

Abstract
Nephrogenic rests are precursor lesions associated with about 40% of Wilms’ tumors. This study identifies genetic steps occurring in the development of Wilms’ tumor. Thirty-four Wilms’ tumors with nephrogenic rests and/or areas of anaplasia were microdissected from paraffin sections to determine whether and at what stage loss of heterozygosity (LOH) occurred, using polymerase chain reaction-based polymorphic markers at 11p13, 11p15, and 16q. LOH at these loci have been identified in Wilms’ tumors and are associated with identified or putative tumor suppressor genes. Three cystic nephromas/cystic partially differentiated nephroblastomas were also examined. LOH was detected in six cases at 11p13 and in six cases at 11p15, and two of these cases had LOH at both loci. All intralobar rests showing LOH also showed LOH in the tumor. A case with a small perilobar rest showed LOH of 11p13 only in the tumor. Five cases showing LOH at 16q were identified (this was identified only in the tumor, and not in the associated rest), and three of these had recurrence of the tumor. Two cases had a WT1 mutation (one germline and the other somatic), as well as LOH in both the intralobar rest and the tumor. A cystic partially differentiated nephroblastoma showed loss at 11p13 and 11p15, as well as at 16q. This study suggests that LOH at 11p13 and 11p15 and WT1 mutations are early events but that LOH at 16q occurs late in the pathogenesis of Wilms’ tumor. Intralobar and perilobar nephrogenic rests are known to have different biological behaviors, and this study suggests that they are genetically different. A multistep model of Wilms’ tumor pathogenesis is supported by these findings.
Wilms’ tumor is a developmental tumor of the kidney and is one of the commonest solid tumors of childhood, usually presenting between the ages of 1 and 5 years old. 1 Nephrogenic rests are associated with 40% or so of Wilms’ tumors, adjacent to or separate from the tumor. They appear to be precursor lesions, 2,3 analagous to colonic adenomatous polyps.
The kidney develops after an interaction between the branching ureteric bud and blastema. Glomeruli develop from the blastemal rind around the lobes of the kidney, and the blastema disappears when nephrogenesis ceases around 36 weeks’ gestation. 4 Nephrogenic rests appear to develop from persisting blastema 3 and consist of blastema and/or epithelial structures and variable amounts of stroma. There are two types of nephrogenic rests: the perilobar and the intralobar types. 3 The perilobar type is located at the edge of the renal lobes and is thought to arise relatively late in renal development (Figure 1A) ▶ . The intralobar rests are sited within the lobe, indicating an origin earlier in development (Figure 1B) ▶ . Perilobar rests may be seen in up to 0.87% of carefully examined infants at postmortem. 3 As rests are rarely seen in adults, most nephrogenic rests must involute with age. Intralobar rests are rarely seen as an isolated finding at postmortem examination in infants. The two types of rest appear to have a different malignant potential. Few of the perilobar rests progress to Wilms’ tumor, whereas progression appears more frequent in intralobar rests. 3 Nephrogenic rests may show areas of hyperplasia and increased cellularity, suggesting clonal progression. 3 The absence of rests in some tumors is explained by the tumor overgrowing the precursor lesion. The presence of a rest separate from the tumor suggests a germline defect or an early embryonic somatic mutation. 5,6 Perilobar nephrogenic rests may also be seen associated with cystic renal dysplasia 7 and congenital mesoblastic nephroma, 8 suggesting that disruption of nephrogenesis may also cause the blastema to persist. Areas within Wilms’ tumors may resemble related benign tumors such as cystic nephroma (CN) or cystic partially differentiated nephroblastoma (CPDN), and these areas may also represent precursor lesions. 9,10 CN and CPDN are closely related to one another, differing by the presence of small amounts of blastema in the latter. CPDN is distinguished from cystic Wilms’ tumors only by the presence of macroscopic nodules of blastema. Five percent of Wilms’ tumors may contain diffuse or focal areas of anaplasia, 11-13 which is associated with p53 mutations. 14,15 Renal cell carcinomas may arise in or follow the diagnosis of Wilms’ tumors, suggesting another possible progression in some tumors. 16,17
Figure 1.
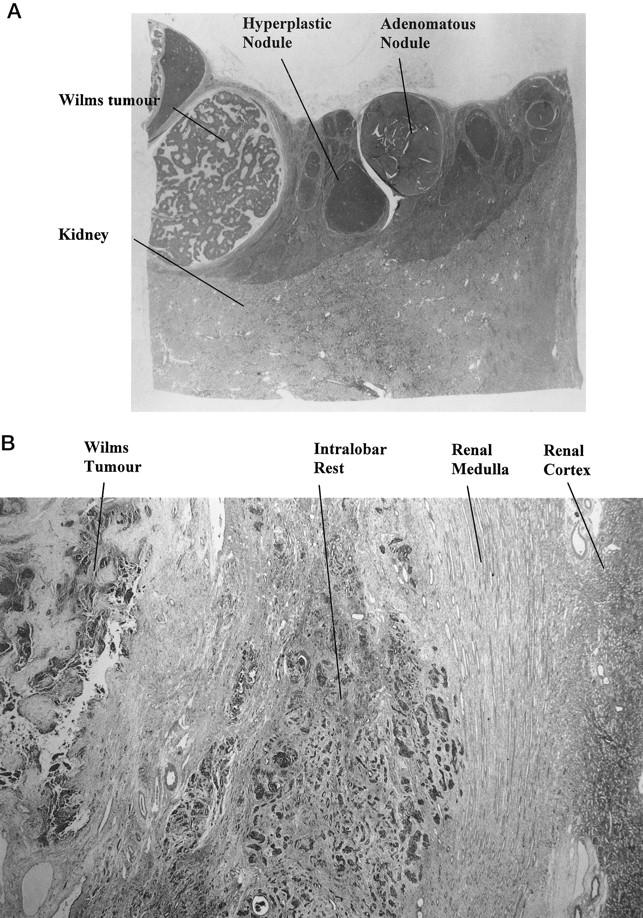
A (case B36): Low power view of renal cortex, with a rind of perilobar nephroblastomatosis involving the upper half of the tissue. There are several hyperplastic nodules, an adenomatous nodule, and a small Wilms’ tumor (8 mm in diameter) with serpiginous blastema and pale stroma on the left. B (case B9): Low power view of intralobar rest in the middle of the field. The rest has a diffuse infiltrative pattern and is sited in the renal medulla. Renal cortex appears in the far right, and Wilms’ tumor in the upper left.
Knudson and Strong 18 suggested that a two-hit genetic pathway may be occurring in Wilms’ tumors. However, familial Wilms’ tumors are much less common than this model would suggest, and a more complicated pathway seems likely. Wilms’ tumors have an age:incidence curve, suggesting that two separate biological mechanisms are occurring. The increasing incidence from infancy until the peak incidence at 3 to 4 years represents the increased chance of accruing the necessary mutations to develop a tumor with passing time. The declining number of cases after this age is due to the target cells maturing or dying before acquiring the necessary mutations for malignancy to develop.
Wilms’ tumor is genetically heterogeneous, with at least four separate candidate “tumor suppressor genes,” presumably involved early in the pathogenesis of Wilms’ tumors: 19 the WT1 gene at 11p13, 20,21 a probable gene (or genes) at 11p15 22 (WT2; see later), another at 7p15, 23 and several (at least one at 17q24) associated with familial Wilms’ tumors. 25-27 The WT1 gene product is a zinc finger protein particularly involved in the development of the urogenital tract 28 and is expressed in many Wilms’ tumors, 29 suggesting a more complex role than just that of a tumor suppressor function. 30,31 Mutation of the WT1 gene itself is only demonstrated in 10% of Wilms’ tumors 32 (usually stroma rich). 32,33 WT2 at 11p15 has not been characterized. 19 Of sporadic tumors, 10 to 25% show abnormalities of chromosome 16q, 34 and these tumors appear to have a higher rate of recurrence. 35,36 The younger age of presentation in boys than in girls suggests a role for the sex chromosomes. 37 Wilms’ tumors have also been associated with trisomy 18 38 and a somatic overgrowth syndrome involving a gene at Xp26. 39
Patients with Wilms’ tumor syndromes 40 tend to have different types of rests. 3 Intralobar rests are associated with germline mutations involving the WT1 gene at 11p13, such as the Denys-Drash (Wilms’ tumors, genitourinary abnormalities, and glomerulosclerosis) 41,42 and WAGR (for Wilms’ tumor, aniridia, genitourinary abnormalities, and mental retardation) syndromes. Perilobar rests are associated with hemihypertrophy and the Beckwith-Wiedemann syndrome, 2 which involves either a loss of imprinting at 11p15 or uniparental (paternal) disomy. 43
This is the first large-scale study to try to identify at what morphological stage loss of heterozygosity (LOH) occurs in Wilms’ tumor progression. We used microdissected areas from paraffin sections, and polymerase chain reaction (PCR) techniques, to detect LOH using polymorphic markers located at 11p13, 11p15, and 16q. We show that LOH at 11p13 and 11p15 appears to be an early event, being present in all nephrogenic rests apart from one perilobar rest. However, 16q LOH was confined to tumors and therefore appears to be a late event.
Materials and Methods
Tumors
All renal tumors received at the Royal Hospital for Sick Children (Bristol, UK) between 1980 and 1997 were reviewed. One hundred five Wilms’ tumors were identified. An additional 34 Wilms’ tumors were obtained from the files of the Cardiff Royal Infirmary, giving 139 tumors in all. Wilms’ tumors showing evidence of nephrogenic rests, focal anaplasia, or CN-like areas within the tumor were identified. Those cases with adequate material for analysis were examined for LOH with PCR techniques. The tumors were examined by AKC, and anaplasia, perilobar, and intralobar rests and CN-like areas were identified in 49 cases. Three cases of CN/CPDN were also included. These cases were reviewed by two observers (AKC and PJB), and agreement on classification was reached.
Microdissection
Of the cases identified above, 37 were selected for microdissection. The cases not examined included referred cases for which there was no material or for which no rests were identified in the recut sections. Material was selected from the Wilms’ tumor or from any separate tumor if more than one tumor was present from areas of anaplasia or CN-like areas within the Wilms’ tumor, from any rest, and from different areas within the rest showing morphological changes. Up to eight areas were microdissected, although often only three or four areas were selected. Sections (5 μm) on slides were dewaxed by heating to 60°C for 30 minutes followed by three 5-minute washes in xylene and then three 5-minute washes in 100% ethanol. After air drying, the slides were lightly stained with nuclear red (0.1% in 5% aluminum sulfate). The slides were then placed on a dissecting microscope stage. Buffer (100 μl; 20 mmol/L Tris-HCl, 1 mmol/L ethylenediaminetetraacetic acid, pH 8.6, and 0.05% Tween 20) was placed in a sterile Eppendorf tube, and 10 to 20 μl of this was pipetted on the slide over the area of interest with a micropipette. The pipette tip was scraped over the target areas (usually 1 to 2 mm in diameter), and the cells were then aspirated into the pipette tip and placed in a 0.5-ml Eppendorf tube. Adjacent kidney was used as control. This left the slides available for examination and confirmation of the areas taken. Proteinase K (5 μl of 20 mg/ml concentration) in water was added, and the Eppendorf tube was agitated and placed in a heated block at 55°C overnight. The proteinase K was inactivated by heating the tube in a boiling water bath for 5 minutes. The tube was centrifuged briefly and the supernatant used for the PCR reaction.
PCR
PCR was performed on all of the rests and tumors using two pairs of primers for 11p13, one pair for 11p15, one pair for 16q, and two pairs for 7p15 as listed in Table 1 ▶ . The mix consisted of 2.0 μl of 10× Supertaq buffer (HT Biotechnology, Cambridge UK; 10× = 100 mmol/L Tris, 15 mmol/L MgCl2, 500 mmol/L KCl, 1% Triton X-100, and 0.1% w/v gelatin, pH 9.0), 2.5 μl of deoxynucleotide triphosphate mix (2 mmol/L each deoxynucleotide triphosphate), 1.0 μl of each primer (25 μmol/L stock), 8.5 μl of H2O, and 5 μl of sample for each tube, covered by 50 μl of mineral oil. A manual hot start technique was used with an initial denaturing step of 5 minutes at 94°C. Five μl of warmed Taq mix (consisting of 0.5 μl of 10× Supertaq buffer, 4.5 μl of H2O, and 0.04 U Supertaq polymerase (HT Biotechnology) per tube) was added at the annealing temperature. A negative (no DNA template) control was used for each run, and a control using the proteinase K solution and the Tris buffer confirmed that there was no contamination.
Table 1.
PCR Details
| Locus | Primer sequences (5′–3′) | PCR conditions (all 45 cycles, hot start) | Reference |
|---|---|---|---|
| 11p13 (a), WT1 dinucleotide repeat | AATGAGACTTACTGGGTGAGG, TTACACAGTAATTTCAAGCAACGG | 94°C (1 minute), 55°C (1 minute, 30 seconds), 72°C (2 minutes) | Haber et al 57 |
| 11p13 (b), D11S192, tetranucleotide repeat | TTGCATCCATACGGAAAGTC, ACATCTGAGACTTGTAGTAGAAGGC | 94°C (1 minute), 55°C (1 minute, 30 seconds), 72°C (2 minutes) | GDB no. G00-228-905 |
| 11p15 (T), TH, tetranucleotide repeat | GGGTATCTGGGCTCTGGGGT, GGTCACAGGGAACACAGACTCC | 94°C (1 minute), 60°C (1 minute, 30 seconds), 72°C (2 minutes) | Hearne et al 58 |
| 11p15 (G), GATA23FO6 | TACATGGCAGCAGGCATATA, GAGTAAACAAGATTGCTAGATAGGC | 94°C (1 minute), 55°C (1 minute), 72°C (2 minutes) | GDB no. GATA23F06 |
| 16q D16S265, dinucleotide repeat | CCAGACATGGCAGTCTCTA, AGTCCTCTGTGCACTTTGT | 94°C (1 minute), 55°C (1 minute, 30 seconds), 72°C (2 minutes) | Weber et al 59 |
| WT1, exon 7 | GACCTACGTGAATGTTCACATG, CTCTTGAACCATGTTTGCCCAAGAC | 94°C (1 minute), 60°C (1 minute), 72°C (1 minute) | Baird et al 60 |
| WT1, exon 6 | CAGAACCCTGCATCTAAAGTG, CCCAAAGAGTCCATCAGTAA | 94°C (1 minute), 55°C (1 minute), 72°C (1 minute) | Brown et al, unpublished |
GDB, GenBank database.
After PCR (conditions given in Table 1 ▶ ), 10 μl of product was run on a 6% nondenaturing acrylamide gel except for the 16q products, which were run on a 2% Nusieve, 1% agarose gel with a 100-bp marker. The products were visualized by ultraviolet transillumination after ethidium bromide staining. The CA repeat sequences were examined on a midigel (20 cm); the tetrameric repeats were examined initially on a minigel (10 cm) but were checked on a midigel. The gels were examined to see that the different cases generally showed different allelic sizes and that these concurred with the expected size of the alleles. In cases in which there appeared to be only one allele in the normal kidney, the products were loaded onto a 30-cm 8% acrylamide gel for improved resolution. Runs in which the control showed contamination were repeated, and if necessary the specimen was redissected from the paraffin sections. Positive results were repeated at least once, and unsuccessful amplifications were repeated to try to obtain a result.
Anticontamination measures were used. The microdissection was performed in a separate laboratory, where no PCR has been performed. The PCR master mix and the Supertaq solution were prepared in a separate room from where the PCR products were opened. The PCR master mix and the Supertaq solution were pretreated with 5 minutes of ultraviolet light. The PCR products were run out and kept in a separate room from the preparation and the PCR area. Stock solutions were prepared in a laminar flow cabinet.
WT1 Mutation
Examination of the tumor, rest, and kidney of one case previously found to have a mutation of WT1 exon 7 in the tumor, and another case with a WT1 germline mutation in exon 6 (Miyagawa et al, unpublished results) was carried out using similar methods using the primers and conditions shown in Table 1 ▶ .
Results
Forty-nine Wilms’ tumors showed histological evidence of clonal progression with either nephrogenic rests, areas of CN, or anaplasia. The frequency of rests is similar to those found in previous studies. Nephrogenic rests were seen in 42 cases, 22 perilobar, 17 intralobar, and an additional 3 cases with both types of rest (Figure 1, A and B) ▶ . Three Wilms’ tumors had CN-like areas. There were 7 cases with anaplasia; 2 of these cases had diffuse anaplasia. Multifocal disease was seen in 26 cases, 9 cases with bilateral lesions and 9 cases with lesions confined to one kidney. In the remaining cases there was no information about the other kidney.
Clinical Details of Tumors
One case (B19) was a male with bilateral tumors, an intralobar rest, and a history of undescended testes and hypospadias. This patient had a germline 8-bp deletion in exon 6 of WT1 (Brown, unpublished results). Two patients (B6 and B9) had aniridia with intralobar rests. One case (B25) had Beckwith-Wiedemann syndrome with intralobar rests, a few subcapsular scars (probable involuted rests), and focal anaplasia. One child (B12) had hemihypertrophy and an intralobar rest, and another (B13) had trisomy 18 and perilobar nephroblastomatosis. Case B15 with bilateral radial aplasia, small perilobar nephrogenic rests, and a constitutional translocation t(1;7)(q42;p15) has been reported previously. 23
PCR
The results are discussed below; representative results are shown in Figure 2 ▶ and summarized in Table 2 ▶ . Overall, the technique was successful in about 80% of the samples. A few remained refractory, although good results were obtained from paraffin blocks up to 16 years old. LOH at one or more loci examined was found in 13 of the 37 patients (35%). All of the rests and tumors were examined, as sequential examination of the rest after the tumor would have introduced a bias in the results. LOH at 11p was seen in 10 of the 36 informative cases (27%). In no case was a loss found in the rest and not the tumor.
Figure 2.

a (case B14): 11p15 tetranucleotide repeat. Shown is LOH in the intralobar rest (R), as well as in the tumor (T), compared with that in the kidney (K). The heteroduplexes are clearly seen. A1, larger allele; A2, smaller allele; H, heteroduplexes. b (case B14): Acrylamide gel with products of WT1 exon 7 PCR. The tumor (T) and the rest (R) produced only a single product smaller than the product from the normal kidney (K). This demonstrates loss of both normal alleles, and that there is a mutation present in both the rest and the tumor producing the smaller product. N, normal allele; M, mutant allele. c (case B19): Acrylamide gel showing the 8-bp deletion in the normal tissue (K), rest (R), and both the left and right (bilateral) tumors (LT and RT, respectively). There is clearly loss of the normal allele in the rest and the tumors. N, normal allele; M, mutant allele. d (case B38): 16q dinucleotide repeat; agarose gel. Two alleles are clearly seen in the kidney (K), both perilobar rests (R1 and R2), and one tumor (T1). The other tumor (T2) showed LOH. M, 100 bp marker. e (case B32): 16q dinucleotide repeat. Agarose gel showing two alleles (A1 and A2) in the kidney (K) and in the dormant (D) and hyperplastic (H) rests, and only one (A2) allele in one area of the tumor (Ta). M, 100 bp marker. f (case B32): 16q dinucleotide repeat. Repeat of the PCR reaction as shown in e. Other areas of the tumor (Tb, Tc, and Td) show loss of the smaller product, as does the brain metastasis (B).
Table 2.
Cases Examined with LOH Results or Other Genetic Information
| Case | Age (years) | Ana- plasia | Multi- focal* | Syndromic | Area examined† | p13a‡ | p13b‡ | p15T‡ | p15G‡ | 16q‡ | WT1 | Cytogenetics (of tumors unless shown); other notes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Perilobar rest | ||||||||||||
| B1 | 2 | No | Yes | K, Rpl, T | N | N | N | |||||
| B2 | 4 | No | Yes | K, Rpl, T | N | N | N | |||||
| B5 | 4 | No | Yes | K, Rpl, T | N | N | N | |||||
| B10 | 3 | No | Yes | K, Rpl, T | N | N | N | N | ||||
| B13 | 2 | No | Yes | Trisomy 18 constitutional | ||||||||
| B15 | 5 | No | Yes | Radial aplasia | 1q, 7p translocation (de novo constitutional) | |||||||
| B16 | No | Yes | K, Rpl | X | N | N | Hyperplastic rest only | |||||
| B20 | 4.8 | No | Yes | K, Rhy, T | N | N | N | Treated | ||||
| B31 | 4 | No | Yes | K, Rpl, T | N | Y(T) | N | Y(T) | ||||
| B32 | 4.75 | No | Yes | K, Ri, Rh, Ta–d, M | N | N | Y(T) | See Figure 2F ▶ | ||||
| B38 | 5 | No | Yes | K, Rl, Rr, Tl, Tr | N | NI | N | Y(T) | Bilateral tumors | |||
| B40 | 4 | No | Yes | K, Rpl, T | N | NI | NI | N | ||||
| C3 | No | No | K, Rpl, T | N | N | N | ||||||
| B36 | 3 | Focal | Yes | K, Rpl, Rad, Tt, T(1), T(2) | N | NI | N | NI | ||||
| B37 | 4 | Diffuse | Yes | K, Rpl, Tr, Tl | N | N | NI | Left: 44–48 XX,+6, t(7:21)(q22;q22),+8, del(11)(q23), add (12)(q2),+13,−16, add(17)(p) Right: 44–50, XX, add(4)(q35),−10,+13,−16, +der(16)t(1;16)(q21;q12),−17,+18 | ||||
| B41 | 4 | Diffuse | Yes | L/N, K, T(an) | X | X | N | N | Tiny perilobar rests | |||
| Both peri- and intralobar rests | ||||||||||||
| B34 | 3 | No | Yes | K, Rpl, Ril, T | N | N | N | 46 XY: add 1(q1), add 3(p13),−7, −16+mar, | ||||
| B25 | 16 | Focal | Yes | BWS | K, Ril, T, T(an)§ | N | NI | X | N | 48 XX: 8,i10, q10, 12; constitutional uniparental disomy at 11p15 (Brown, unpublished) | ||
| Intralobar rest | ||||||||||||
| B4 | 15 | No | No | K, Ril, T | N | N | N | N | ||||
| B6 | 1 | No | Yes | Aniridia | K, R(1), R(2), T(1), T(2) | N | N | N | N | 11p, constitutional | ||
| B7 | 14 | No | No | K, Ril, T | N | Y | NI | N | N | |||
| B9 | 1.67 | No | No | Aniridia | K, Ril, T | N | N | N | N | |||
| B12 | 4 | No | No | Hemihyper- trophy | K, Ril, CNL, T | Y | Y | NI | NI | NI | ||
| B14 | 1 | No | No | K, Ril, T | N | Y | N | Exon 7 | Exon 7 mutation in rest and tumor (not germline) | |||
| B17 | 0.84 | No | No | K, Ril, T | N | N | N | X | No growth previous 48 XX+13; no 11p changes | |||
| B18 | 1 | No | No | XY, 6, 7, 13, m1, m2/i, 2, 6, 7, 12, 13, 20 | ||||||||
| B19 | 1 | No | Yes | Abnormal genital | K, Ril, T(1), T(2) | N | NI | Y | N | Exon 6 | Germline 8-bp deletion in exon 6 | |
| B22 | 1 | No | No | K, Ril, T | N | NI | N | N | ||||
| B24 | 1 | No | No | 46 XX | ||||||||
| B27 | 2 | No | No | K, Ril, CNL, T | N | N | Y(T) | Complex cytogenetics, with trisomy for chromosome 16 | ||||
| B30 | 2 | No | Yes | K, Ril T(1), T(2) | X | Y | NI | |||||
| B33 | 4.5 | No | No | K, Ril, T | N | N | N | 52 XX+del(1)(p13),+2, i(7)(q10), +10, add | ||||
| B23 | 5 | Yes | Yes | K, Ril, T(1), T(2)(an) | N | N | NI | NI | NI | Separate smaller tumor T2, anaplastic | ||
| Cystic nephromalike areas | ||||||||||||
| B8 | 4 | No | No | K, CNL, T | N | Y | N | 46 XY | ||||
| B21 | 2 | No | No | K, CNL, T | Y | NI | Y | NI | ||||
| B28 | 1 | No | No | K, CNL, T | N | N | NI | NI | NI | |||
| Cystic nephroma/cystic partially differentiated nephroblastoma | ||||||||||||
| B42 | 6 mo. | No | No | K, CN | N | N | NI | |||||
| B43 | 1 | No | No | K, CPDN | Y | Y | Y | 52 XY,+2, 5, 7, 8, 17, 18 | ||||
| B44 | 1 | No | No | K, CN | N | N | N | 46 XY | ||||
| Anaplasia alone | ||||||||||||
| B29 | 4 | Focal | No | K, T, T(an) | N | N | N | N | 46 XX, 47 XX+8 | |||
| C8 | Focal | No | K, T, T(an) | Y | N | X |
*Multifocal, at least two separate lesions present, ie, tumor and separate rest or two tumors with an adjacent rest.
†K, kidney; R, rest (the type according to the grouping above); Rpl, perilobar; Ril, intralobar; R(1), associated with tumor T(1); Ri, involuting rest; Rad, adenomatous rest; Rh, hyperplastic rest, Rr, rest from right kidney; Rl, rest from left kidney; CNL, cystic nephroma-like area in a Wilms’ tumor; T, Wilms’ tumor; T(1) and T(2), separate Wilms’ tumors; Tl and Tr, tumors from the left or right kidney in bilateral cases; Ta–d, separate areas of main tumor microdissected; Tt, microscopic Wilms’ tumor (tumourlet; see Figure 1A ▶ ); T(an), anaplastic Wilms’ tumor.
‡Y, LOH in rest and tumor; Y(T), LOH in tumor only; N, no LOH; NI, noninformative; X, incomplete PCR result obtained, despite repeats.
§Case B25 had small hypocellular subcortical scars, presumed to be sclerosed perilobar rests.
11p15
LOH at 11p15 was found in 6 out of 32 informative cases (19%) (a representative example is shown in Figure 2A ▶ ). In all cases, LOH was observed in both the rest or cystic area and the tumors, including case B19 with bilateral tumors and an intralobar rest (and the WT1 mutation described above), and case B30, in which both tumors and the rest showed the loss. The other cases included one with intralobar rests, two with CN-like areas, and one CPDN. Two cases showed loss at 11p13 as well as 11p15; the other three showed loss only at 11p15. One case was noninformative at 11p13; in another, the 11p13 would not produce a result.
11p13
Homozygous WT1 mutation or LOH at 11p13 was found in 8 of 34 informative cases (24%) with one or another primer pair (also at 11p15 in 4 cases and noninformative at 11p15 in 1 case). In one patient (B31) with a perilobar rest, the loss was seen in the tumor only and not in the rest. Although this rest was small and the negative result could be an artifact caused by contamination with normal tissue, this is unlikely, as it did not occur with the intralobar rests of similar size and cellularity. Two cases (B7 and B12) had intralobar rests and showed LOH in the tumor and the rest. Both of these were noninformative for the 11p15 locus, so it was not possible to tell whether LOH extended to 11p15. Another case with 11p15 loss had a CN area (B21). One case (C8) showed LOH in the areas of focal anaplasia, as well as the Wilms’ tumor; this case had no rests.
WT1 mutation
One case (B14) previously demonstrated to have a WT1 mutation with a 5-bp deletion in exon 7 (Miyagawa et al, unpublished results), was shown to have the same mutation in the rest and the tumor (Figure 2B) ▶ . Another case (B19) was shown to have a heterozygous germline 8-bp deletion in exon 6 (Figure 2C) ▶ . Both cases showed loss of the normal allele in the tumor and the rest and also showed LOH at 11p15 in the rest and the tumor, indicating that two hits had occurred at the intralobar rest stage.
16q
Loss was established in 4 of 23 informative cases (17%). In all cases, LOH was only seen in the tumors and not in the associated rests. One case (B31) with a perilobar rest had 11p13 loss in the tumor (but not the rest) also had LOH at 16q only in the tumor. In one case (B27), the loss was seen in the main tumor, but not in a CN-like area. Karyotype of this tumor was complex but showed trisomy of chromosome 16. The patient had a recurrence in the abdominal scar. Case B38 had multifocal perilobar rests and several tumors. There was LOH at 16q only in one of the tumors (Figure 2D) ▶ . There has been an early local tumor recurrence. Case B32 presented with bilateral tumors and lung metastases, and after chemotherapy he had bilateral partial nephrectomies. One Wilms’ tumor showed a good response to chemotherapy, with no residual viable tumor. The other tumor showed extensive viable tumor with an adjacent perilobar nephrogenic rest, containing foci of hyperplasia. From one slide, normal kidney, dormant nephrogenic rest, hyperplastic rest, and Wilms’ tumor were sampled. LOH for 16q was seen in the Wilms’ tumor but not in the rests (Figure 2, E and F) ▶ . Subsequently, this patient developed a brain metastasis, confirmed histologically, and this also showed only a single 16q allele, but this was the other allele from that identified in the tumor. Microdissecting both from the original slide and from other slides from the same renal tumor showed that different areas within this tumor had lost different 16q alleles. In view of the surprising nature of these results, the whole process including microdissection was rechecked, with identical results. The CPDN also showed a loss at 16q. This case has now developed a contralateral tumor with widespread metastases, but no biopsy material has been taken.
Discussion
This study examined whether a sequential process in the development of Wilms’ tumors could be demonstrated. We have correlated genetic events and morphology in a much larger series of Wilms’ tumors and nephrogenic rests than reported previously. 44,45 The results presented in this paper include several new observations supporting a multistep model in Wilms’ tumor. We have demonstrated that if LOH is seen in a rest, it is also present in the associated Wilms’ tumor, suggesting that the rest does represent a precursor lesion. We have also shown that LOH at 11p is seen in intralobar rests and CN-like areas within Wilms’ tumors, as well as their associated Wilms’ tumors. However, one case with a Wilms’ tumor showing LOH at 11p and having a perilobar rest showed no LOH in the perilobar rest. LOH for 16q demonstrated in four tumors was seen only in the tumor and not the associated rests. In one case, there appears to be loss of different 16q alleles in different areas of the tumor. A previous study by Park et al 44 reported a homozygous WT1 mutation in an intralobar nephrogenic rest and a heterozygous mutation in a hyperplastic perilobar rest. Nordenskjold et al 45 reported a single case of 11p LOH in a perilobar rest. Cui et al 46 have also demonstrated inactivation of H19 in perilobar rests. These cases, together with our findings, support the view that 11p events occur early in Wilms’ tumor development. In a recent study of 46 Wilms’ tumors, using comparative genomic hybridization and LOH, Steenman et al 47 examined six cases with nephrogenic rests, although the type of rest was not clearly identified. Some genetic abnormalities were shared, although more changes were seen in the tumors. 47
Recent molecular and epidemiological studies have demonstrated that the pathogenesis of Wilms’ tumors is complex and that different Wilms’ tumors have different genetic changes. 48 Wilms’ tumors presumably arise from persistent blastema, and the early steps are probably related to failure of the blastema to disappear resulting in nephrogenic rests, which then accrue further genetic changes and become malignant. The frequent association of nephrogenic rests (which may show hyperplastic or adenomatous foci) with Wilms’ tumors suggests multistep genetic progression, 2 analagous to the Fearon-Vogelstein model of colonic carcinoma. 49 As only some Wilms’ tumors show nephrogenic rests, there may be different pathogenetic routes, some involving only a two-step process and others involving several steps. The four or five stages seen in one case (case B36; see Figure 1 ▶ ), suggests a high mutation rate and, perhaps, that an early genetic event increases the rate of further mutations. There is some evidence that loss of both alleles is not a random event 50 and that a mutation of one allele may be transferred to the other allele during mitosis. 5
The number of steps may be different for different syndromes. The age of children presenting with Wilms’ tumors in patients with hemihypertrophy is the same as for sporadic Wilms’ tumors, 37 suggesting that the number of acquired mutations is the same in this syndrome as in the sporadic cases. The increased risk of tumors in hemihypertrophy and the fact that children with sporadic Wilms’ tumors tend to be heavier at birth than controls suggest a nonspecific growth promoter effect, and increased numbers of target cells. 51 The fact that WAGR and Beckwith-Wiedemann patients present with their tumors at a younger age suggests that the germline mutation is the first step in the oncogenetic pathway in these tumors. 52
Wilms’ tumors appear to be genetically heterogeneous. Some Wilms’ tumors show no apparent cytogenetic abnormality, suggesting that changes may be subtle, including alterations of imprinting and small mutations. WT1 is mutated in only 10 to 20% or so of Wilms’ tumors, but LOH of the short arm of chromosome 11 occurs in 30 to 40% of tumors. 53 LOH at 11p may be found in apparently normal kidney adjacent to a Wilms’ tumor, suggesting that LOH alone is not sufficient for a tumor to develop. 5 The role ofWT1 in renal development and maturation of blastemal cells suggests that abnormalities of this gene may be involved in the persistence of blastema and, hence, have a role in the early pathogenesis of Wilms’ tumors. Other genes, including the putative WT2 gene and H19 at 11p15, a gene at 17q (involved in some familial tumors), and the gene at 7p15, are likely to be involved in the pathogenesis of different Wilms’ tumors. IGF2 (located at 11p15) is overexpressed in the Beckwith-Wiedemann syndrome and in some Wilms’ tumors as a result of loss of imprinting 54 and is expressed in blastema of the developing kidney, suggesting a role in promoting blastema. The p57KIP2 is also located at 11p15, and p57 knockout mice may be a partial mouse model of Beckwith-Wiedemann syndrome. 55 At the molecular level, the various gene products may be involved in a single developmental or pathogenetic pathway.
The results presented in this paper add further support for a multistep model. In all cases in which intralobar rests showed LOH of 11p13 or 11p15, the corresponding Wilms’ tumor lost the same allele. This confirms both that intralobar rests are related to the tumor and that both alleles of a tumor suppressor gene are inactivated before the development of an intralobar rest. Evidence supporting inactivation of both alleles of WT1 in the intralobar rest is demonstrated in case B14, in which both the mutation and LOH were demonstrated, and in case B19 with the germline WT1 mutation. This was also demonstrated by Park et al 44 in their finding of an intralobar rest with a homozygous WT1 mutation. These results imply that a further genetic event is required for the Wilms’ tumor to develop from the intralobar rest.
The tumor with the perilobar rest showing loss of 11p13 (and 16q) in the tumor only suggests that only one hit may be required for a perilobar rest to develop, and this is also supported by the findings of Cui et al. 46 Although a perilobar rest with a heterozygous WT1 mutation was described by Park et al, 44 this was a hyperplastic rest and therefore morphologically had already progressed from a dormant perilobar rest. Nordenskjold et al 45 reported 11p LOH in a perilobar rest, but the exact histology was not discussed. It is possible that mutations are not always required, as perilobar rests may be seen to be associated with cystic renal dysplasia and congenital mesoblastic nephromas, suggesting that a physical alteration of the blastemal environment alone may sometimes lead to persistence of the blastema beyond 36 weeks’ gestation. 7,8,56
There is clearly a difference between the two types of nephrogenic rests in view of their different biological behavior. It seems likely that the perilobar rests have not progressed as far along the pathogenetic pathway to Wilms’ tumors as intralobar rests. The fewer cases of LOH of 11p seen in the perilobar (1 of 12) compared with the intralobar rests (5 of 14) also suggests that the pathogenesis of the two rests is different. The fewer cases of LOH seen between kidney and Wilms’ tumor associated with perilobar rest compared with the intralobar rests could also be due to the apparently normal kidney showing LOH, as demonstrated by Chao et al. 5 We may have detected further cases in which the LOH involved a small area of 11p15 using additional markers. Perilobar rests may arise after loss of imprinting 54 rather than LOH at 11p.
Whether the CN-like areas of Wilms’ tumor represent a residual precursor lesion is not clear, but the bland cytology of the cells, often with well developed glomeruloid areas, suggest that they are. The fact that case B7 had extensive intralobar rest with cystic areas suggests a close relationship between intralobar rest and these cystic nephromatous areas. The LOH seen in this area in cases B7 and B21 shows that they are closely related to Wilms’ tumor and intralobar rests. This is also suggested by the CPDN in case B43. A similar conclusion has been reported previously. 9
The results in this paper suggest that the genetic changes at 16q are a late event, although others have suggested it may be an early change. 34 An informative tumor was case B32, which was demonstrated to have a 16q loss, and this patient subsequently developed a brain metastasis. The identification of different clones within the tumor showing loss of different alleles is strong evidence that this is a late event. Failure to find parts of the tumor showing both alleles may be explained by previous chemotherapy destroying those tumor cells. Another case with multiple rests and tumors showed no loss in the rests or one of the tumors, but loss in one other tumor also suggests a late event (B38). The tumors showing LOH at 16q did not show anaplasia. Three of these tumors have recurred. 35 LOH for 16q and the genetic changes underlying anaplasia (possibly p53 mutations) are separate events. Grundy et al 36 suggested that LOH for 16q is a late event, as they found only partial LOH for 16q in several of their tumors.
The similar genetic changes seen in this study and others suggests that there is a close relationship between the rests and the tumors. Two models are possible. One would be that an early stem cell with a mutation then disseminated daughter cells throughout the kidney, giving rise to rests and tumors. Another model, already proposed by Beckwith 3 and others, 2 is of a progression from rests to tumors (Figure 3) ▶ . This is suggested by the morphology of cases, such as B36 (Figure 1A) ▶ , in which Wilms’ tumors appear to be developing from a rest. In this model, metanephric stem cells persist beyond 36 weeks as a nephrogenic rest due to a germline mutation, an acquired mutation (of, eg, the WT1, WT2, 7p15, or familial gene), change of imprinting (in the perilobar rest and kidney) or possibly a physical disruption. Genes such as IGF2 at 11p15 may act as a growth promoter, amplifying the number of target cells. Intralobar rests appear to require two hits to develop. For a Wilms’ tumor to develop probably requires a further mutation in one of these stem cells. Further genetic events, including mutations of p53 or chromosome 16q, could occur later in the development of the Wilms’ tumor, giving the tumor cells a growth advantage and/or chemotherapy resistance.
Figure 3.

Possible model of Wilms’ tumor development via intralobar or perilobar rests. Note that 16q loss and anaplasia are probably late events and independent of each other. p53 abnormalities and anaplasia appear related. It is possible that there may be no rest stage in the development of some tumors.
Acknowledgments
Adrian Charles was a Smith and Nephew Foundation Research Fellow and gratefully acknowledges this support. The authors wish to thank Dr. Gordan Vujanic (Cardiff, UK) for allowing access to his slides and for his free help, time, and interest; The Cancer and Leukaemia in Childhood Research Trust, in whose laboratory this work was undertaken; Miss C. Anderson for section cutting and slide preparation; and colleagues and laboratory staff of the Department of Pediatric Pathology, St. Michael’s Hospital. We are grateful for the oncologists and surgeons of the Royal Hospital for Sick Children (Bristol) for their support and the cytogenetic unit at Southmead Hospital (Bristol) for the cytogenetics.
Footnotes
Address reprint requests to Dr. Adrian K. Charles, Department of Pediatric Pathology, St. Michael’s Hospital, Southwell Street, Bristol BS2 8EG, United Kingdom. E-mail: adrian.charles@bris.ac.uk.
AKC was Smith and Nephew Foundation Research Fellow.
References
- 1.Murphy WM, Beckwith JB, Farrow GM: Tumors of the kidney. Atlas of Tumor Pathology: Tumors of the Kidney, Bladder, and Related Urinary Structures. 1994:pp 1-192 Armed Forces Institute of Pathology, Washington DC
- 2.Bove KE, McAdams AJ: Multifocal nephroblastic neoplasia. J Natl Cancer Inst 1978, 61:285-294 [PubMed] [Google Scholar]
- 3.Beckwith JB, Kiviat NB, Bonadio JF: Nephrogenic rests, nephroblastomatosis, and the pathogenesis of Wilms’ tumour. Pediatr Pathol 1990, 10:1-36 [DOI] [PubMed] [Google Scholar]
- 4.Bard JBL, McConnell JE, Davies JA: Towards a genetic basis for kidney development. Mech Dev 1994, 48:3-11 [DOI] [PubMed] [Google Scholar]
- 5.Chao LY, Huff V, Tomlinson G, Riccardi VM, Strong LC, Saunders GF: Genetic mosaicism in normal tissues of Wilms’ tumour patients. Nat Genet 1993, 3:127-131 [DOI] [PubMed] [Google Scholar]
- 6.Birch JM, Breslow N: Epidemiologic features of Wilms tumour. Hematol Oncol Clin North Am 1995, 9:1157-1178 [PubMed] [Google Scholar]
- 7.Gaddy CD, Gibbons MD, Gonzalez ET, Finegold MJ: Obstructive uropathy, renal dysplasia and nodular renal blastema: Is there a relationship to Wilms tumor? J Urol 1985, 134:330-333 [DOI] [PubMed] [Google Scholar]
- 8.Vujanic GM, Sandstedt B, Dijoud F, Harms D, Delemarre JFM: Nephrogenic rest associated with a mesoblastic nephroma: what does it tell us? Pediatr Pathol 1995, 15:469-475 [DOI] [PubMed] [Google Scholar]
- 9.Joshi VV, Beckwith JB: Multilocular cyst of the kidney (cystic nephroma) and cystic, partially differentiated nephroblastoma: terminology and criteria for diagnosis. Cancer 1989, 64:466-479 [DOI] [PubMed] [Google Scholar]
- 10.Walford N, Delemarre JF: Wilms’ tumour associated with deep cystic nephroma-like changes: three cases of a putative Wilms’ tumour precursor. Histopathology 1991, 18:123-131 [DOI] [PubMed] [Google Scholar]
- 11.Beckwith JB, Palmer NF: Histopathology and prognosis of Wilms tumor: results of the National Wilms Tumor Study. Cancer 1978, 41:1937-1948 [DOI] [PubMed] [Google Scholar]
- 12.Schmidt D, Beckwith JB: Histopathology of childhood renal tumors. Hematol Oncol Clin North Am 1995, 9:1179-1200 [PubMed] [Google Scholar]
- 13.Faria P, Beckwith JB, Mishra K, Zuppan C, Weeks DA, Breslow N, Green DM: Focal versus diffuse anaplasia in Wilms tumor: new definitions with prognostic significance. Am J Surg Pathol 1996, 20:909-920 [DOI] [PubMed] [Google Scholar]
- 14.Malkin D, Sexsmith E, Yeger H, Williams BR, Coppes MJ: Mutations of the p53 tumour suppressor gene occur infrequently in Wilms’ tumour. Cancer Res 1994, 54:2077-2079 [PubMed] [Google Scholar]
- 15.Bardeesy N, Falkoff D, Petruzzi MJ, Nowak N, Zabel B, Adam M, Aguiar MC, Grundy P, Shows T, Pelletier J: Anaplastic Wilms’ tumor, a subtype displaying poor prognosis, harbours p53 gene mutations. Nat Genet 1994, 7:91-97 [DOI] [PubMed] [Google Scholar]
- 16.Kodet R, Marsden HB: Papillary Wilm’s tumour with carcinoma-like foci and renal cell carcinoma in childhood. Histopathology 1985, 9:1091-1102 [DOI] [PubMed] [Google Scholar]
- 17.Allsbrook WC, Boswell WC, Takahashi H, Pantazis C, Howell CG, Martinez JE, Beck JR: Recurrent renal cell carcinoma arising in Wilms’ Tumour. Cancer 1991, 67:690-695 [DOI] [PubMed] [Google Scholar]
- 18.Knudson AG, Strong LC: Mutation and cancer: a model for Wilms’ tumour of the kidney. J Natl Cancer Inst 1972, 8:313-324 [PubMed] [Google Scholar]
- 19.Grundy P, Coppes MJ, Haber D: Molecular genetics of Wilms tumor. Haematol Oncol Clin North Am 1995, 9:1201-1215 [PubMed] [Google Scholar]
- 20.Call KM, Glaser T, Ito CY, Buckler AJ, Pelletier J, Haber DA, Rose EA, Kral A, Yeger H, Lewis WH, Jones C, Housman DE: Isolation and characterization of a zinc finger polypeptide gene at the human chromosome 11 Wilms’ tumour locus. Cell 1990, 60:509-520 [DOI] [PubMed] [Google Scholar]
- 21.Gessler M, Poustka A, Cavenee W, Neve RL, Orkin SH, Bruns GA: Homozygous deletion in Wilms tumours of a zinc-finger gene identified by chromosome jumping. Nature 1990, 343:774-778 [DOI] [PubMed] [Google Scholar]
- 22.Mannens M, Slater RM, Heyting C, Bliek J, de Kraker J, Coad N, de Pagter-Holthuizen P, Pearson PL: Molecular nature of genetic changes resulting in loss of heterozygosity of chromosome 11 in Wilms’ tumours. Hum Genet 1988, 81:41-48 [DOI] [PubMed] [Google Scholar]
- 23.Wilmore HP, White GF, Howell RT, Brown KW: Germline and somatic abnormalities of chromosome 7 in Wilms’ tumor. Cancer Genet Cytogenet 1994, 77:93-98 [DOI] [PubMed] [Google Scholar]
- 24.Rahman N, Arbour L, Tonin P, Renshaw J, Pelletier J, Baruchael S, Pritchard-Jones K, Stratton MR, Narod SA: Evidence for a familial Wilms tumour gene (FWT1) on chromosome 17q12–q21. Nat Genet 1996, 13:461-463 [DOI] [PubMed] [Google Scholar]
- 25.Grundy P, Koufos A, Morgan K, Li FP, Meadows AT, Cavenee WK: Familial predisposition to Wilms’ tumour does not map to the short arm of chromosome 11. Nature 1988, 336:374-376 [DOI] [PubMed] [Google Scholar]
- 26.Huff V, Reeve AE, Leppert M, Strong LC, Douglass EC, Geiser CF, Li FP, Meadows A, Callen DF, Lenoir G, Saunders GF: Nonlinkage of 16q markers to familial predisposition to Wilms’ tumour. Cancer Res 1992, 52:6117-6120 [PubMed] [Google Scholar]
- 27.Breslow NE, Olson J, Moksness J, Beckwith JBB, Grundy P: Familial Wilms’ tumour: a descriptive study. Med Pediatr Oncol 1996, 27:398-403 [DOI] [PubMed] [Google Scholar]
- 28.Pritchard-Jones K, Fleming S, Davidson D, Bickmore W, Porteous D, Gosden C, Bard J, Buckler A, Pelletier J, Housman D, van Heyningen V, Hastie H: The candidate Wilms’ tumour gene is involved in genitourinary development. Nature 1990, 346:194-197 [DOI] [PubMed] [Google Scholar]
- 29.Pritchard-Jones K, Fleming S: Cell types expressing the Wilms’ tumour gene (WT1) in Wilms’ tumours: implications for tumour histogenesis. Oncogene 1991, 6:2211-2220 [PubMed] [Google Scholar]
- 30.Reddy JC, Licht JD: The WT1 Wilms’ tumour suppressor gene: how much do we really know? Biochim Biophys Acta 1996, 1287:1-28 [DOI] [PubMed] [Google Scholar]
- 31.Lamond AI: Wilms’ tumour: the splicing connection. Curr Biol 1995, 5:862-865 [DOI] [PubMed] [Google Scholar]
- 32.Hastie ND: The genetics of Wilms’ tumour: a case of disrupted development. Annu Rev Genet 1994, 28:523-558 [DOI] [PubMed] [Google Scholar]
- 33.Schumacher V, Schneider S, Figge A, Wildhardt G, Harms D, Schmidt D, Weirich A, Ludwig R, Royer-Pokora B: Correlation of germ-line mutations and two-hit inactivation of the gene with Wilms tumors of stromal-predominant histology. Proc Natl Acad Sci USA 1997, 94:3972-3977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Austruy E, Candon S, Henry I, Gyapay G, Tournade MF, Mannens M, Callen D, Junien C, Jeanpierre C: Characterization of regions of chromosomes 12 and 16 involved in nephroblastoma tumorigenesis. Genes Chromosomes Cancer 1995, 14:285-294 [DOI] [PubMed] [Google Scholar]
- 35.Maw MA, Grundy PE, Millow LJ, Eccles MR, Dunn RS, Smith PJ, Feinberg AP, Law DJ, Paterson MC, Telzerow PE, Callen DF, Thompson AD, Richards RI, Reeve AE: A third Wilms’ tumor locus on chromosome 16q. Cancer Res 1992, 52:3094-3098 [PubMed] [Google Scholar]
- 36.Grundy PE, Telzerow PE, Breslow N, Moksness J, Huff V, Paterson MC: Loss of heterozygosity for chromosomes 16q and 1p in Wilms’ tumours predicts an adverse outcome. Cancer Res 1994, 54:2331-2333 [PubMed] [Google Scholar]
- 37.Breslow N, Olshan A, Beckwith JB, Green DM: Epidemiology of Wilms tumour. Med Pediatr Oncol 1993, 21:172-181 [DOI] [PubMed] [Google Scholar]
- 38.Olson JM, Hamilton A, Breslow NE: Non-11p constitutional chromosome abnormalities in Wilms’ tumour patients. Med Pediatr Oncol 1995, 24:305-309 [DOI] [PubMed] [Google Scholar]
- 39.Neri G, Marini R, Cappa M, Borrelli P, Opitz JM: Simpson-Golabi-Behmel Syndrome. Am J Med Genet 1988, 30:287-299 [DOI] [PubMed] [Google Scholar]
- 40.Clericuzio CL, Johnson C: Screening for Wilms tumour in high-risk individuals. Hematol Oncol Clin North Am 1995, 9:1253-1265 [PubMed] [Google Scholar]
- 41.Baird PN, Santos A, Groves N, Jadresic L, Cowell JK: Constitutional mutations in the WT1 gene in patients with Denys-Drash syndrome. Hum Mol Genet 1992, 1:301-305 [DOI] [PubMed] [Google Scholar]
- 42.Hastie ND: Dominant negative mutations in the Wilms tumour (WT1) gene cause Denys-Drash syndrome: proof that a tumour-suppressor gene plays a crucial role in normal genitourinary development. Hum Mol Genet 1992, 1:293-295 [DOI] [PubMed] [Google Scholar]
- 43.Elliott M, Maher ER: Beckwith-Wiedemann syndrome. J Med Genet 1994, 31:560-564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Park S, Bernard A, Bove KE, Sens DA, Hazen-Martin DJ, Garvin AJ, Haber DA: Inactivation of WT1 in nephrogenic rests, genetic precursors to Wilms’ tumour. Nat Genet 1993, 5:363-367 [DOI] [PubMed] [Google Scholar]
- 45.Nordenskjold A, Friedman E, Sandstedt B, Soderhall S, Anvret M: Constitutional and somatic mutations in the WT1 gene in Wilms’ tumor patients. Int J Cancer 1995, 63:516-522 [DOI] [PubMed] [Google Scholar]
- 46.Cui H, Hedborg F, He L, Nordenskjold, Sandstedt B, Pfeifer-Ohlsson S, Ohlsson R: Inactivation of H19, an imprinted and putative tumor suppressor gene, is a preneoplastic event during Wilms’ tumorigenesis. Cancer Res 1997, 57:4469-4473 [PubMed] [Google Scholar]
- 47.Steenman M, Redecker B, de Meulemeester M, Wiesmeijer K, Voûte PA, Westerveld A, Slater R, Mannens M: Comparative genomic hybridization analysis of Wilms tumours. Cytogenet Cell Genet 1997, 77:296-303 [DOI] [PubMed] [Google Scholar]
- 48.Coppes MJ, Haber DA, Grundy PE: Genetic events in the development of Wilms’ tumour. N Engl J Med 1994, 331:586-590 [DOI] [PubMed] [Google Scholar]
- 49.Fearon ER, Vogelstein B: A genetic model of colorectal tumorigenesis. Cell 1990, 61:759-767 [DOI] [PubMed] [Google Scholar]
- 50.Zhu X, Dunn JM, Goddard AD, Squire JA, Becker A, Phillips RA, Gallie BL: Mechanisms of loss of heterozygosity in retinoblastoma. Cytogenet Cell Genet 1992, 59:248-252 [DOI] [PubMed] [Google Scholar]
- 51.Leisenring WM, Breslow NE, Evans IE, Beckwith JB, Coppes MJ, Grundy P: Increased birth weights of National Wilms’ Tumour Study patients suggest a growth factor excess. Cancer Res 1994, 54:4680-4683 [PubMed] [Google Scholar]
- 52.Nordling CO: A new theory on the cancer-inducing mechanism. Br J Cancer 1953, 7:68-72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coppes MJ, Bonetta L, Huang A, Hoban P, Chilton-MacNeill S, Campbell CE, Weksberg R, Yeger H, Reeve AE, Williams BR: Loss of heterozygosity mapping in Wilms tumor indicates the involvement of three distinct regions and a limited role for nondisjunction or mitotic recombination. Genes Chromosomes Cancer 1992, 5:326-334 [DOI] [PubMed] [Google Scholar]
- 54.Issa JP, Baylin SB: Epigenetics and human disease. Nat Med 1996, 2:281-282 [DOI] [PubMed] [Google Scholar]
- 55.Zhang P, Liegeois NJ, Wong C, Finegold M, Hou H, Thompson JC, Silverman A, Harper JW, DePinho RA, Elledge SJ: Altered Cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith Wiedemann syndrome. Nature 1997, 387:151-158 [DOI] [PubMed] [Google Scholar]
- 56.Charles AK: Nephrogenic rest and mesoblastic nephroma. Pediatr Pathol Lab Med 1996, 16:695-696 [PubMed] [Google Scholar]
- 57.Haber DA, Buckler AJ, Glaser T, Call KM, Pelletier J, Sohn RL, Douglass EC, Housman DE: An internal deletion within an 11p13 zinc finger gene contributes to the development of Wilms’ tumor. Cell 1990, 61:1257-1269 [DOI] [PubMed] [Google Scholar]
- 58.Hearne CH, Ghosh S, Todd JA: Microsatellites for linkage analysis of genetic traits. Trends Genet 1992, 8:288-294 [DOI] [PubMed] [Google Scholar]
- 59.Weber JL, Kwitek AE, May PE: Dinucleotide repeat polymorphisms at the D16S260, D16S261, D16S265, D16S266, and D16S267 loci. Nucleic Acids Res 1990, 18:4034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Baird PN, Groves N, Haber DA, Housman DE, Cowell JK: Identification of mutations in the WT1 gene in tumors from patients with the WAGR syndrome. Oncogene 1992, 7:2141-2149 [PubMed] [Google Scholar]