

Abstract
Subacute hyperoxia may cause basement membrane disruption and subsequent fibrosis. To test the role of extracellular matrix degradation in hyperoxic damage, we analyzed the expression of gelatinases A and B and tissue inhibitors of metalloproteinases (TIMP)-1 and TIMP-2 in rats exposed to 85% O2. Oxygen-exposed rats were studied at 1, 3, 5, and 7 days, and compared with air-breathing rats. Lung mRNAs assayed by Northern and in situ hybridization showed an up-regulation of lung gelatinases A and B from the 3rd day on. Gelatinase A was localized in alveolar macrophages and in interstitial and alveolar epithelial cells. Gelatinase B mRNA and protein were localized in macrophages and bronchiolar and alveolar epithelial cells. Increased gelatinase A and B activities were demonstrated in bronchoalveolar lavage. TIMP-1 and TIMP-2 were constitutively expressed, and only TIMP-1 displayed a moderate increase with hyperoxia. To elucidate transcriptional mechanisms for increased gelatinase B expression after hyperoxia, nuclear transcription factor-κβ activation was explored. Oxidative stress significantly increased the lung expression of nuclear transcription factor-κβ (p65) protein, and nuclear transcription factor-κβ activation and increased levels of gelatinases A and B were found in isolated type II alveolar cells obtained from hyperoxic rats. Conceivably, subacute hyperoxia induces excessive gelatinase activity, which may contribute to lung damage.
Subacute exposure to elevated concentrations of O2 induces oxidative damage that leads to endothelial and epithelial injury, and eventually to lung fibrosis. 1,2 The sequence of pathological changes includes severe disruption of the alveolar-capillary barrier, leading to pulmonary edema. Often, parenchymal cell injury is followed by an inflammatory response and later by fibroblast proliferation and collagen accumulation, with the consequent distortion of the lung architecture. 3 All of these events modulate the remodeling of the surrounding extracellular matrix, including the basement membrane.
The basement membrane plays a dynamic role in maintaining the integrity and differentiation of the alveolar epithelium. It has been suggested that early disruption of this structure may participate in the pathogenesis of lung fibrosis, enhancing the migration of fibroblasts and deposit of interstitial collagens into the alveolar spaces. 4 In addition, disrupted basement membrane may also contribute to the failure to replace damaged alveolar type I epithelial cells after severe injury, which appears to be an important condition contributing to the progression to fibrosis. 5,6
The lung basement membrane is a complex structure that includes type IV collagen, laminin, entactin, fibronectin, and heparan sulfate/chondroitin proteoglycans. 7 The in vivo turnover of basement membrane has not been elucidated. At least two members of the matrix metalloproteinases family (MMPs), gelatinases A and B (MMP-2 and MMP-9), have been shown to degrade several components of the basement membrane. 8,9 They are plausible candidates for its remodeling under physiological and pathological conditions. The substrate specificity of gelatinases includes denatured collagens (gelatin), native type IV collagen, fibronectin, and elastin.8–10. Characteristically, gelatinases are secreted as latent zymogens that can be activated in vitro by several proteases, including other MMPs such as stromelysin, matrilysin, and the membrane type 1 MMP, which has been shown to be a progelatinase A activator. 11-13 MMPs are inhibited by specific tissue inhibitors of metalloproteinases (TIMPs), and in addition, progelatinase A and progelatinase B form complexes with TIMP-2 and TIMP-1, respectively. The regulation of gelatinases by TIMPs is complex and occurs at different levels via not yet elucidated mechanisms. 14,15 MMPs are expressed at low levels in normal adult tissues, and its up-regulation in several pathological conditions is modulated by a variety of cytokines through transcriptional and posttranscriptional mechanisms. 16
Reactive oxygen species, as occurs in hyperoxia-induced damage, modulate the function of some transacting molecules including nuclear transcription factor-κB (NF-κB). NF-κB attaches to DNA in the promoter regions of target genes as a dimer composed of two Rel family proteins, p50 (NF-κB1) and Rel-A (p65). 17,18 Under unstimulated conditions, NF-κB is sequestered in the cytoplasm through its interaction with the inhibitors IκB-α and IκB-β, or IκB-ε. 17 Once activated, NF-κB is able to regulate a wide variety of inflammatory genes that may influence extracellular matrix remodeling. 19,20
To identify some of the molecular events associated with sublethal hyperoxic injury, we set out to determine the activation of NF-κB and the extent of participation of MMP-2 and MMP-9, as well as of TIMP-1 and TIMP-2, in rat lungs exposed to 85% oxygen.
We reasoned that excessive production of gelatinases might contribute to the pathogenesis of subacute hyperoxia-induced lung injury through the basement membrane breakdown.
Materials and Methods
Experimental Model
Pathogen-free male Sprague Dawley rats were purchased from Charles River Laboratories (Raleigh, NC). Twenty-four adult Sprague Dawley rats weighing 200 to 250 g were exposed to 85% O2 and maintained in a 68 × 99 × 83-cm forced-air environmental chamber with a 12:12-h light-dark cycle. Food and water were available ad libitum. Oxygen concentration in the chamber was continuously monitored with Oxycheck Critikon (McNeil Laboratories, Irvine, CA).
Six rats were sacrificed at 1, 3, 5, and 7 days, respectively. Room air-breathing rats were used as controls. Animals were anesthetized, and the right lung was instilled with 4% paraformaldehyde and used for in situ hybridization and immunohistochemistry. The left lung was used for RNA extraction and Northern blot analysis.
Alveolar Epithelial Cell Isolation
In a parallel experiment, lungs from experimental and control rats were lavaged with 15 ml of sterile saline solution. Bronchoalveolar lavage fluid (BALF) was obtained to analyze gelatinolytic activity by zymography and by measuring [3H]gelatin degradation. Additionally, rat alveolar type II pneumocytes (AT2 cells) were obtained from rats exposed for 7 days to 85% O2 rats and from control animals as previously described. 22,23 Briefly, the lungs were perfused via the pulmonary artery, lavaged, and digested with elastase, 30 U/ml (Worthington Biochemical, Freehold, NJ), for 20 minutes at 37°C. The tissue was minced and filtered through sterile gauze and 70-μm nylon mesh. The cell population was enriched for type II pneumocytes by panning the cellular suspension over a surface coated with rat immunoglobulin G to remove cells with Fc receptors. Viability and purity of the final cell preparation exceeded 90%. Fresh type II cells from experimental and control rats were used to prepare cytosolic and nuclear extracts for Western blot analysis and for electrophoretic mobility assays. The rest of the AT2 cells were cultured in Dulbecco’s minimal essential medium containing 2 mmol/L glutamine, 100 U/ml penicillin G, 100 μg/ml streptomycin, and 10% fetal calf serum. Cells were seeded on tissue culture dishes at 2.5 × 10 5 cells/cm 2 and incubated at 37°C in 5% CO2, 95% air. After 3 days in culture, cells were changed to serum-free medium for 24 h. Conditioned media (CM) were collected and stored at −20°C until assayed.
Gelatin Zymography
Sodium dodecyl sulfate (SDS) polyacrylamide gels containing gelatin (1 mg/ml) were used to identify proteins with gelatinolytic activity from the BALF and from the alveolar epithelial cell CM. After electrophoresis, gels were placed in a solution of 2.5% Triton X-100 (two times for 15 minutes each), washed extensively with water, and incubated overnight at 37°C in glycine 100 mmol/L, pH 8.0, containing 10 mmol/L CaCl2 and 50 nmol/L ZnCl2. Identical gels were incubated but in the presence of 20 mmol/L ethylenediaminetetraacetic acid (EDTA). Gels were stained with Coomassie Brilliant Blue R250 and destained in a solution of 7.5% acetic acid and 5% methanol. Zones of enzymatic activity appeared as clear bands against a blue blackground. Molecular weight of the gelatinolytic bands were estimated by using prestained molecular weight marker (Bio-Rad, Richmond, CA).
Gelatinolytic Activity Assay
Gelatinase activity was assayed as previously described 24 using as substrate guinea pig skin collagen labeled with [3H]acetic anhydride denatured to gelatin by heating to 60°C for 20 minutes. Samples were activated with 1 mmol/L p-aminophenylmercuric acetate (APMA) (Sigma Chemical Co., St. Louis, MO) and incubated for 18 hours at 37°C. Gelatinolytic activity was calculated after background subtraction and is expressed as μg of gelatin degraded at 37°C for 18 hours.
Western Blot Assay
Cytosolic and nuclear extracts from fresh AT2 cells were prepared as previously described. 25 Protein was quantified by the Bradford method, and equal amounts were loaded in a denaturing 8% polyacrylamide gel. Proteins were electrotransferred to polyvinylidene difluoride membranes and, after nonspecific sites were blocked with 5% nonfat milk in phosphate-buffered saline (PBS), the proteins were incubated with 1 μg/ml of p65 rabbit polyclonal antibody (Santa Cruz Biotechnology, Santa Cruz, CA) for 2 hours. The membranes were incubated with anti-rabbit immunoglobulin G-horseradish peroxidase antibody (Amersham, Arlington Heights, IL) for 1 hour as recommended by the manufacturer, and antibody binding was determined using enhanced chemiluminscence (Amersham).
Electrophoretic Mobility Shift Assay
Nuclear extracts from freshly obtained AT2 cells derived from control and hyperoxic rats were used. The probe used for this assay was a double-stranded 29-bp oligonucleotide derived from the reported human gelatinase B promoter. 26 The sequence includes the NF-κB binding site (TGGAATTCCCC) common to human, mouse, and rabbit genes (the rat promoter for this gene has not been cloned) and adjacent nucleotides (from −607 to −578). The probe was labeled using a fill-in reaction with a Klenow fragment of DNA polymerase I using [32P]dCTP and purified with conventional spin-column chromatography.
To visualize DNA-protein complexes, binding reactions were carried out in a 20-μl volume of buffer B [20 mmol/L Hepes, pH 7.9, 60 mmol/L KCl, 20% glycerol, 0.25 mmol/L EDTA, 0.125 mmol/L ethylene glycol tetraacetic acid, 1 mmol/L dithiothreitol, 4 mmol/L MgCl2, 4 mmol/L spermidine, 200 mmol/L NaCl, 25 mg/ml bovine serum albumin, and 2 μg poly(deoxyinosinic-deoxycytidylic acid)] with 10 μg of nuclear extract on ice for 10 minutes. One ng of labeled double-stranded oligonucleotide (approximately 100,000 cpm) was added, and the reaction was incubated 30 minutes on ice. Complexes were resolved by electrophoresis on a 6% nondenaturing polyacrylamide gel. The gel was vacuum dried and exposed to film at room temperature for 12 hours. Competition was performed by adding 2.5, 25, and 250 mol/L excess of cold oligonucleotide sequence to binding reactions.
In Situ Hybridization
Riboprobes for in situ hybridization were generated as previously described 27 from human cDNA gelatinase A provided by G. I. Goldberg (Washington University, St. Louis MO), a rat cDNA gelatinase B for the 3′ region donated by J. Windsor (University of Alabama at Birmingham, Birmingham, AL), and mouse TIMP-1 and TIMP-2 cDNA provided by Dylan Edwards (University of Calgary, Calgary, Alberta, Canada).
In situ hybridization was performed on 4-μm sections as previously described. 27 Briefly, the sections mounted on silanized slides were incubated in 0.001% proteinase K (Sigma Chemical Co.) for 20 minutes at 37°C. After acetylation with acetic anhydride, the sections were prehybridized for 1 hour at 45°C in a hybridization buffer. The sections were incubated with the digoxigenin-labeled probes at 45°C overnight. Some sections were hybridized with digoxigenin-labeled sense RNA probe. After hybridization, the sections were rinsed in 2× standard saline citrate (SSC) for 1 hour at room temperature, 0.5× SSC with pancreatic RNase A at 65°C for 30 minutes, and 0.5× SSC for 1 hour at room temperature. The tissues were rinsed in PBS and incubated with a polyclonal sheep anti-digoxigenin antibody coupled to alkaline phosphatase (Boehringer Mannheim Co., Indianapolis IN) for 1 hour at room temperature. The color reaction was performed by incubation in 100 mmol/L Tris-HCl, 50 mmol/L MgCl2, pH 9.5, with 0.1 mmol/L levamisole, 0.338 mg/ml 4-nitroblue tetrazolium chloride, and 0.173 mg/ml 5-bromo 4-chloro 3-indolyl-phosphate or Fast Red chromogen (Biomeda Corp., Foster City, CA). Sections were lightly counterstained with eosin or hematoxylin.
Immunohistochemistry
Mouse monoclonal antibody against rat 95-kd type IV collagenase 28 was kindly provided by J. Windsor (University of Alabama at Birmingham). Polyclonal antibody directed against an epitope of the carboxy terminus of the Rel-A (p65) protein (C-20) and polyclonal antibody directed against an epitope of the amino terminal of the IκB-α protein (C-15) were used. (Santa Cruz Biotechnology). Polyclonal antibody to type IV collagen from human placenta, which cross-reacts with rat type IV collagen, was used to visualize basement membranes (BioGenex, San Ramon, Ca) .
Tissue sections were deparaffinized and then rehydrated, and endogenous peroxidase was blocked by preincubation with 0.45% H2O2 in methanol for 30 minutes. Antigen retrieval was performed with 10 mmol/L citrate buffer, pH 6.0, for 5 minutes in microwave. For type IV collagen, tissue sections were digested with pepsin (BioGenex EK000-5K) at 37°C for 5 minutes instead of microwave antigen retrieval. Tissue sections were incubated with an antibody diluent with background-reducing components (DAKO, Carpinteria, CA) diluted 1:100 in PBS for 45 minutes. The sections were then incubated with 50 μg/ml of primary antibodies at 4°C overnight. A secondary biotinylated antibody (Vector Laboratories, Burlingame, CA), diluted 1:150, and avidin-biotin peroxidase complex were applied sequentially for 30 minutes followed by 3,3′-diaminobenzidine in PBS containing 0.045% H2O2 for 5 minutes. The sections were counterstained with hematoxylin. PBS was used to dilute the antibodies and to rinse sections between steps. The primary antibody was replaced by normal rabbit or mouse sera for negative control slides.
Before counterstaining, immunoreactive Rel-A and IκB-α intensities were quantified using an image analyzer (Foster and Findley Associates) with the PC Image version 2.1 software. After gray calibration, 10 fields at ×40 were analyzed, and the gray weight in arbitrary was units determined. The statistical analysis was performed using an unpaired Student’s t-test.
RNA Isolation and Northern Blot Analysis
Total RNA from lung specimens was isolated with Trizol reagent (Life Technologies, Grand Island NY) according to the specifications of the manufacturer. Total RNA (15 μg/lane) was separated in 1% agarose-formaldehyde gels and transferred to Nytran membranes (Schleicher & Schuell, Keene, NH). The membranes were prehybridized at 42°C for 16 hours in 5× SSC, 50% formamide, 5× Denhardt’s solution, and 0.5% SDS containing 100 μg/ml of denatured salmon sperm DNA. Hybridization was carried out at 42°C for 16 hours in hybridization buffer containing 50% dextran sulfate plus heat denatured 32P-labeled probe. Membranes were washed in 2× SSC-0.1% SDS at 42°C, followed by 0.25× SSC-0.1% SDS at 55°C, and 0.1× SSC-0.1% SDS at 65°C. Membranes were exposed to Kodak BIOMAX MS film at −70°C with an intensifying screen. Loading of RNA samples was monitored by assessing the mRNA level of glyceraldehyde-3-phosphate-dehydrogenase (GAPDH, obtained from the American Type Culture Collection, Manassas, VA). Gelatinase A, gelatinase B, TIMP-1, TIMP-2, and GAPDH probes were radiolabeled with [32P]dCTP to specific activity of 200 × 10 6 dpm/mg using a multiprime DNA labeling kit (NEP-103; Dupont, Wilmington, DE).
Results
After 85% exposure to oxygen for up to 7 days, rats did not have noticeable respiratory distress. BALF protein concentration increased significantly at 3, 5, and 7 days of hyperoxic exposure reaching a concentration of 206 ± 76 μg/ml, 236 ± 141 μg/ml, and 287 ± 161 μg/ml, respectively, which was statistically significant when compared with 59 ± 16 μg/ml in controls (P < 0.001, 0.008, and 0.005, respectively).
BALF Gelatinolytic Activity
Gelatinolytic activity assessed on [3H]gelatin in APMA-activated BALF was significantly increased at 5 and 7 days in 85% oxygen-exposed rats (P < 0.01). In Figure 1 ▶ it can be observed that BALF from hyperoxic rats digested gelatin approximately 2.5-fold more compared with the controls. Gelatinolytic activity was inhibited up to 78% by 80 mmol/L 1,10-o-phenantroline.
Figure 1.

Gelatinolytic activity of BALF obtained from rats exposed to 85% O2. Samples activated with 1 mmol/L APMA were incubated with 100 μg of [3H]gelatin for 18 hours at 37°C. Results are presented as mg of gelatin degraded at at 37°C for 18 hours. Values represent the mean ± SD of six animals at each point. *P < 0.01.
Gelatin zymography performed with nonreduced BALF fluid revealed quantitative and qualitative differences among oxygen-exposed and control animals. Samples derived from normal lungs displayed proteolytic bands mainly of 72 kd representing the proenzyme form of gelatinase A (Figure 2 ▶ , lanes C). BALF obtained at 3, 5, and 7 days of 85% oxygen exposure exhibited an increased intensity of this gelatinolytic band together with a 66-kd proteolytic activity representing the active form of gelatinase A. Maximum activities were reached at 5 and 7 days (Figure 2) ▶ .
Figure 2.

Identification of gelatinolytic enzymes in BALF by SDS-polyacrylamide gel electrophoresis gelatin zymography. Lanes C: Control rats; lanes 3 , 5, and 7: 85% oxygen-exposed animals after 3, 5, and 7 days; left and right lanes: prestained molecular weight standards. Zones of enzymatic activity appear as clear bands over a dark background. Gelatinolytic activity bands were inhibited by EDTA.
Additionally, gelatinolytic activities with apparent molecular weights of 95 and 88 kd, which are the appropriate sizes to be progelatinase B and its activated form, respectively, were clearly observed at 5 and 7 days of hyperoxic injury (Figure 2) ▶ . In some BALF samples obtained after 5 and 7 days of oxygen exposure, several higher molecular weight bands of gelatinolytic activity were also evident, presumably representing lipocalin-associated latent forms, specific to neutrophils, and/or dimers of the gelatinases. 22,29 All gelatinolytic activity bands were inhibited by EDTA (not shown).
Gelatinase A and B Expression
Northern Blot Analysis
To define the time-course expression of MMP-2 and MMP-9, membranes with total RNAs prepared from lung extracts were hybridized with the specific cDNAs and the housekeeping gene GAPDH. Animals exposed to 85% oxygen exhibited an increased expression of MMP-2 and MMP-9 mRNAs from the 3rd day on, as compared with controls. As seen in Figure 3, a ▶ marked up-regulation was noticed, especially on days 5 and 7, both for gelatinases A and B. When the levels of these gelatinase transcripts were standardized by the level of GAPDH mRNA, a∼ 4-fivefold increase was noted for both MMPs.
Figure 3.

Northern blot of rat lung RNA. Lanes 1, 3, 5, and 7: Total RNA (15 μg/lane) from lung tissues of rats exposed for 1, 3, 5, and 7 days to 85% oxygen. Lane C: Total RNA from unexposed animals. Membranes were hybridized with specific cDNA probes for gelatinase A (MMP-2), gelatinase B (MMP-9), and GAPDH as a normalization control.
Gelatinase A and B Localization
The cellular source of gelatinase A and B mRNAs in the rat lung after hyperoxic exposure was examined by in situ hybridization at days 3, 5, and 7. Likewise, immunoreactive gelatinase B was analyzed by immunohistochemistry.
In lung sections from untreated controls, few cells were positive for gelatinase A mRNA (Figure 4A) ▶ . Animals sacrificed at 3 days of oxygen exposure showed no apparent differences with normal rats. At 5 days, but mostly at 7 days of oxygen exposure, lungs exhibited a strong signal for gelatinase A (Figure 4B) ▶ . MMP-2 mRNA transcript was observed in an interstitial localization, mainly in areas of alveolar wall thickening. Alveolar macrophages were positively stained, and additionally, label was also noticed in cells located in the corners of alveoli, protruding to the alveolar spaces, which were thus putatively identified as type II pneumocytes (Figure 4, B and C) ▶ .
Figure 4.
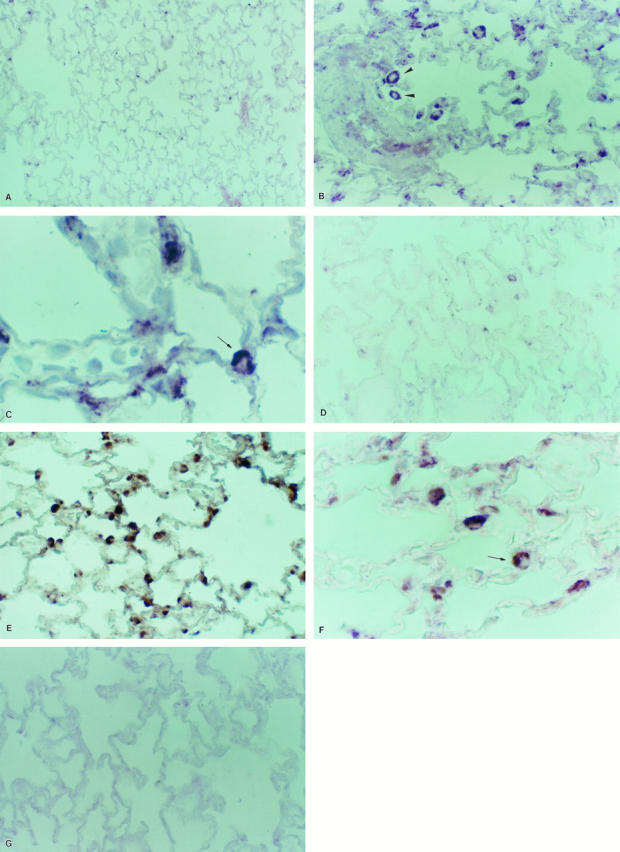
In situ hybridization for MMP-2 (A to C) and MMP-9 (D to F). Tissue samples were processed for in situ hybridization with digoxigenin-labeled riboprobes and after that with anti-digoxigenin antibody coupled to alkaline phosphatase. The color reaction was performed with nitroblue tetrazolium chloride. Sections were lightly counterstained with eosin. Gelatinase A antisense RNA probe: A: Normal lung; original magnification, ×35. B: Lung exposed 7 days to hyperoxia, exhibiting strong MMP-2 up-regulation; arrowheads indicate alveolar macrophage; ×70. C: Lung exposed 7 days to hyperoxia; arrow indicates a putative type 2 pneumocyte; ×350. Gelatinase B antisense riboprobe: D: Normal lung; ×70. E: Lung exposed 7 days to hyperoxia, showing cytoplasmic label in epithelial cells and alveolar macrophages; ×70. F: High magnification of sample of lung that was hyperoxic for 7 days; arrowhead indicates a putative type 2 pneumocyte; ×350. G: Sense riboprobe; ×70.
Gelatinase B mRNA expression was observed in scattered cells in control lungs (Figure 4D) ▶ . As appreciated for gelatinase A, an overexpression was noted at 5 and mainly at 7 days of exposure. The enzyme transcript was predominantly localized in alveolar macrophages and alveolar epithelial cells (Figure 4, E and F) ▶ . Tissue sections did not show any signal when they were hybridized with the sense probes nor when they were treated with RNase before hybridizing with the antisense probes (Figure 4G) ▶ .
Using a specific monoclonal gelatinase B antibody, paraffin-embedded tissues were also examined to immunolocalize MMP-9 protein. Gelatinase B protein was barely detectable in lung sections from control rats (Figure 5A) ▶ . By contrast, a clear immunoreactivity was observed at 5 and 7 days of 85% oxygen exposure, thus paralleling in situ hybridization results. Gelatinase B protein was observed in alveolar epithelial cells (Figure 5B) ▶ , and a strong signal was observed in areas of hyperplastic type 2 pneumocytes (Figure 5C) ▶ . Additionally, prominent labeling was seen in the bronchiolar epithelium, localized in nonciliated bronchiolar cells (Figure 5D) ▶ . Controls using nonspecific antisera showed no reactivity (Figure 5E) ▶ . Tissue sections from rats exposed to 7 days of hyperoxia revealed some discrete areas suggestive of basement membrane rupture when explored with anti-type IV collagen antibody (Figure 5F) ▶ .
Figure 5.
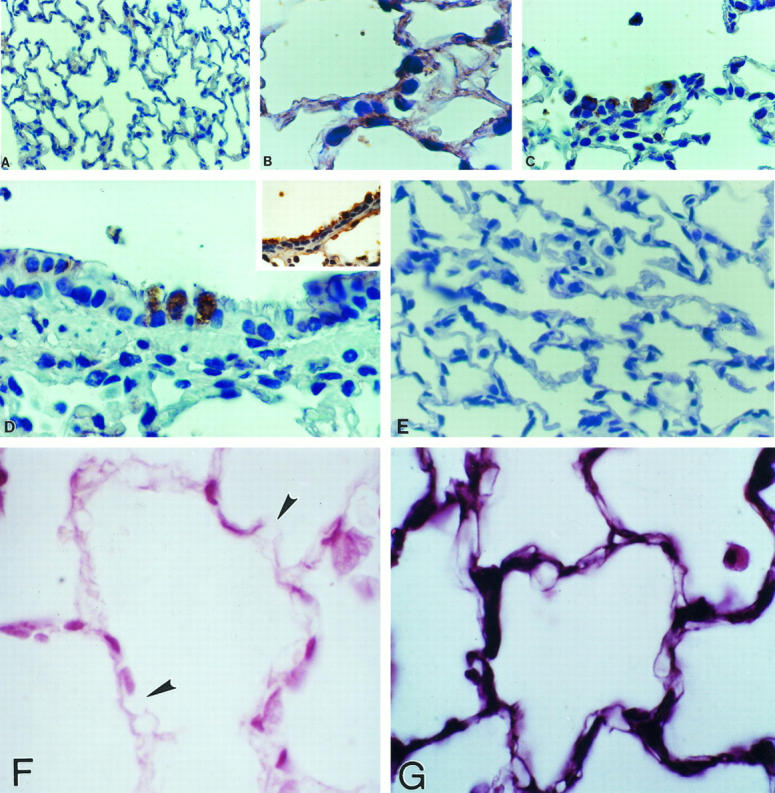
Immunohistochemical localization of gelatinase B (MMP-9) in lung tissue sections. Immunoreactive gelatinase B was revealed by peroxidase reaction. Sections were lightly counterstained with hematoxylin. A: Normal lung (magnification, ×70). B to D: Lung of rat exposed 7 days to hyperoxia, at higher magnification (×350), showing alveolar (B and C) and bronchiolar (D) epithelial cells. D (inset) shows bronchiolar epithelial cells stained with cytokeratin. E: Control lung after 7 days of hyperoxia with primary antibody omitted (×70). F: Lung tissue from a rat exposed 7 days to hyperoxia, stained with anti-type IV collagen; arrows indicate areas of basement membrane disruption (×350). G: Normal lung tissue stained with anti-type IV collagen.
Type IV Collagenases/Gelatinases in Isolated Type 2 Cells
To further examine the effect of 85% O2 on type 2 pneumocyte gelatinase production, alveolar cells from controls and 5 and 7 days hyperoxia-exposed animals were obtained. Gelatin zymography of nonreduced serum-free CM revealed a constant major ∼72-kd proteolytic band, the latent form of gelatinase A, both in control and experimental type II primary cultures (Figure 6 ▶ , lanes 1 to 4). When the control CM was treated with APMA, the MMP-2/gelatinase A activity was partially converted to a 66-kd active form (Figure 6 ▶ , lane 2). Primary cultures of control type II pneumocytes also expressed low levels of a ∼92-kd gelatinase B activity (Figure 6 ▶ , lane 1), which was transformed to an 88-kd active form after APMA activation (Figure 6 ▶ , lane 2). After 85% exposure, an increase in 92-kd gelatinolytic band was observed in AT2 cell-derived CM from lungs exposed to oxygen for 5 and 7 days (Figure 6 ▶ , lanes 3 and 4). CM of normal human lung fibroblasts used as control showed 72-kd gelatinolytic activity (Figure 6 ▶ , lane 5). All gelatinolytic bands were inhibited by 20 mmol/L EDTA (data not shown).
Figure 6.

Gelatinase production by isolated type II cells. CM derived from primary cultures of type II pneumocytes of controls, and those exposed 5 and 7 days to 85% oxygen were resolved by SDS-polyacrylamide gel electrophoresis using a 7.5% gel copolymerized with 1 mg/ml gelatin. Lane 1: Control sample. Lane 2: Control sample preincubated with 1 mmol/L APMA. Lane 3: Pneumocytes exposed 5 days to hyperoxia. Lane 4: Pneumocytes exposed 7 days to hyperoxia. Lane 5: Rat fibroblast CM. Gelatinolytic activity was inhibited by EDTA.
TIMP-1 and TIMP-2 Expression
Northern Blot Analysis
TIMP-2 mRNA was constitutively expressed in control lungs revealing two transcripts of ∼1.0 and ∼3.5 kb. No major changes were observed in the time course injury induced by hyperoxic exposure (Figure 7) ▶ .
Figure 7.

Northern blot of rat lung RNA. Lanes1, 3, 5, and 7: Total RNA (15 μg/lane) from lung tissues of rats exposed for 1, 3, 5, and 7 days at 85% oxygen. Lane C: Total RNA from lung tissues of unexposed animals. Membranes were hybridized with specific cDNA probes for TIMP-1, TIMP-2, and GAPDH.
TIMP-1 mRNA was also expressed in controls, and a moderate increase of this transcript was observed with the hyperoxic injury (Figure 7) ▶ .
TIMP-1 and TIMP-2 Localization
In normal lungs, TIMP-1 and TIMP-2 transcripts were mainly localized in alveolar and bronchiolar nonciliated epithelial cells, and occasionally in interstitial cells (Figure 8, A and B) ▶ . In oxygen-exposed rats, mainly at 3 days of exposure, both inhibitors were additionally strongly expressed by alveolar macrophages (Figure 8 ▶ C and D). No signal was observed when tissue samples were hybridized with the sense probes (Figure 8E) ▶ .
Figure 8.

In situ hybridization for TIMP-1 and TIMP-2 in controls and in 85% oxygen-exposed rats from 3 days. Tissue samples were processed for in situ hybridization with digoxigenin-labeled riboprobes and after that with anti-digoxigenin antibody coupled to alkaline phosphatase. The color reaction was performed with Fast Red chromogen, and tissues were slightly counterstained with hematoxylin. A: TIMP-1 expression in bronchiolar cells from a normal lung; original magnification, ×70. B: TIMP-2 expression in alveolar epithelial cells in a normal lung; ×210. C: TIMP-1 expression in lungs hyperoxic for 3 days; positive alveolar macrophages are indicated by arrowheads; ×210. D: TIMP-2 expression in rats exposed 3 days to hyperoxia, exhibiting label in macrophages (arrowhead) and alveolar epithelial and interstitial cells; ×210. E: Sense riboprobe; 140×.
NG-κB (p65) and IκB-α Protein Detection
Quantification of immunoreactive Rel-A (p65) protein with an image analyzer revealed a significant and progressive increase from the 3rd day on. As shown in Figure 9 ▶ , the protein increased from 35% at 3 days after hyperoxia exposure to 454% at the last time point analyzed (P = 0.003, 0.0006, and 0.0005 at 3, 5, and 7 days, respectively). NF-κB was observed mainly in epithelial cells and macrophages, thus exhibiting a pattern similar to that observed for gelatinase B (not shown).
Figure 9.

NF-κB (p65) protein graph of immunoreactive protein. Lung tissue sections of normal and hyperoxia-exposed rats were treated with a polyclonal antibody directed against an epitope of the carboxy terminus of the Rel-A (p65) protein. Immunoreactive protein was revealed by peroxidase reaction. Quantification of Rel-A (p65) protein was performed with an image analyzer. P = 0.003, 0.0006, and 0.0005 at 3, 5, and 7 days, respectively.
By contrast, no significant differences were observed with IκB-α when a polyclonal antibody directed against an epitope of the amino terminal protein was used (data not shown).
To correlate these changes with the increase in the 92-kd gelatinase activity, Western blot analysis of p65 protein was performed in freshly isolated type II cells. As shown in Figure 10 ▶ , p65 protein levels were increased in AT2 cells derived from 85% oxygen-exposed rats. This increment was present in both cytosolic and nuclear fractions, reflecting an increase in the total p65 protein levels. Also, the increase in the nuclear p65 suggests the presence of active NF-κB complexes. Electrophoretic mobility shift assay was performed using an oligonucleotide with the NF-κB site from the reported gelatinase B promoter. As shown in Figure 11, a ▶ specific increase in the protein-DNA binding was demonstrated in freshly isolated type II cells from 85% hyperoxic rats.
Figure 10.

Western blot analysis for p65 protein. Cytosolic extracts of type II alveolar cells freshly isolated from unexposed animals (lane 1) and from rats exposed for 7 days to 85% oxygen (lane 2). Nuclear extracts of type II cells isolated from unexposed control rats (lane 3), and from animals exposed to 85% oxygen (lane 4). Molecular weight markers in kd are shown to the left.
Figure 11.

Electrophoretic mobility shift assay for NF-κB site derived from gelatinase B promoter. Lanes 1 and 2: Nuclear extracts of type II cells isolated from control rats (lane 1) and from rats exposed for 7 days to 85% oxygen (lane 2). Lanes 3 to 5: Nuclear extracts from hyperoxic injured rats incubated with a 2.5-times excess (lane 3), a 25-times excess (lane 4), and a 250-times excess (lane 5) cold oligonucleotide to demonstrate specific binding. Lane 6: 25-times cold excess heterologous oligonucleotide (p53-binding site).
Discussion
Sustained exposure to sublethal concentrations of oxygen cause lung injury that may lead to fibroblast hyperplasia and abnormal extracellular matrix accumulation. 1-3,30
In the well characterized rat model of 85% O2 exposure, in which animals suffer diffuse pulmonary injury, our findings revealed a time-dependent increase in the expression of mRNA for MMP-2 and MMP-9 throughout the lung, where particularly high levels were noted at 5 and 7 days of hyperoxia exposure. By contrast, TIMP-1 and TIMP-2 mRNAs were constitutively expressed, and only TIMP-1 exhibited a moderate increase in the lungs after hyperoxic injury as revealed by Northern blot. Increased expression of TIMP-1 has been previously reported in rabbit and murine lungs after 100% O2. 31,32
The much greater increase in the abundance of the mRNA for gelatinases (from almost undetectable to strongly positive) than TIMPs suggests a major alteration in the balance between inhibitors and enzymes, thus favoring the degradation of a number of matrix macromolecules such as type IV collagen, elastin, and fibronectin. Supporting the presence of active gelatinases during sublethal hyperoxic injury is the finding that BALF revealed not only a clear increase of both gelatinases A and B, but a noteworthy abundance of the activated enzymes. Interestingly, it has been recently demonstrated that superoxide-derived metabolites increase gelatinase A activity. 33
The metalloproteinases genes are stimulated by a variety of biologically active agents, including growth factors, oncogenes, and cytokines. 34 During hyperoxia exposure, several cytokines and growth factors are up-regulated, such as tumor necrosis factor-α and interleukin-1β, which, among other actions, can activate NF-κB, an oxidative stress-responding factor and an immediate early mediator of immune and inflammatory responses. 17,35,36 Accumulation of p65 during lethal concentrations of hyperoxia has been reported in vitro by Li et al, 37 and the present study extends this in vitro observation showing that at 7 days of sublethal hyperoxia, a severalfold increase in the in vivo lung expression of the p65 subunit of NF-κB occurs. Additionally, an increase in the total p65 content in type II cells isolated from hyperoxic rats was also found. Although the levels of the inhibitor IκB-α were not significantly modified, it could be argued that other members of the family could be participating in this model given that it has been shown that IκB-β and not IκB-α is responsible for modulating NF-κB activation in long-term activation. 38
The activation of NF-κB by continuous hyperoxia could be modulating directly gelatinase B expression, given that an NF-κB-like site is found in all of the gelatinase B promoters cloned to date, which includes the human, rabbit, and mouse genes. 39-41 This site has been found to cooperate with an activator protein-1-like element in determining the full response to phorbol 12-myristate 13-acetate and cytokines in various cell lines. 42 In this study we found an increment in DNA-protein binding to the gelatinase B promoter NF-κB site, supporting the notion of direct participation of this transcriptional factor in the 92-kd gelatinase induction.
Gelatinase A promoter seems to be very different from gelatinase B promoter and from other metalloproteinases. No NF-κB site has been identified in gelatinase A promoter, but recently a p53 binding site has been reported, which can mediate gelatinase A transactivation after DNA damage. 43 We can hypothesize that, because NF-κB can also transactivate p53, 44,45 it would be able to indirectly induce gelatinase A transcription. Interestingly, exposure of mice to 95% hyperoxia has been shown to induce p53 expression, particularly in distal bronchiolar airways and in AT2 cells, supporting a putative participation of p53 transactivating function in the hyperoxic injury. 46
Our results support the notion that hyperoxia could affect cytokine gene expression, and consequently MMP levels, via activation of NF-κB/Rel nuclear regulatory factors. Activated NF-κB, in a positive feedback regulation, may up-regulate cytokines such as interleukin-1, interleukin-6, and tumor necrosis factor-α, which in turn influence MMP expression. Actually, it has been demonstrated that 100% oxygen injury activates NF-κB, which precedes the hyperoxia-induced increase in tumor necrosis factor-α and interferon-γ transcription. 47 Tumor necrosis factor-α is able to induce gelatinase B production, contributing to increased vascular endothelial permeability. 48 Likewise, it has been reported that short-term exposure to α- and γ-interferons can induce gelatinases A and B. 49
Under pathological conditions, a number of different cells have been shown to contribute to the in vivo MMP production in lung parenchyma. 25,50,51 During 85% lung exposure, we observed that gelatinase A mRNA was expressed mainly by interstitial and alveolar epithelial cells, whereas gelatinase B was localized in alveolar macrophages, nonciliated bronchiolar cells, and alveolar epithelial cells. The ability of type 2 pneumocytes to produce gelatinases was additionally assessed by obtaining alveolar epithelial cells from 85% O2-exposed rats. Our results showed that hyperoxia in vivo increases gelatinase production and corroborated previous findings demonstrating that type 2 pneumocytes obtained from normal rats secrete in vitro gelatinases A and B. 22,52
Excessive gelatinase activity in the local lung microenvironment may result in abnormal extracellular matrix remodeling and basement membrane disruption, enhancing edema formation, inflammation, and fibroblast migration to the alveolar spaces. Actually, alveolar-capillary membrane remodeling, followed by alveolar wall thickening and interstitial fibrosis, is a well known sequela of hyperoxia-induced lung injury.
In human acute lung injury, an increase in BALF gelatinases A and B has been suggested to play a role in basement membrane disruption. 53-55 Likewise, we have demonstrated that lethal exposure to 100% oxygen provokes a remarkable increase of gelatinolytic and collagenolytic activities both in BALF and in lung tissue. 27 In the present study, some areas exhibiting basement membrane disruption were observed after 7 days of 85% hyperoxic injury. Interestingly, after 60 hours of 100% hyperoxia exposure, 27 an up-regulation of gelatinases A and B is also revealed in a pattern similar to that observed after 7 days with 85% oxygen exposure, although the clinical and pathological behavior is different. Lung damage with 100% oxygen represents a lethal model of acute lung injury with denudation of the alveolar epithelium and edema. The 85% oxygen damage produces moderate morphological changes, with proliferation of the alveolar epithelial type II cells, with which animals survive, but a number of them may develop pulmonary fibrosis. Therefore, in two different lung responses that share O2 as the initial pathogenetic mechanism, an overexpression of gelatinases A and B occurs, suggesting that these metalloproteinases participate in some shared events. We postulate that abnormal degradation of basement membrane could be one of these events.
In this context, disruption of basement membrane appears to play a critical role in many human and experimentally induced diffuse lung disorders. Thus, in idiopathic pulmonary fibrosis and in adult respiratory distress syndrome, gaps in the distribution of type IV collagen and laminin, supporting basement membrane disruption, have been described early in the disease course. 4 Moreover, morphometric studies of lungs with acute and proliferative stages of diffuse alveolar damage have shown that intraalveolar fibrosis develops in the early proliferative stage, in which activated myofibroblasts migrate into intraalveolar spaces through gaps in the epithelial basement membrane. 56 Likewise, in bleomycin-induced lung damage, the presence of buds of intraalveolar fibrosis has been associated with discontinuous epithelial basement membranes. 57
The findings of the present work support that both MMPs are up-regulated during a sublethal oxidative stress, which suggests an eventual pathway for lung fibrosis. The lung injury and repair process is a complex interplay between epithelial, endothelial, mesenchymal, and inflammatory cells, in which cytokines, growth factors, extracellular matrix, signals for programmed cell death, metalloproteinases and TIMPS actively participate, that is ultimately characterized by new extracellular matrix formation and tissue remodeling.
Clearly, no single mechanism is responsible for the progression of diffuse alveolitis to pulmonary fibrosis. A network of complex different factors must interact in the local microenvironment, finally resulting in exaggerated fibroblast proliferation and collagen accumulation. Our results suggest that the alteration in the balance of gelatinases A and B and of TIMP-1 and TIMP-2 may be part of this process.
Footnotes
Address reprint requests to Annie Pardo, Ph.D., Facultad de Ciencias, UNAM, Apartado Postal 21–630, Coyoacán, México DF, CP 04000, México. E-mail: aps@hp.fciencias.unam.mx.
Supported by PUIS & PAPIT grant IN202697 (UNAM), grant HL-48129 from the Department of Medicine, Michael Reese Hospital, and CONACYT grant F643-M9406.
References
- 1.Gould VE, Tosco R, Wheelis RF, Gould NS, Kapanci Y: Oxygen pneumonitis in man: ultrastructural observations on the development of alveolar lesions. Lab Invest 1972, 26:499-508 [PubMed] [Google Scholar]
- 2.Durr RA, Dubaybo BA, Thet LA: Repair of chronic hyperoxic lung injury: changes in lung ultrastructure and matrix. Exp Mol Pathol 1987, 47:219-240 [DOI] [PubMed] [Google Scholar]
- 3.Katzenstein ALA, Bloor CM, Liebow AA: Diffuse alveolar damage-the role of oxygen, shock, and related factors. Am J Pathol 1976, 85:210-224 [PMC free article] [PubMed] [Google Scholar]
- 4.Raghu G, Striker LJ, Hudson LD, Striker GE: Extracellular matrix in normal and fibrotic human lungs. Am Rev Respir Dis 1985, 131:281-289 [DOI] [PubMed] [Google Scholar]
- 5.Adamson IYR, Young L, Bowden DH: Relationship of alveolar epithelial injury and repair to the induction of pulmonary fibrosis. Am J Pathol 1988, 130:377-383 [PMC free article] [PubMed] [Google Scholar]
- 6.Uhal BD, Joshi I, Mundle S, Raza A, Pardo A, Selman M: Fibroblasts isolated after fibrotic lung injury induce apoptosis of alveolar epithelial cells in vitro. Am J Physiol 1995, 269:L819-L828 [DOI] [PubMed] [Google Scholar]
- 7.Yurchenco PD, Schittny JG: Molecular architecture of basement membranes. FASEB J 1990, 4:1577-1590 [DOI] [PubMed] [Google Scholar]
- 8.Collier IE, Wilhem SM, Eisen AZ, Marmer BL, Grant GA, Seltzer JL, Kromberger A, Bauer EA, Goldberg GI: H-ras oncogene-transformed human bronchial epithelial cells (TBE-1) secrete a single metalloproteinase capable of degrading membrane collagen. J Biol Chem 1988, 263:6579-6587 [PubMed] [Google Scholar]
- 9.Wilhem SM, Collier IE, Marmer BL, Eisen AZ, Grant GA, Goldberg GI: SV40-transformed lung fibroblasts secrete a 92 kd type IV collagenase which is identical to that secreted by normal macrophages. J Biol Chem 1989, 264:17213-17221 [PubMed] [Google Scholar]
- 10.Senior RM, Griffin GL, Fliszar CJ, Shapiro SD, Goldberg GI, Welgus HG: Human 92-, and 72-kilodalton type IV collagenases are elastases. J Biol Chem 1991, 266:7870-7875 [PubMed] [Google Scholar]
- 11.Ogata Y, Enghild JJ, Nagase H: Matrix metalloproteinase 3 (stromelysin) activates the precursor for the human matrix metalloproteinase 9. J Biol Chem 1992, 267:3581-3584 [PubMed] [Google Scholar]
- 12.Abramson SR, Conner EG, Nagase H, Nehaus I, Woessner JF: Characterization of rat uterine matrilysin and its cDNA: relationship to human pump-1 and activation of procollagenases. J Biol Chem 1995, 270:1606-1622 [DOI] [PubMed] [Google Scholar]
- 13.Sato H, Takino T, Okada Y, Kao J, Shinagawa A, Yamamoto E, Seiki M: A matrix metalloproteinase expressed on the surface of invasive tumour cells. Nature 1994, 370:61-65 [DOI] [PubMed] [Google Scholar]
- 14.Strongin AY, Collier I, Bannikov G, Marmer BL, Grant GA, Goldberg GL: Mechanism of cell surface activation of 72-kd type IV collagenase: isolation of the activated form of the membrane metalloproteinase. J Biol Chem 1995, 270:5331-5338 [DOI] [PubMed] [Google Scholar]
- 15.Ogata Y, Ito Y, Nagase H: Steps involved in activation of the pro-matrix metalloproteinase 9 (progelatinase B)-tissue inhibitor of metalloproteinases-1 complex by aminophenylmercuric acetate and proteinases. J Biol Chem 1995, 270:1850-18511 [DOI] [PubMed] [Google Scholar]
- 16.Birkedal-Hansen H, Moore WGI, Bodden MK, Windsor LJ, Birkedal-Hansen B, DeCarlo A, Engler JA: Matrix metalloproteinases: a review. Crit Oral Biol Med 1993, 4:197-250 [DOI] [PubMed] [Google Scholar]
- 17.Blackwell TS, Christman JW: The role of nuclear factor-κB gene regulation. Am J Respir Cell Mol Biol 1997, 17:3-9 [DOI] [PubMed] [Google Scholar]
- 18.Schmitz ML, Baeuerle PA: The p65 subunit is responsible for the strong transcription activating potential of NF-κB. EMBO J 1991, 10:3805-3810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheskrkalberman K, Baeuerle PA: Nuclear factor κB: an oxidative stress-response transcription factor of eukaryotic cell. Free Radical Res Commun 1992, 17:221-237 [DOI] [PubMed] [Google Scholar]
- 20.Schmidt KN, Amstad P, Cerutti P, Baeuerle PA: The roles of hydrogen peroxide and superoxide as messengers in the activation of transcription factor NF-κB. Chem Biol 1995, 2:13-22 [DOI] [PubMed] [Google Scholar]
- 21.Sato H, Seiki M: Regulatory mechanism of 92 kd type IV collagenase gene expression which is associated with invasiveness of tumour cells. Oncogene 1993, 8:395-405 [PubMed] [Google Scholar]
- 22.Pardo A, Ridge K, Uhal B, Sznajder JI, Selman M: Lung alveolar epithelial cells synthesize interstitial collagenase and gelatinases A and B in vitro. Int J Biochem Cell Biol 1997, 29:901-910 [DOI] [PubMed] [Google Scholar]
- 23.Dobbs LG, Williams MC, González R: Monoclonal antibodies specific to apical surfaces of rat alveolar type I cells binds to surfaces of cultured, but not freshly isolated, type II cells. Biochim Biophys Acta 1988, 970:146-156 [DOI] [PubMed] [Google Scholar]
- 24.Pardo A, Ramírez R, Gutiérrez L, Mendoza F, Bauer E, Selman M: Purification of a procollagenase-activator present in medium of cultured guinea pig carrageenin granuloma. Connect Tissue Res 1991, 26:259-269 [DOI] [PubMed] [Google Scholar]
- 25.Maldonado V, Meléndez-Zajgla J, Ortega A: Modulation of NF-κB, p53 and Bcl-2 in apoptosis induced by cisplatin in HeLa cells. Mutat Res 1997, 381:67-75 [DOI] [PubMed] [Google Scholar]
- 26.Huhtala P, Tuuttila A, Chow LT, Lohi J, Kesk-Oja J, Tryggvason K: Complete structure of the human gene for 92-kd type IV collagenase. J Biol Chem 1991, 266:16485-16490 [PubMed] [Google Scholar]
- 27.Pardo A, Selman M, Ridge K, Barrios R, Sznajder JI: Increased expression of gelatinases and interstitial collagenase in rat lungs exposed to 100% oxygen. Am J Respir Crit Care Med 1996, 154:1067-1075 [DOI] [PubMed] [Google Scholar]
- 28.Salo T, Lyons JG, Rahemtulla F, Birkedal-Hansen H, Lasjava H: Transforming growth factor-B1 upregulates type IV collagenase expression in cultured human keratinocytes. J Biol Chem 1991, 266:11436-11441 [PubMed] [Google Scholar]
- 29.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N: Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem 1993, 268:10425-10432 [PubMed] [Google Scholar]
- 30.Moore AM, Buch S, Han RNN, Freeman BA, Post M, Tanswell AK: Altered expression of type I collagen, TGF-β1, and related genes in rat lung exposed to 85% O2. Am J Physiol 1995, 268:L78-L84 [DOI] [PubMed] [Google Scholar]
- 31.Horowitz S, Shapiro DL, Finkelstein JN, Notter RH, Johnston CJ, Quible DJ: Changes in gene expression in hyperoxia-induced neonatal lung injury. Am J Physiol 1997, 258:L107-L111 [DOI] [PubMed] [Google Scholar]
- 32.Piedboeuf B, Johnston CJ, Watkins RH, Hudak BB, Lazo JS, Cherian MG, Horowitz S: Increased expression of tissue inhibitor of metalloproteinases (TIMP-1) and metallothionein in murine lungs after hyperoxic exposure. Am J Respir Cell Mol Biol 1994, 10:123-132 [DOI] [PubMed] [Google Scholar]
- 33.Owens MW, Milligan SA, Jourd’Heuil D, Grisham MB: Effects of reactive metabolites of oxygen and nitrogen on gelatinase A activity. Am J Physiol 1997, 273:L445-L450 [DOI] [PubMed] [Google Scholar]
- 34.Mauviel A: Cytokine regulation of metalloproteinase gene expression. J Cell Biochem 1993, 53:288-295 [DOI] [PubMed] [Google Scholar]
- 35.Schreck R, Aldermann K, Baeuerle X: NF-κB: an oxidative stress-responsive transcription factor of eukaryotic cells. Free Radical Res Commun 1992, 7:221-231 [DOI] [PubMed] [Google Scholar]
- 36.Horinouchi H, Wang CC, Shepherd KE, Jones R: TNF α gene and protein expression in alveolar macrophages in acute and chronic hyperoxia-induced lung injury. Am J Respir Cell Mol Biol 1996, 14:548-555 [DOI] [PubMed] [Google Scholar]
- 37.Li Y, Zhang W, Mantell LL, Kazzaz JA, Fein AM, Horowitz S: Nuclear factor-κB is activated by hyperoxia but does not protect from cell death. J Biol Chem 1997, 272:20646-20649 [DOI] [PubMed] [Google Scholar]
- 38.Thompson JE, Phillips RJ, Erdjument-Bromage H, Temkpst P, Ghosh S: IkBb regulates the persistent response in a biphasic activation of NF-κB. Cell 1995, 80:573-582 [DOI] [PubMed] [Google Scholar]
- 39.Sato H, Seiki M: Regulatory mechanism of 92 kd type IV collagenase gene expression. Oncogene 1992, 8:395-405 [PubMed] [Google Scholar]
- 40.Huhtala P, Tuuttila A, Chow LT, Lohi J, Keski-Oja J, Tryggvason K: Complete structure of the human gene for 92 kd type IV collagenase. J Biol Chem 1991, 266:16485-16490 [PubMed] [Google Scholar]
- 41.Fini ME, Bartlett JD, Matsubara M, Rinehart WB, Mody MK, Girard MT, Rainville M: The rabbit gene for 92-kd matrix metalloproteinase. J Biol Chem 1994, 269:28620-28628 [PubMed] [Google Scholar]
- 42.Sato H, Kita M, Seiki M: v-src activates the expression of 92 kd type IV collagenase gene through the AP-1 site and the GT box homologous to retinoblastoma control elements. J Biol Chem 1993, 268:23460-23468 [PubMed] [Google Scholar]
- 43.Bian J, Sun Y: Transcriptional activation by p53 of the human type IV collagenase (gelatinase A or matrix metalloproteinase 2) promoter. Mol Cell Biol 1997, 17:6330-6338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu H, Lozano G: NF-κB activation of p53: a potential mechanism for suppressing cell growth in response to stress. J Biol Chem 1994, 269:20067-20074 [PubMed] [Google Scholar]
- 45.Chen W, Cooper NR: Epstein-Barr virus nuclear antigen 2, and latent membrane protein independently transactivate p53 through induction of NF-κB activity. J Virol 1996, 70:4849-4853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O’Reilly MA, Staversky RJ, Stripp BR, Finkelstein JN: Exposure to hyperoxia induces p53 expression in mouse lung epithelium. Am J Respir Cell Mol Biol 1998, 18:43-50 [DOI] [PubMed] [Google Scholar]
- 47.Shea LM, Beehler C, Schwartz M, Shenkar R, Tuder R, Abraham E: Hyperoxia activates NF-κB and increases TNFα and IFNγ gene expression in mouse pulmonary lymphocytes. J Immunol 1996, 157:3902-3908 [PubMed] [Google Scholar]
- 48.Partridge CA, Jeffrey JJ, Malik AB: A 96-kd gelatinase induced by TNF-α contributes to increased microvascular endothelial permeability. Am J Physiol 1993, 265:L438-L447 [DOI] [PubMed] [Google Scholar]
- 49.Huajanen ES, Vaisanen A, Zheng A, Tryggvason, Turpeenniemi-Hujanen: Modulation of 72,000 and 92000 type-IV collagenase (gelatinase A and B gene expression by interferons α and γ in human melanoma. Int J Cancer 1994, 58:582-586 [DOI] [PubMed] [Google Scholar]
- 50.Selman M, Montaño M, Ramos C, Vanda B, Becerril C, Delgado J, Sansores R, Barrios R, Pardo A: Tobacco smoke-induced lung emphysema in guinea pigs is associated with increased interstitial collagenase. Am J Physiol 1996, 271:L734-L743 [DOI] [PubMed] [Google Scholar]
- 51.Hayashi T, Stetler-Stevenson WG, Fleming MV, Fishback N, Koss MN, Liotta LA, Ferrans VJ, Travis WD: Immunohistochemical study of metalloproteinases and their tissue inhibitors in the lungs of patients with diffuse alveolar damage and idiopathic pulmonary fibrosis. Am J Pathol 1996, 149:1241-1256 [PMC free article] [PubMed] [Google Scholar]
- 52.D’Ortho MP, Clerici C, Yao PM, Delacourt C, Delclaux C, FrancoMontoya ML, Harf A, Lafuma C: Alveolar epithelial cells in vitro produce gelatinases and tissue inhibitor of matrix metalloproteinase-2. Am J Physiol 1997, 273:L663-L675 [DOI] [PubMed] [Google Scholar]
- 53.Ricou B, Nicod L, Lacraz S, Welgus H, Sutter P, Dayer JM: Matrix metalloproteinases and TIMP in acute respiratory distress syndrome. Am J Respir Crit Care Med 1996, 154:346-352 [DOI] [PubMed] [Google Scholar]
- 54.Tori K, Iida KI, Miyasaki Y, Saga S, Kondoh Y, Tanigushi H, Taki F, Takagi K, Matsuyama M, Susuki R: Higher concentrations of matrix metalloproteinases in bronchoalveolar lavage fluid of patients with adult respiratory distress syndrome. Am J Respir Crit Care Med 1997, 155:43-46 [DOI] [PubMed] [Google Scholar]
- 55.Delclaux C, D’Ortho MP, Delacourt C, Lebargy F, Brun-Buisson C, Brochard L, Lemaire F, Lafuma C, Harf A: Gelatinases in epithelial lining fluid of patients with adult respiratory distress syndrome. Am J Physiol 1999, 272:L442-L451 [DOI] [PubMed] [Google Scholar]
- 56.Fukuda Y, Ishizaki M, Masuda Y, Kimura G, Kawanami O, Masugi Y: The role of intraalveolar fibrosis in the process of pulmonary structural remodeling in patients with diffuse alveolar damage. Am J Pathol 1987, 126:171-182 [PMC free article] [PubMed] [Google Scholar]
- 57.Usuki J, Fukuda Y: Evolution of three patterns of intra-alveolar fibrosis produced by bleomycin in rats. Pathol Int 1995, 45:552-564 [DOI] [PubMed] [Google Scholar]