD4+ T cells expressed perforin and thus presumably behaved as CTLs. In view of the central role of T cells, we sought to test the effects of cyclosporine A (CsA) and rapamycin in this model. We show that CsA alone reduces but does not prevent lymphocyte-derived cytokine release, assessed indirectly by vascular cell adhesion molecule 1 (VCAM-1) expression by endothelial cells or human leukocyte antigen-DR expression by keratinocytes. Moreover, CsA alone had no measurable effect on human lymphocyte infiltration of the allograft nor on the extent of microvascular injury. On the other hand, treatment with CsA plus rapamycin was very effective at reducing infiltration of the graft by allogeneic lymphocytes and reducing injury of the graft microvasculature.
Materials and Methods
Animals
C.B-17 SCID and C.B-17 SCID/beige mice (Harlan, Indianapolis, IN) were used at ages 5 to 8 weeks. Both types of mice are severely deficient in T- and B-cell function. Animals with the beige mutation are, in addition, deficient in natural killer (NK) cell function. The animals were housed in microisolator cages and were fed sterilized water and mouse chow. All experimental protocols were approved by the Yale Animal Care and Use Committee.
Antibodies
Rabbit anti-asialo GM1 for mouse NK cell depletion was purchased from Wako Chemicals (Richmond, VA). The following antibodies were used for immunohistochemistry: rabbit polyclonal anti-human T cell (anti-CD3), mouse anti-human monocyte (anti-CD68), mouse anti-human platelet endothelial cell adhesion molecule (anti-CD31, all from DAKO, Carpinteria, CA), mouse anti-human human leukocyte antigen-DR (LB3.1, a gift from J. Strominger, Harvard University, Cambridge, MA), OKT4 mouse anti-human CD4 and OKT8 mouse anti-human CD8 (American Type Culture Collection, Manassas, VA), mouse anti-human VCAM-1, (E1/6, a gift from M. P. Bevilacqua, Amgen, Denver, CO), mouse anti-human perforin (T Cell Diagnostics, Cambridge, MA), hamster anti-mouse CD3, rat anti-mouse NK cell clone DX5 (Pharmingen, San Diego, CA), rat anti-mouse macrophage (clone F4/80, American Type Culture Collection), and peroxidase-conjugated rabbit anti-mouse C3 (Cedarlane, Hornby, Ontario, Canada). The following antibodies were used for fluorescence flow cytometry: fluorescein isothiocyanate-conjugated mouse anti-human CD3, fluorescein isothiocyanate-conjugated mouse anti-human CD4, phycoerythrin-conjugated mouse anti-human CD8 (Exalpha, Cambridge, MA), and Quantum Red-conjugated rat anti-mouse CD45 (Sigma Chemical Co., St. Louis, MO).
Skin Engraftment
Discarded normal adult human breast skin was obtained through the respective institutions’ Department of Pathology under a protocol approved by the ethics review board. The superficial portions of the skin were harvested in 500 to 700-μm-thick sheets using a Goulian dermatome knife, gauge size .016 (Weck, Research Triangle Park, NC), and cut in approximately 7 × 7-mm pieces, which were kept in RPMI 1640 (Life Technologies, Inc.) at 4°C until transplantation (no longer than 6 hours). SCID mice were anesthetized by inhalation of methoxyflurane (Pitman-Moore, Mundelein, IL). The skin was shaved, two 5 × 5-mm skin segments were excised from the back of each mouse, and the defects were covered immediately with the human skin grafts and fixed with disposable skin staples (3M, St. Paul, MN). Grafts were allowed to heal for 2 to 3 weeks before each experiment was initiated.
Human Leukocyte Isolation and Engraftment
PBMCs were isolated from adult volunteer donors by leukopheresis under a protocol approved by the respective institutions’ ethical review boards and were purified using LSM lymphocyte separation medium according to the manufacturer’s instructions (Organon Technika, Durham, NC). Cells (3 × 108) were injected i.p. into each mouse. C.B-17 SCID mice were pretreated by i.p. injection of 50 μl anti-asialo GM1 antibody 24 hours before PBMC engraftment to deplete NK cells. In experiments using C.B-17 SCID/beige mice, pretreatment with anti-asialo GM1 antibody was omitted. In the experiments described in this report, PBMCs were injected 2 to 3 weeks after skin grafting, and all times hereafter refer to days postinoculation with leukocytes.
Where indicated, B cells were depleted from PBMCs by magnetic cell sorting using anti-CD19 microbeads (Miltenyi Biotec, Auburn, CA) according to the directions of the manufacturer, before injection of PBMCs into the mice. After negative selection by magnetic cell sorting, less than 1% CD19+ cells remained in the inoculum as determined by fluorescent flow cytometry.
In some experiments, CD8+ or CD4+ T cells were depleted from the PBMCs by negative selection using anti-mouse immunoglobulin G (IgG)-conjugated immunomagnetic beads loaded with either OKT8 or OKT4 monoclonal antibody, respectively, according to the instructions of the manufacturer (Immunotech, Westbrook, ME). Analysis of the resulting cell population by direct immunofluorescence flow cytometry showed <1% contaminating CD8+ or CD4+ T cells. In pilot studies, C.B-17 SCID mice were injected with either 2 × 108 CD8-depleted or CD4-depleted cells/mouse. No circulating CD3+ T cells were detected in the peripheral blood of mice reconstituted with the CD4-depleted PBMCs. However, the frequency of mice with human CD3+ cells and the fraction of CD3+ cells in the peripheral blood of mice injected with either whole PBMCs or CD8-depleted PBMCs was indistinguishable.
Enzyme-Linked Immunosorbent Assay and Flow Cytometry
Mouse or human IgG levels were quantitated by a sandwich enzyme-linked immunosorbent assay using capture reagents and standards from Cappel (Durham, NC). Mice with murine IgG levels greater than 1 μg/ml were excluded from the studies. Human IgG was quantitated in serum obtained 7 days after injection of human PBMCs, or human B cell-depleted PBMCs, as indicated. The efficiency of engraftment of human lymphocytes in SCID mice was determined from heparinized blood collected by venipuncture. The frequency of circulating human T lymphocytes as a proportion of total blood leukocytes was determined by direct immunofluorescence flow cytometry (FACSort, Becton Dickinson, Mountain View, CA) on blood harvested between days 3 and 7 after human PBMC injection. A live gate was established in the lymphocyte region, and at least 10,000 events were recorded for analysis. Human PBMC engraftment was considered successful if a distinct population consisting of greater than 0.5% of circulating leukocytes expressed human CD3.
Immunosuppressant Drugs
CsA in polyoxyethylated castor oil vehicle was purchased from Sandoz (East Hanover, NJ) and diluted in 100 μl of sterile normal saline immediately before injection. Rapamycin was obtained as a gift from Dr. F. Bach (New England Deaconess Hospital, Boston, MA) and was prepared in 100 μl of carboxymethylcellulose vehicle (Sigma Chemical Co.) for injection. Control animals received injections of normal saline or carboxymethylcellulose vehicle alone. CsA was administered subcutaneously daily, and the first dose was given immediately after i.p. injection of the human PBMCs. Whole-blood CsA levels were measured by the Yale New Haven Hospital Organ Transplant Center by the TDX method (Abbott Laboratories, North Chicago, IL) with a monoclonal CsA-specific antibody. Where indicated, rapamycin was started 7 days after injection of PBMCs.
Histology and Immunohistology
Skin grafts were harvested at time intervals up to 21 days after inoculation of PBMCs, using the same anesthesia protocol as for graft placement. Each skin sample was divided in two and used to prepare 3-μm paraffin-embedded sections stained with hematoxylin and eosin (H&E) and 4-μm cryostat sections used for immunohistochemical staining. Negative controls were performed using species-matched nonbinding control mAbs instead of specific mAbs. Peroxidase-conjugated goat anti-mouse IgG and goat anti-rabbit IgG secondary antibodies were obtained from Jackson Immunoresearch Laboratories (West Grove, PA). Binding of the antibodies was detected using the Vectastain kit (Vector Laboratories, Burlingame, CA). The peroxidase label was developed using 3-amino ethyl carbazole, and the alkaline phosphatase label was developed using Fast Blue RR salt as described. 23
Data Analysis
All skin specimens were evaluated and scored by a dermatopathologist (JMM) who was blinded to the treatment protocol. The degree of leukocytic infiltration was scored on H&E-stained paraffin-embedded sections using the following system: grade 0, rare leukocytes comparable to normal skin; grade 1, sparse perivascular leukocytes; grade 2, dense perivascular leukocytes; grade 3, dense perivascular leukocytes with modest infiltration of the surrounding dermis; and grade 4, dense infiltrate filling the dermis. Dermal microvessel injury was scored separately on the same sections by evidence of endothelial sloughing and/or intravascular thrombosis. Immunostained frozen sections were evaluated in parallel for evidence of human CD3 (a T-cell marker), human CD31 (an endothelial cell marker), and perforin (a marker of cytolytic T cells).
Statistical Analysis
Evaluation of the intensity of mononuclear cell infiltration of the skin graft specimens was done using analysis of variance. Evaluation of the frequency of adhesion molecule upregulation, mononuclear cell infiltration, and microvascular injury was done using Fisher’s exact test. The analyses were performed using the SPSS 6.1 program (SPSS Inc., Chicago, IL).
Results
Histopathologic Characterization of Skin Grafts
The basic features of the skin graft changes were presented in our original description of the model. 23 In brief, split-thickness human skin grafts placed on C.B-17 SCID or SCID/beige mice healed in approximately 2 to 3 weeks. The healed grafts showed no evidence of inflammation in the epidermis or dermis and displayed essentially normal keratinocyte maturation. The majority of the microvessels in the dermis were lined by UEA-1+, CD31+, and MHC class I+ and II+ human endothelial cells. There was no evidence of endothelial sloughing or thrombosis, and the vessels contained erythrocytes, indicating perfusion by the mouse circulation. Such grafts were essentially unchanged for at least 2 months, the longest time examined.
Once the skin grafts were healed, some mice in each group were inoculated with PBMCs. Animals inoculated with PBMCs allogeneic to the skin graft showed intense mononuclear cell infiltrates, initially (at 10 to 12 days postinoculation) in a perivascular distribution, and evolving (by 16 to 21 days) to a more diffuse distribution throughout the human dermis. These cells were predominantly human CD3+ T cells and were composed of approximately equal numbers of CD4+ and CD8+ subsets. Some specimens were stained for the presence of murine CD3+ T cells or for NK cells using the DX5 mAb, and none were found. Rare murine macrophages stained with the F4/80 mAb were detected. Human CD68+ macrophages were observed, but the number seemed no greater than in skin from animals not injected with human PBMCs. Few, if any, human CD16+ NK cells were noted. In heavily infiltrated specimens, a few lymphocytes crossed into the epidermis and cause isolated epithelial cell apoptosis. This finding was variable. Many of the lymphocytes in the dermis appeared activated, and some (up to 10%) stained positive for human perforin in a granular cytoplasmic pattern. All of the perforin-positive cells also expressed CD3 in double-label immunohistochemical analysis. Beginning by about day 10, endothelial sloughing and thrombus formation were noted. The number of vascular structures stained by UEA-1 or human CD31 was progressively diminished, whereas vessels lined by mouse CD31+ cells were preserved or even increased. The presence of perforin-positive, CD3+ T cells and the paucity of human macrophages led us to favor the interpretation that human microvessel injury was mediated by CTLs and not delayed hypersensitivity.
We have extended these findings to explore the role of human antibody. By immunoassay, we observed that human antibody was routinely detected in the circulation of animals receiving human PBMCs, whether or not skin grafts were placed. The values in animals receiving 2 to 3 × 108 PBMCs were typically more than 100 μg human IgG/ml of blood and can reach over 1000 μg/ml. The human and mouse tissues in every animal receiving human PBMCs were diffusely infiltrated by human IgG as detected by immunoperoxidase staining. Antibody staining was not concentrated at specific anatomical sites. Immunohistochemical staining for mouse C3 revealed deposition in a diffuse pattern concentrated on the human dermal microvessels in animals not receiving human PBMCs. The intensity of staining was actually diminished in those animals receiving human PBMCs and essentially absent on vessels showing evidence of endothelial loss (Figure 1) ▶ . These observations do not rule out a role for antibody and/or complement in human microvessel injury, but suggest that mouse complement alone is not sufficient to account for the findings. In light of these results, we investigated the role of human alloantibody in greater detail.
Figure 1.
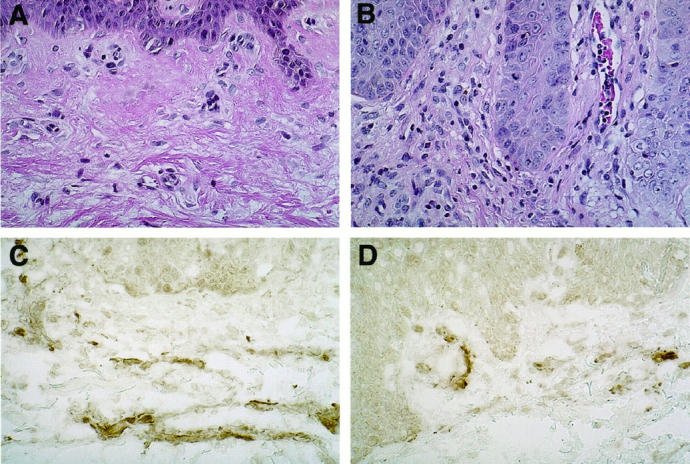
The deposition of murine complement on dermal microvessels in human split-thickness skin grafts. Grafts harvested from mice that received either i.p. injections of saline (A and C) or human PBMCs (B and D) were stained with H&E (A and B, ×140 magnification) or antibody to mouse C3 (C and D, ×140 magnification).
Effect of B-Cell Depletion on Microvessel Injury
To further examine the role of human B cells and alloantibodies as mediators of human microvessel injury in the allogeneic dermis, we reconstituted SCID/beige mice bearing human skin grafts with either allogeneic whole PBMCs or with B cell-depleted PBMCs. Human CD3+ lymphocytes were identified circulating in the blood of 9 of 10 and 7 of 8 mice reconstituted with either whole or B cell-depleted PBMCs, respectively, in two pooled experiments. Human IgG concentration was 184.8 ± 49.5 versus 0.28 ± 0.16 μg/ml (mean ± SE; P < 0.05) in serum taken from mice injected with whole or B cell-depleted PBMCs, respectively, at 7 days after injection. Tissues of mice receiving B cell-depleted PBMCs were negative for human IgG by immunoperoxidase staining. At day 16, perivascular mononuclear cell infiltrates were absent in skin specimens from mice that did not receive any PBMCs, whereas the extent of T-cell infiltration was indistinguishable between the whole-PBMC and B cell-depleted PBMC groups in mice that successfully reconstituted (Figures 2 and 3) ▶ ▶ . Similarly, microvessel injury, including thrombosis, was seen in 8 of 9 and 6 of 7 specimens in whole-PBMC or B cell-depleted mice, respectively; again, there was no statistically significant difference in the extent of injury observed. Mouse C3 deposition was also indistinguishable in specimens from these two groups. We conclude that human dermal microvascular injury in this model does not depend on B cells or antibodies.
Figure 2.
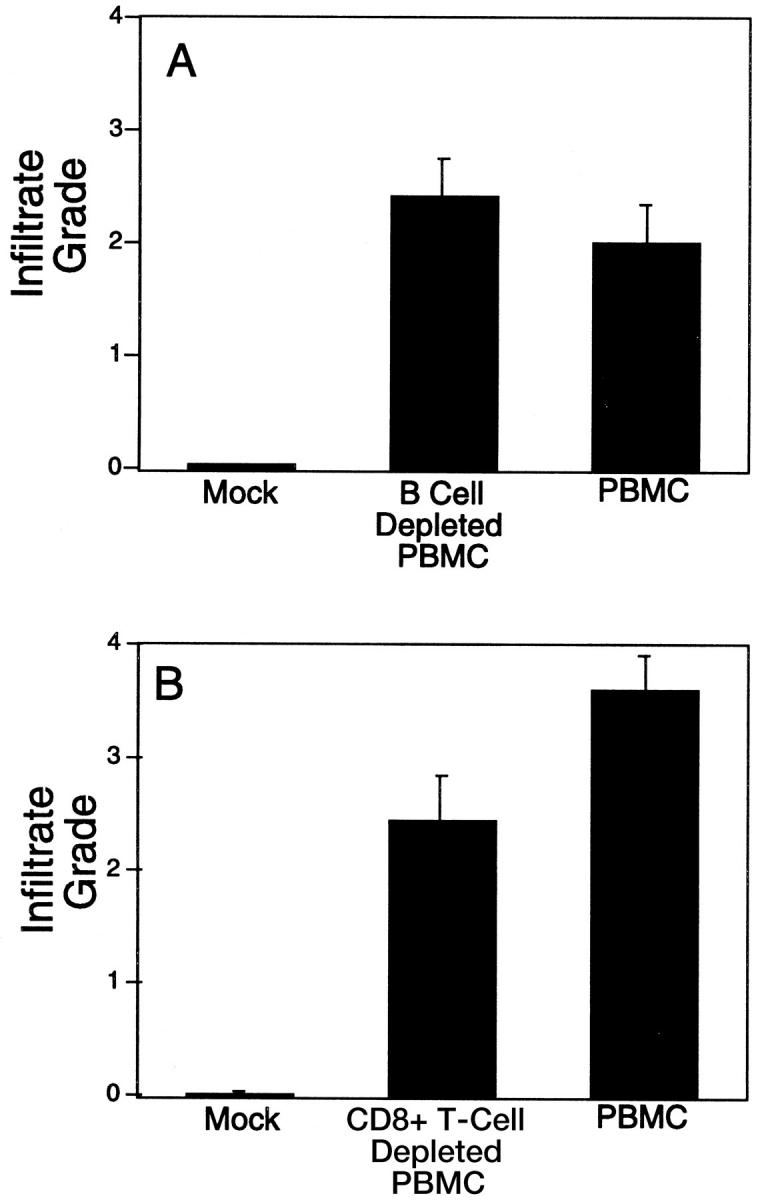
The effect of B-cell (A) or CD8+ T-cell (B) depletion on the development of human lymphocyte infiltration of allogeneic skin grafts in SCID mice. Skin grafts were harvested between days 16 and 21 after i.p. injection of human PBMCs, and then H&E-stained specimens were scored for the degree of mononuclear cell infiltration as described in Materials and Methods. The mean score of mock-treated versus other groups was different (P < 0.05 by analysis of variance). No difference was apparent between either depletion group and the whole-PBMC group (P > 0.05 by analysis of variance).
Figure 3.

The histology of human skin grafts harvested from mice between days 16 and 21 after i.p. injection of whole PBMCs (A), B cell-depleted PBMCs (B), saline (C), or CD8 T cell-depleted PBMCs (D). The specimens are stained with H&E. Magnification: A to D, ×70; insets, ×200.
Effect of CD8+ T-Cell Depletion on Microvessel Injury
To examine the participation of selective T-cell subsets in microvessel injury in this model, we prepared PBMC populations depleted of either CD8+ or CD4+ T cells for injection into C.B-17 SCID mice. In pilot studies, mice reconstituted with CD4+ T cell-depleted PBMCs did not show detectable CD3+ T cells in the peripheral blood and hence were not studied further. Four independent experiments using different leukocyte and skin donor pairs were performed to evaluate the effect of CD8-depleted PBMCs on microvessel injury, and the results were pooled for analysis. Using flow cytometry to analyze blood harvested 7 days after the PBMC injection, all mice (n = 17) injected with CD8-depleted PBMCs had detectable circulating CD4+ T cells, and 13 of 16 mice injected with whole PBMCs had discrete CD4+ and CD8+ T-cell populations. No CD8+ T cells were detected in either peripheral blood or skin grafts harvested from the CD8-depleted PBMC animals.
The extent of graft infiltration by human leukocytes is shown in Figures 2 and 3 ▶ ▶ . Skin harvested from mice that were not injected with human PBMCs showed no significant infiltrate. In contrast, skin grafts taken from mice injected with either whole PBMCs or CD8-depleted PBMCs showed perivascular infiltrates at day 6 after injection and more marked infiltration of the dermis at later times (day 16–21). In each group, the infiltrating cells stained for human CD3 using immunohistochemistry. Small numbers of mouse polymorphonuclear neutrophils in a largely intravascular distribution were present in equal numbers in all specimens. The infiltrating mononuclear cells in grafts taken from mice reconstituted with CD8-depleted PBMCs stained for CD4 (Figure 4) ▶ . In each group, some perivascular cells clustered around injured microvessels stained for both human CD3 and perforin. Examination of serial sections of skin grafts harvested from the CD8-depleted-PBMC animals indicated that a subpopulation of CD4+ T cells expressed perforin.
Figure 4.
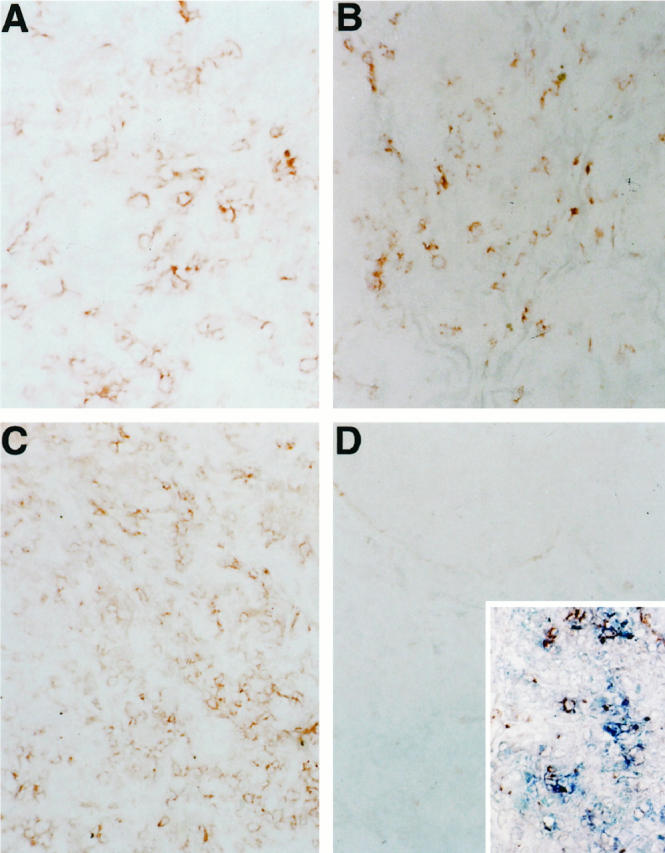
The phenotype of T cells infiltrating the skin grafts. Skin graft specimens were harvested between days 16 and 21 after i.p. injection of whole PBMCs (A and B; magnification ×250) or CD8+ lymphocyte-depleted PBMCs (C and D; magnification ×150) and immunostained for CD4 or CD8 antigens as described in Materials and Methods (C, inset : Magnification ×150). A and C, CD4 stain; B and D, CD8 stain. Some lymphocytes infiltrating skin grafts from mice injected with CD8-depleted PBMC stain for both CD3 (blue) and perforin (red), D, inset.
Finally, we analyzed the injury to the endothelium. No microvessel damage was seen in skin grafts taken from mice that had not received PBMC injections. Endothelial sloughing and thrombosis were seen in 5 of 8 and 9 of 10 grafts harvested from mice in the whole-PBMC or CD8-depleted PBMC groups, respectively (P = not significant). In particular, endothelial injury was noted in all grafts harvested from mice injected with either the whole or the CD8-depleted PBMCs when the mononuclear cell infiltrate was scored greater than 1. Microvessel injury in each group was detected as early as day 6. These experiments indicate that CD8+ T cells were not required to induce microvessel injury and provide evidence for CD4+ CTL-mediated damage.
Determination of CsA Drug Levels
Preliminary experiments were performed to determine doses of CsA, which, when administered subcutaneously once daily, would result in trough whole-blood CsA levels of approximately 300 μg/L and 1000 μg/L (data not shown). Based on these data, the animals were treated with either 10 mg/kg/day or 20 mg/kg/day.
Effects of CsA and Rapamycin on Human PBMC Engraftment
The ability of immunosuppressive drugs to modulate microvessel injury in the allogeneic skin graft was studied. Human CD3+ cells were detected as a discrete population in the peripheral blood in more than 95% of mice reconstituted with human PBMCs. Both mock- and CsA-treated animals showed similar levels of circulating CD3+ lymphocytes in the peripheral blood, but when rapamycin treatment was initiated at the same time the human PBMCs were injected, the frequency of circulating human T cells was reduced or undetectable. In contrast, rapamycin treatment initiated 7 days after PBMC injection appeared to have no effect on circulating T-cell numbers. In subsequent experiments, rapamycin was administered starting on day 7 to test its effect on T-cell infiltration and microvessel injury.
Effect of CsA on Endothelial Cell and Keratinocyte Activation
The production of cytokines by activated T cells was evaluated indirectly by analysis of the de novo expression of 1) human MHC class II molecules by the basal keratinocytes and 2) human VCAM-1 molecules by the human dermal microvascular endothelium in the grafts. For these experiments, skin grafts were harvested at day 6 after reconstitution, and the results of three experiments were pooled for analysis. Skin grafts harvested from animals that did not receive human PBMCs did not express MHC class II on the keratinocytes and, at most, weakly expressed VCAM-1 on a subset of the dermal microvessels, in agreement with previous work. 23 In contrast, grafts taken from mice that received human PBMCs showed strong induction of MHC class II and VCAM-1 expression on the keratinocytes and microvessel endothelial cells, respectively. Treatment of animals with CsA 20 mg/kg/day, but not 10 mg/kg/day, prevented the induction of VCAM-1 and MHC class II molecule expression in about half of the grafts examined (Table 1) ▶ . The effect of rapamycin on these molecules was not examined, because treatment with this drug was initiated at day 7 after PBMC injection, ie, after MHC class II and VCAM-1 were already induced.
Table 1.
The Effect of Cyclosporine and Rapamycin Treatment on the Frequency of Grafts Displaying Induction of Endothelial VCAM-1, or Keratinocyte MHC Class II Molecules at 6 Days
| Treatment group | Number of grafts with induced expression | |
|---|---|---|
| VCAM-1 | HLA-DR | |
| Mock | 8 /9 | 9 /9 |
| Cyclosporine 10 mg/kg | 8 /10 | 9 /10 |
| Cyclosporine 20 mg/kg | 2 /6* | 3 /6* |
HLA-DR, human leukocyte antigen-DR.
*P < 0.05 versus mock-treated group using Fisher’s exact test.
Effect of CsA and Rapamycin on T-Cell Infiltration and Graft Injury
Skin grafts were harvested from mice between days 16 and 21 after the injection of the PBMCs. Data were pooled from eight experiments using different leukocyte and skin donor pairs, each with similar results. As illustrated in Figures 5 and 6 ▶ ▶ , human skin grafts from SCID mice reconstituted with allogeneic human PBMCs showed marked infiltration of the graft with human mononuclear cells. Phenotyping of the infiltrating lymphocytes by immunohistochemistry showed that a mixture of both CD4+ and CD8+ lymphocytes were present. Occasional human CD68+ monocytes were also identified at levels comparable with those in skin from animals that did not receive PBMCs. Treatment of the mice with either CsA 10 mg/kg/day or 20 mg/kg/day did not change the degree of mononuclear cell infiltration (Figure 5) ▶ .
Figure 5.

The effect of CsA and/or rapamycin (Rapa) treatment on lymphocyte infiltration of allogeneic skin grafts. Mice were treated with CsA (starting at day 1) or rapamycin (starting at day 7) alone or in combination as described in Materials and Methods. Skin grafts were harvested between days 16 and 21 after i.p. injection of human PBMCs, and then H&E-stained sections were scored for the degree of mononuclear cell infiltrate as described in Materials and Methods. The mean score of either the CsA + Rapa group versus mock or any single drug treatment group was significant (P < 0.05 by analysis of variance).
Figure 6.

The histology of skin grafts harvested from mice 16 to 21 days after i.p. injection of PBMCs and treated with either saline or CsA 10 mg/kg/dose and rapamycin 3 mg/kg/dose as described in Materials and Methods. A and C: Mock-treated animal. B and D: CsA- and rapamycin-treated animal. Magnification: A and B, ×70; C and D, ×350.
H&E-stained sections were examined for evidence of injury to the human microvessels. Grafts harvested from mice reconstituted with PBMCs consistently showed injury to the human dermal microvasculature, characterized by loss of the endothelial cells, and/or thrombosis of the vessels (Table 2) ▶ . Occasional vessel remnants showed proliferating clusters of endothelial cells. Treatment of the mice with either CsA 10 mg/kg/day or 20 mg/kg/day did not reduce the number of grafts with evidence of microvessel damage (Table 2) ▶ . Immunohistochemistry revealed that human perforin-positive lymphocytes were present in a perivascular distribution at comparable numbers among specimens from mock- and CsA-treated animals (data not shown).
Table 2.
The Effect of CsA and Rapamycin (Rapa) on the Frequency of Grafts with Injured Microvessels at 16 to 21 Days
| Treatment group | No. of injured grafts | % injured grafts |
|---|---|---|
| Mock | 17 /19 | 89† |
| CsA 10 mg/kg | 19 /21 | 90† |
| CsA 20 mg/kg | 8 /10 | 80† |
| CsA 10 mg/kg+ Rapa 1 mg/kg | 1 /4* | 25* |
| CsA 10 mg/kg+ Rapa 3 mg/kg | 2 /9† | 22† |
| Rapa 1 mg/kg | 4 /5 | 80† |
| Rapa 3 mg/kg | 4 /6 | 67† |
*P < 0.05 versus mock-treated group, using Fisher’s exact test.
†P < 0.01 versus mock-treated group, using Fisher’s exact test.
Some mice were treated on an alternate-day schedule with rapamycin 1 mg/kg/dose or 3 mg/kg/dose, starting at day 7 after introduction of the PBMCs into the peritoneum of SCID mice bearing human skin grafts. Treatment of animals with rapamycin alone did not reduce the degree of mononuclear cell infiltration of the graft dermis (Figure 5) ▶ and did not confer protection of the graft microvasculature (Table 2) ▶ . In contrast, treatment with either the low-dose (1 mg/kg) or the high-dose (3 mg/kg) rapamycin plus CsA 10 mg/kg/day regimen had the effect of decreasing the extent of allogeneic T-cell infiltration of the skin graft (Figures 5 and 6) ▶ ▶ . Moreover, treatment with rapamycin and CsA reduced the degree of microvessel injury evident in these specimens compared with specimens from mock-treated control animals or those treated with CsA alone (Table 2) ▶ . Immunohistochemical staining showed that perforin-positive lymphocytes were still present in a perivascular distribution, but the total number of such cells was reduced in specimens taken from animals treated with the combination of rapamycin and CsA compared with the other groups (data not shown).
Discussion
In this report, we have analyzed the mechanism of graft microvessel injury in a human peripheral blood lymphocyte-SCID (huPBL-SCID)/skin allograft model. As we have shown previously, injury of the dermal microvessels is dependent on the introduction of human PBMCs and is accompanied by perivascular infiltration of human T cells. 23 Our experiments suggest that microvessel damage in this system is unlikely to be caused by antibody-dependent mechanisms mediated either through mouse complement or mouse leukocyte antibody-directed cell-mediated cytotoxicity. Specifically, we find that a reconstituted human humoral immune system is not necessary for the microvessel injury seen in the huPBL-SCID/skin graft system, because B-cell depletion of the human PBMCs used to reconstitute the immune system of these mice did not alter the efficient destruction of the skin graft microvasculature when compared with animals reconstituted with the complete PBMC population. The success of the B-cell depletion was confirmed by the absence of human IgG in blood and skin specimens taken from the B cell-depleted PBMC-inoculated group. In addition, immunohistochemical staining of human skin grafts for the mouse complement component C3 was comparable in mice receiving whole PBMCs, B cell-depleted PBMCs, or no human PBMCs. Mouse C3 was deposited along the human microvessels but was, if anything, decreased in the skin from PBMC-injected mice. This suggests that, although the mouse complement system may be activated to a small degree by human endothelium even in the absence of antibody, complement does not mediate widespread injury of the human microvasculature. Similarly, others have reported that mouse C3 is deposited on xenogeneic endothelial cells in vivo in the SCID mouse but is insufficient to mediate injury. 26 Finally, we did not observe a robust mouse neutrophil or NK cell infiltrate, as would be expected if antibody binding to the graft microvessel endothelium led to complement activation or antibody-dependent cell-mediated cytotoxicity, respectively.
Our data are consistent with the interpretation that CTLs mediate the observed microvessel injury, causing both endothelial cell death and thrombosis. The absence of significant numbers of human or mouse macrophages in the skin graft infiltrates argues against delayed hypersensitivity as a major mechanism of vascular injury. Similarly, the lack of damage evident among mouse vessels growing into the dermis of the skin graft suggests that antigen-specific CTLs rather than T cell-derived cytokines or shed fas ligand mediate injury because the mouse vessels are exposed to a proinflammatory cytokine milieu similar to that of the native human microvessels. (In theory, human anti-mouse xenoreactive CTLs could arise in these mice in parallel to the development of human alloreactive CTLs; we speculate that they do not largely because mouse MHC molecules and co-stimulators are too divergent from their human homologs to activate human T cells. Alternatively, such xenoreactive cells may traffic to mouse organs other than skin.) We do not interpret these findings to suggest that only CTLs mediate endothelial injury in human allograft rejection. Rather, our findings suggest that CTL development is reconstituted in this mouse model, whereas other effector mechanisms (eg, delayed hypersensitivity) are not, and that CTLs appear sufficient to cause endothelial injury. We have extended these observations to demonstrate that human CD4+ T cells in the absence of CD8+ T cells are capable of injuring the microvasculature of a human allograft. This population also appears capable of inducing injury through direct cytotoxicity, given that some CD4+ cells express perforin.
Nevertheless, we have been unable to directly test whether the infiltrating lymphocytes can mediate cytotoxicity in vitro. Moreover, we have not formally proven that the observed reaction is initiated or targeted by recognition of alloantigen. The human dermal endothelial cells in this model express both MHC class I and class II molecules and hence may function to present alloantigen to both CD8+ and CD4+ T cells. In vitro, endothelial cells are known to directly present alloantigen to human T cells, but it has not been possible to obtain sufficient quantities of skin from our adult volunteer leukopheresis donors to perform autologous control experiments that would address this point. However, using this model, Petzelbauer et al 27 have demonstrated increased human lymphocyte infiltration in an autologous skin graft injected with tetanus toxoid compared with a saline-injected control graft. 27 Taken together, these data suggest that either the Langerhans cells or microvascular endothelial cells in the dermis of the skin graft are capable of initiating the alloresponse, and the microvascular endothelial cells are subsequently targeted by alloreactive CTLs.
Others have reported evidence of T cell-mediated rejection in variations of the huPBL-SCID/allograft system. Shiroki and colleagues 28 found that huPBL-SCID mice rejected human islet cell allografts injected beneath the kidney capsule. Human c-peptide, a marker for islet graft viability, was maintained in unreconstituted control mice for more than 60 days after human islet cells were injected but could not be detected past day 21 in huPBL-SCID mice. Graft-infiltrating lymphocytes were isolated and shown to lyse islet target cells of the same, but not a third-party, donor. Similarly, sensitized lymphocytes are capable of injuring allogeneic human pancreatic grafts, 29 and SCID mice reconstituted with human splenocytes rejected allogeneic human skin grafts, but skin graft rejection was abrogated after injection with mAb directed to CD3 on human T cells. 30 Finally, human lymphocytes injected into SCID mice preferentially injured allogeneic human rather than xenogeneic mouse tissues, 23,31 indicating antigen specificity. Taken together, these data argue that human T lymphocytes remain functional in SCID mice for at least the duration of these experiments and are capable of initiating destruction of allogeneic human grafts.
The restriction of the immune damage to microvessel injury in this model is similar to the pattern of injury of allogeneic human skin grafts in the initial phase of rejection in humans or graft-versus-host disease of the skin after allogeneic bone marrow transplantation. 21,22 In contrast to allogeneic skin graft rejection in the human, however, we do not observe extensive necrosis of the epithelial compartment of the graft. However, in some grafts, small numbers of CD3+ T cells invade the epidermis subjacent to the basal layer of keratinocytes. The foci of localized epithelial cell injury are similar to that seen in graft-versus-host disease of bone marrow transplant recipients. We speculate that the lack of widespread injury of the epidermis may be due to the dual blood supply nourishing the epidermis, which is derived in part through the human microvessel array and in part through new vessels of mouse origin that grow into the graft dermis.
Because our data suggested that a T-cell effector mechanism mediates microvessel injury, we tested whether T cell-directed immunosuppressive agents would afford protection. We observed a dramatic effect when CsA was combined with rapamycin. The action of CsA is probably explained by the known ability of this agent to inhibit cytokine synthesis, an effect we could begin to observe at high-dose treatment with CsA alone, judged by reduced levels of VCAM-1 and human leukocyte antigen-DR expression. The basis for the protective effect of adding rapamycin to CsA treatment in the huPBL-SCID mouse/human skin graft model is not at all clear. This agent is thought to act primarily by preventing T-cell proliferation. However, we are uncertain whether T-cell proliferation plays any role in the development of infiltrates or injury in this model. Although rapamycin can reduce antibody production, reduction of alloantibody cannot explain the observed protective effect, because B cell-depleted PBMCs, which produce no antibody, destroyed the graft microvasculature as effectively as did whole PBMCs. It is more likely that the combination of rapamycin plus CsA acts primarily to inhibit more fully cytokine production by alloreactive T cells. Our previous work has shown that rapamycin can inhibit interleukin 2 secretion by normal human T cells and, more significantly, prevents the development of CsA-resistant interleukin 2 secretion observed when T cells are activated in the presence of endothelial cells. 32 Other possible targets of rapamycin action include inhibition of endothelial functions (eg, local vasodilatation, expression of endothelial adhesion molecules, and local synthesis of chemokines and other leukocyte activators) or inhibition of leukocyte responsiveness to the actions of chemokines and related signals (reviewed in Ref. 6 ). It remains to be determined which of these steps, if any, is most susceptible to the pharmacological suppression we used.
An additional mechanism of protection provided by CsA and/or rapamycin may be inhibition of the development of cytolytic function by the infiltrating T cells. CsA has been found to inhibit the function of cytolytic T cells, 33,34 and rapamycin has been reported to inhibit some actions of interleukin 12 on T cells, 35 suggesting that cytolytic T-cell development may also be impeded. We observed perforin-positive cells within the sparse infiltrates that developed in drug-treated animals, but we do not know whether full cytolytic potential is expressed.
The final point to be drawn from this study is that it provides a new means to test immunosuppressive agents on a human anti-human allogeneic injury. This is a unique advantage of the huPBL-SCID mouse/human skin graft model compared with conventional animal models in which the species’ susceptibility to drugs may differ from human responses. The model is limited in that some agents may impair lymphoid reconstitution, and not all mechanisms of allograft injury are recapitulated. In addition, drug clearance mechanisms in this model still depend on mouse metabolic functions, which may differ from those of humans. Despite these limitations, the present study illustrates that the huPBL-SCID mouse can be used to assess the potential benefit of new immunosuppressive agents for preventing allograft injury. For example, this model has been used to demonstrate the effect of interruption of the CD58/CD2 co-stimulator pathway using species-specific reagents, 36 an experiment not possible in rodents that lack CD58. This model may also permit the study of immunosuppressive reagents, such as mAbs or recombinant proteins, which may specifically block human cytokine receptor, co-stimulatory molecule, or adhesion molecule interactions in a species-specific fashion that could not be tested in rodent models.
Footnotes
Address reprint requests to Dr. Jordan S. Pober, Molecular Cardiobiology Program, Yale University School of Medicine, PO Box 9812, New Haven, CT 06510-0812. E-mail: jordan.pober@yale.edu.
Support for this work was derived from grants from the National Kidney Foundation of Canada (to AGM), National Institutes of Health grants NIH-HL-51014 and NIH-HL-43364 (to JSP) and AI-26689 (to PWA), and from a pilot grant from the Yale Skin Disease Research Center (AR41494). The Molecular Cardiobiology Program was established with a grant from the Lederle Medical Research Division.
Diane E. Epperson’s present address: Laboratory of Molecular Immunology, National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, MD.
Christopher C. W. Hughes’ present address: Department of Molecular Biology and Biochemistry, University of California, Irvine, Irvine, CA.
References
- 1.Lafferty KJ, Prowse SJ, Simeonovic CJ: Immunobiology of tissue transplantation: a return to the passenger leukocyte concept. Annu Rev Immunol 1983, 1:143-173 [DOI] [PubMed] [Google Scholar]
- 2.Pober JS, Orosz CG, Rose ML, Savage CO: Can graft endothelial cells initiate a host anti-graft immune response? Transplantation 1996, 61:343-349 [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg AS, Singer A: Evidence that the effector mechanism of skin allograft rejection is antigen-specific. Proc Natl Acad Sci USA 1988, 85:7739-7742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenberg AS, Singer A: Cellular basis of skin allograft rejection: an in vivo model of immune mediated tissue destruction. Annu Rev Immunol 1992, 10:333-358 [DOI] [PubMed] [Google Scholar]
- 5.Sirak J, Orosz CG, Wakely E, VanBuskirk AM: Alloreactive delayed-type hypersensitivity in graft recipients: complexity of responses and divergence from acute rejection. Transplantation 1997, 63:1300-1307 [DOI] [PubMed] [Google Scholar]
- 6.Pober JS, Cotran RS: Immunologic interactions of T-lymphocytes with vascular endothelium. Adv Immunol 1991, 50:261-302 [DOI] [PubMed] [Google Scholar]
- 7.Fabre JW, Barclay AN, Mason DW: Class II major histocompatibility complex antigens expressed on blood vascular endothelial cells. Transplantation 1983, 36:597-598(letter) [PubMed] [Google Scholar]
- 8.Ferry B, Halttunen J, Leszczynski D, Schellekens H, van de Meide PH, Hayry P: Impact of class II major histocompatibility complex antigen expression on the immunogenic potential of isolated rat vascular endothelial cells. Transplantation 1987, 44:499-503 [DOI] [PubMed] [Google Scholar]
- 9.Hirschberg H, Bergh OJ, Thorsby E: Antigen-presenting properties of human vascular endothelial cells. J Exp Med 1980, 152:249-255 [PubMed] [Google Scholar]
- 10.Wagner CR, Vetto RM, Burger DR: The mechanism of antigen presentation by endothelial cells. Immunobiology 1984, 168:453-469 [DOI] [PubMed] [Google Scholar]
- 11.Pryce G, Male D, Sedgwick J: Antigen presentation in brain: brain endothelial cells are poor stimulators of T-cell proliferation. Immunology 1989, 66:207-212 [PMC free article] [PubMed] [Google Scholar]
- 12.Savage CO, Hughes CC, McIntyre BW, Picard JK, Pober JS: Human CD4+ T cells proliferate to HLA-DR+ allogeneic vascular endothelium: identification of accessory interactions. Transplantation 1993, 56:128-134 [DOI] [PubMed] [Google Scholar]
- 13.Epperson DE, Pober JS: Antigen-presenting function of human endothelial cells: direct activation of resting CD8 T cells. J Immunol 1994, 153:5402-5412 [PubMed] [Google Scholar]
- 14.Adams PW, Lee HS, Ferguson RM, Orosz CG: Alloantigenicity of human endothelial cells. II. Analysis of interleukin 2 production and proliferation by T cells after contact with allogeneic endothelia. Transplantation 1994, 57:115-122 [DOI] [PubMed] [Google Scholar]
- 15.Page CS, Holloway N, Smith H, Yacoub M, Rose ML: Alloproliferative responses of purified CD4+ and CD8+ T cells to endothelial cells in the absence of contaminating accessory cells. Transplantation 1994, 57:1628-1637 [PubMed] [Google Scholar]
- 16.Murray AG, Libby P, Pober JS: Human vascular smooth muscle cells poorly co-stimulate and actively inhibit allogeneic CD4+ T cell proliferation in vitro. J Immunol 1995, 154:151-161 [PubMed] [Google Scholar]
- 17.deWaal RMW, Bogman MJJ, Maass CN, Cornelissen LMH, Tax WJM, Koene RAP: Variable expression of Ia antigens on the vascular endothelium of mouse skin allografts. Nature 1983, 303:426-428 [DOI] [PubMed] [Google Scholar]
- 18.Fabry Z, Waldschmidt MM, Moore SA, Hart MN: Antigen presentation by brain microvessel smooth muscle and endothelium. J Neuroimmunol 1990, 28:63-71 [DOI] [PubMed] [Google Scholar]
- 19.Fabry Z, Sandor M, Gajewski TF, Herlein JA, Waldschmidt MM, Lynch RG, Hart MN: Differential activation of Th1 and Th2 CD4+ cells by murine brain microvessel endothelial cells and smooth muscle pericytes. J Immunol 1993, 151:38-47 [PubMed] [Google Scholar]
- 20.Briscoe DM, DesRoches LE, Kiely JM, Lederer JA, Lichtman AH: Antigen-dependent activation of T helper cell subsets by endothelium. Transplantation 1995, 59:1638-1641 [PubMed] [Google Scholar]
- 21.Dvorak HF, Mihm MC, Dvorak AM, Barnes BA, Manseau EJ, Galli SJ: Rejection of first-set skin allografts in man. J Exp Med 1979, 150:322-337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dumler JS, Beschorner WE, Farmer ER, Di Gennaro KA, Saral R, Santos GW: Endothelial-cell injury in cutaneous acute graft-versus-host disease. Am J Pathol 1989, 135:1097-1103 [PMC free article] [PubMed] [Google Scholar]
- 23.Murray AG, Petzelbauer P, Hughes CCW, Costa J, Askenase P, Pober JS: Human T-cell mediated destruction of allogeneic dermal microvessels in a severe combined immunodeficient mouse. Proc Natl Acad Sci USA 1994, 91:9146-9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dorschkind K, Keller GM, Phillips RA, Miller RG, Bosma GC, O’Toole M, Bosma MJ: Functional status of cells from lymphoid organs and myeloid tissues in mice with severe combined immunodeficiency disease. J Immunol 1984, 132:1804-1808 [PubMed] [Google Scholar]
- 25.Fulop GM, Phillips RA: The scid mutation in mice causes a general defect in DNA repair. Nature 1990, 347:479-482 [DOI] [PubMed] [Google Scholar]
- 26.Naziruddin B, Shiroki R, Shishido S, Howard T, Mohanakumar T: Biochemical and functional characterization of xenoreactive natural antibodies in hu-PBL-SCID mice. J Clin Invest 1996, 97:1267-1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Petzelbauer P, Groger M, Kunstfeld R, Petzelbauer E, Wolff K: Human delayed-type hypersensitivity reaction in a SCID mouse engrafted with human T cells and autologous skin. J Invest Dermatol 1996, 107:576-581 [DOI] [PubMed] [Google Scholar]
- 28.Shiroki R, Poindexter NJ, Woodle ES, Hussain MS, Mohanakumar T, Scharp DW: Human peripheral blood lymphocyte reconstituted severe combined immunodeficient (hu-PBL-SCID) mice: a model for human islet allograft rejection. Transplantation 1994, 57:1555-1562 [PubMed] [Google Scholar]
- 29.MacKenzie DA, Sollinger HW, Hullett DA: Acute graft rejection of human fetal pancreas allografts using donor-specific human peripheral blood lymphocytes in the SCID mouse. Transplantation 1996, 61:1461-1464 [DOI] [PubMed] [Google Scholar]
- 30.Alegre ML, Peterson LJ, Jeyarajah DR, Weiser M, Bluestone JA, Thistlethwaite JR: Severe combined immunodeficient mice engrafted with human splenocytes have functional human T cells and reject human allografts. J Immunol 1994, 153:2738-2749 [PubMed] [Google Scholar]
- 31.Huppes W, Hoffman-Fezer G: Peripheral blood leukocyte grafts that induce human to mouse graft-versus-host disease reject allogeneic human skin grafts. Am J Pathol 1995, 146:1708-1714 [PMC free article] [PubMed] [Google Scholar]
- 32.Karmann K, Pober JS, Hughes CC: Endothelial cell-induced resistance to cyclosporin A in human peripheral blood T cells requires contact-dependent interactions involving CD2 but not CD28. J Immunol 1994, 153:3929-3937 [PubMed] [Google Scholar]
- 33.Dutz JP, Fruman DA, Burakoff SJ, Bierer BE: A role for calcineurin in degranulation of murine cytotoxic T lymphocytes. J Immunol 1993, 150:2591-2598 [PubMed] [Google Scholar]
- 34.Kaiser M, Brooks-Kaiser J, Fitzpatrick L, Bleackley RC, Hoskin DW: Cytotoxic cell proteinase gene expression and cytolytic activity by anti-CD3-activated cytotoxic T lymphocytes is sensitive to cyclosporin A but is not dependent on interleukin-2 synthesis. J Leukocyte Biol 1993, 54:458-464 [DOI] [PubMed] [Google Scholar]
- 35.Bertagnolli MM, Yang L, Herrmann SH, Kirkman RL: Evidence that rapamycin inhibits interleukin-12-induced proliferation of activated T lymphocytes. Transplantation 1994, 58:1091-1096 [PubMed] [Google Scholar]
- 36.Sultan P, Schechner JS, McNiff JM, Hochman PS, Hughes CC, Lorber MI, Askenase PW, Pober JS: Blockade of CD2-LFA-3 interactions protects human skin allografts in immunodeficient mouse/human chimeras. Nat Biotechnol 1997, 15:759-762 [DOI] [PubMed] [Google Scholar]