

Abstract
The cyclin-dependent kinase inhibitor p27 is a negative regulator of the cell cycle and a potential tumor suppressor gene. Because we had previously demonstrated that loss of p27 protein is associated with aggressive behavior in colorectal adenocarcinomas, we used immunohistochemistry and in situ hybridization to evaluate the potential role of alterations in p27 expression in primary and metastatic colorectal adenocarcinomas. Parallel immunostaining was performed for Ki-67 and p53. We evaluated 13 cases of metachronous and 23 cases of synchronous primary and metastatic colorectal tumor pairs. In the synchronous subgroup (Stage IV tumors), 57% of the primary tumor and metastases pairs did not express p27 protein and the remainder were low expressors. In the metachronous subgroup, 54% of the primary tumors were low expressors and the remainder high expressors of p27 protein. There was a significant reduction in the expression of p27 in the metachronous metastases (mean positive cells: 14.5%) when compared to the corresponding primary tumors (mean positive cells: 41.8%), P = 0.0023. All the primary and metastatic tumors in the metachronous subgroup showed high levels of p27 mRNA expression. There was no association between loss of p27 and either Ki-67 count or p53 expression. Because p27 is known to be up-regulated when epithelial cells are grown in suspension, the down-regulation of p27 in circulating tumor cells may confer the ability to grow in an environment of altered extracellular matrix or intercellular adhesion properties, two situations which may facilitate metastases.
Progression through the cell cycle involves sequential activation and inactivation of cyclin-dependent kinases (CDK). CDKs are activated though association with positive regulators called cyclins and inactivated by cyclin-CDK inhibitors (CKI). 1,2 CKIs can be divided into two groups, the ink4 family and the cip/kip family. 1 The ink4 proteins (p15, p16, p18, and p19) specifically inhibit the cyclin D-CDK4/6 complexes. Point mutations and deletions or inactivation by methylation of p16 and p15 genes have been reported in various human malignancies and transformed cell lines. 1,3
The cip/kip proteins (p21, p27, and p57) share partial structural homology, although p27 seems to target CDK2 preferentially. 1,3 Overexpression of p27 protein in mammalian cells induces a G1 block of the cell cycle. 4,5 Furthermore, the high levels of p27 found in quiescent cells suggests that it also plays a role in maintaining cells in G0. 6-8 It is of interest that no homozygous deletions and only rare mutations of the p27 gene have been found in human tumors. 9-12 We and other investigators have shown that absent or low p27 protein expression is a powerful negative prognostic marker in breast, 13-15 colorectal, 16 gastric, 17 prostate, 18,19 esophageal, 20 and pulmonary carcinomas. 21,22 Furthermore, in both colorectal and non-small-cell lung carcinomas, we demonstrated that low levels of p27 in tumor cells were due to tumor-specific, enhanced, proteosome-mediated protein degradation. 16,21
The aim of this study was to characterize the expression of p27 in primary and metastatic (both synchronous and metachronous) colorectal adenocarcinomas and to determine whether alterations in levels of p27 protein expression confer metastatic potential on tumors. Since p53 mutation is a late event in colorectal carcinogenesis and may be associated with the development of metastases from colorectal cancers, we also evaluated the expression of its gene product. 23,24 In addition, we compared p27 expression to that of Ki-67, a marker of proliferation.
Materials and Methods
Patient Population
We performed a retrospective search through the surgical pathology files of the Beth Israel Deaconess Medical Center (West Campus) from 1974 to 1997 to identify patients who had undergone colon resections for Stage I-III adenocarcinomas and subsequently underwent liver (n = 12) or pulmonary (n = 1) resection for metastatic colon adenocarcinomas. 25,26 Another group of patients with synchronous metastases (Stage IV) was used as a control. Paraffin-embedded tissue was available from 13 patients with metachronous and 23 patients with synchronous metastases. Metachronous metastases were defined as metastases occurring more than 6 months after resection/treatment of the primary tumor. Synchronous metastases were defined as metastases found at the time of resection of the primary tumor.
Age at diagnosis of the primary tumor, sex, pathological stage (using the American Joint Committee on Cancer Staging protocol), 27 histopathological grade, and interval until development of metastases were recorded.
The demographic data for patients in the metachronous and synchronous subgroups were as follows, respectively: mean age, 62.5 and 61.5 years; male: female ratio, 6:7 and 15:8. In the metachronous subgroup there were 7 Stage III, 5 Stage II, and 1 Stage I primary tumors. Mean interval to metastases was 27.7 months (range, 6 to 60 months). Eleven primary tumors were moderately differentiated and 2 were poorly differentiated. One patient (Table 1 ▶ , patient 4) presented with pulmonary metastasis 35 months after resection of the primary colorectal tumor. Another patient (Table 1 ▶ , patient 3) initially developed hepatic metastasis that was resected and subsequently presented with brain metastasis, which was also resected. Ten patients received chemotherapy (5-fluorouracil alone or with levamisole). In the synchronous subgroup all tumors were by definition Stage IV. All patients in this subgroup already had liver metastases at the time of resection of their primary colorectal tumors. In this group, 21 tumors were moderately differentiated and 2 were poorly differentiated. Thirteen patients received chemotherapy (5-fluorouracil alone or with levamisole), 3 declined treatment, and follow-up was unavailable on 7 patients.
Table 1.
Immunohistochemical Analysis of p27 Protein Expression in Primary Tumor-Metachronous Metastases Pairs
| Patient no. | Stage | Chemotherapy | Interval to metastases (months) | p27 level (%) | |
|---|---|---|---|---|---|
| in primary tumor | in metastases | ||||
| 1 | III | + | 35 | 67 | 3 |
| 2 | III | − | 36 | 60 | 2 |
| 3 | III | + | 60 | 0 | 7 |
| 4 | I | − | 35 | 38 | 13 |
| 5 | II | + | 36 | 17 | 32 |
| 6 | II | + | 18 | 54 | 28 |
| 7 | II | + | 21 | 0 | 0 |
| 8 | III | + | 17 | 63 | 7 |
| 9 | II | + | 19 | 48 | 11 |
| 10 | II | − | 6 | 69 | 26 |
| 11 | III | + | 21 | 67 | 34 |
| 12 | III | + | 20 | 18 | 14 |
| 13 | III | + | 27 | 41 | 11 |
| Mean ± SEM | 41.7± 7.0 | 14.5± 3.2 (p = 0.0023) |
Immunohistochemistry
Tissue sections 5 μm thick were cut from paraffin-embedded blocks, placed on charged glass slides, deparaffinized in xylene, and rehydrated through graded alcohol. Immunohistochemistry was performed as previously described. 13,16
Briefly, after antigen retrieval by microwave irradiation (10 mmol/L sodium citrate buffer, pH 6.0, (Biogenex, San Ramos, CA)) in a pressure cooker at 750 W for 30 minutes, mouse monoclonal antibodies against p27 (Transduction Laboratories, Lexington, KY), anti-Ki-67 (Immunotech, Westbrook, ME), and anti-p53 (Calbiochem, Cambridge, MA) were applied on the slides at dilutions of 1:400, 1:100, and 1:500, respectively, in phosphate-buffered saline (PBS). Immunohistochemistry was performed by an automated processor (Ventana ES, Ventana Medical Systems, Tucson, AZ). Steps performed by the instrument included blocking with normal horse serum, application of secondary antibody conjugated to the avidin-biotin peroxidase complex and visualization with 3,3-diaminobenzidine as a substrate with standardized development times. Identical reaction times permitted accurate comparison of all samples. The slides were lightly counterstained with hematoxylin. A mixture containing antibodies with no known human recognition site was used as a negative control. An osteosarcoma cell line MG-63 (obtained from the American Type Culture Collection, Rockville, MD) was used as a positive control for p27. After 48 hours of serum starvation (necessary to increase the levels of p27), cells from two confluent flasks were harvested, fixed in neutral buffered formalin for 8 hours, and paraffin-embedded. Five-micrometer-thick sections were used in each immunohistochemistry run as a positive control.
In Situ Hybridization
In situ hybridization for p27 mRNA was performed in all primary and corresponding metastatic tumors of the metachronous subgroup. One μg of recombinant plasmid pCR™II (Invitrogen, San Diego, CA), containing the full-length human p27 gene was linearized using BamHI and XbaI to generate sense and antisense transcripts, respectively. Digoxigenin-labeled riboprobes were made with T7 and SP6 polymerase for 1 hour at 37°C in 1 × transcription buffer (Promega Corp., Madison, WI), 10 mmol/L dithiothreitol, 40 U of ribonuclease inhibitor, adenosine, cytosine, and guanosine triphosphates (1 mmol/L each) and a mixture of cold uridine triphosphate and digoxigenen-uridine triphosphate (6.5 and 3.5 mmol/L, respectively, for a total concentration of 1 mmol/L) (Boehringer Mannheim, Indianapolis, IN)). Slide sections were digested with proteinase K (20μg/ml) in 1 mol/L Tris-EDTA buffer, pH 8.0, for 8 minutes at 37°C and then washed in PBS. Hybridization was performed at 42°C for 3 hours with the application of 10 pmol/L digoxigenin-labeled riboprobe in 100μl of hybridization buffer (50% deionized formamide, 2 × sodium chloride/sodium citrate (SSC), 50% dextran sulfate, 10% SDS and denatured herring sperm DNA 10 mg/ml) per slide under liquid coverslip (Ventana Medical Systems, Tuscon, AZ). Four washes of SSC at 55°C, the most stringent of which was at 0.1 × SSC, followed hybridization. Anti-digoxigenin antibody (1:500) was applied for 28 minutes at 37°C followed by detection with nitroblue tetrazolium and 5-bromo-4-chloro-3-indolyl phosphate for 12 minutes. The slides were counterstained with methyl green. Sense probes were used as controls. Duration and temperature of all steps were standardized by the automated in situ hybridization instrument.
Immunohistochemical (p27, p53, Ki-67) Scoring for Statistical Evaluation
Two pathologists without knowledge of the clinical and pathological parameters of the cases independently evaluated and scored them for percentage of cells expressing strong nuclear staining for p27. At least 10 high-power fields were chosen at random and 1000 cells were counted. Of these cases, 25% were chosen at random and scored a third time by another pathologist (ML). There was >98% concordance among observers’ scores. The percentage of p27 positive cells was expressed as a ratio of positive cells to the total number of cells counted. The percentage of p53 and Ki-67 cells per total population was obtained similarly. Based on prior reports of p27 expression in colorectal and breast cancers, cases were classified as high or low p27 expressors. 15,16 Patients with low p27 expression had ≤50% of the nuclei in the specimen staining positive, while high expressors had >50% p27 positive nuclei. For p53 and Ki-67 the cutoff used was ≤20% for nonexpressors and >20% for expressors.
Statistical Methods
The difference in p27, p53, and Ki-67 expression was assessed by the paired t-test using absolute percentage of positive cells for the individual tumor/metastases pairs in both the synchronous and metachronous subgroups. In addition p27, p53, and Ki-67 expression was compared in the same groups using the cutoffs described above and McNemar’s paired comparison test for binary data. In the metachronous subgroup, the effect of chemotherapy on p27 expression was analyzed using the Mann-Whitney rank sum test (for two categories). The Pearson correlation coefficient was used to test the strength of association between continuous variables. A P value ≤0.05 was required for significance.
Results
Primary tumors with metachronous metastases were available in 13 cases. Fifty-four percent of the primary tumors in this subgroup were low expressors of p27 (≤50% of tumor cells expressing p27), while the remainder were high expressors (>50% of cells expressing p27). The distribution of p27 was consistent with our previous analysis of a larger database. 16 There was a striking reduction in p27 protein nuclear expression in the metastases (mean = 14.5%) compared to the primary tumors (mean = 41.8%); (P = 0.0023 by paired t-test or P = 0.03 by McNemar’s paired comparison test for binary data). (Table 1 ▶ and Figure 1, A and B ▶ ). In contrast to the protein level, p27 mRNA as assessed by in situ hybridization (ISH) was expressed in all of the primary tumor/metastasis pairs in the metachronous subgroup (Figure 2) ▶ .
Figure 1.

Nuclear localization of p27 by immunohistochemistry in a primary colorectal adenocarcinoma (a) with subsequent down-regulation/loss of expression in the corresponding metachronous metastasis (b). In the synchronous subgroup, there is low expression in both the primary tumor (c) and synchronous metastasis (d). Stromal lymphocytes are strongly stained in both subgroups. Hematoxylin counterstain. Magnification, × 250.
Figure 2.
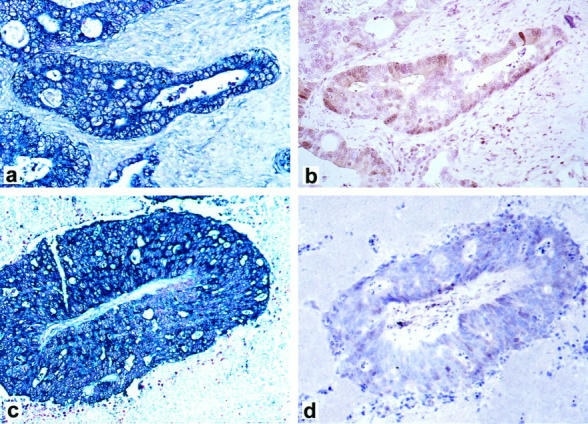
Primary colorectal adenocarcinoma from the metachronous subgroup showing high levels of p27 mRNA by in situ hybridization (a) and p27 protein expression by immunohistochemistry (b). The subsequent metachronous metastasis expresses high levels of p27 mRNA (c) but corresponding immunohistochemistry demonstrates lack of p27 protein expression (d). Magnification, × 250.
In the metachronous subgroup, the mean value of percentage of positive cells in the primary tumors and metastases for p53 was 61 ± 10.6 and 50.6 ± 10.7, respectively (P = 0.41 by paired t-test or P = 0.75 by McNemar’s paired comparison test for binary data). There was no correlation between p53 and p27 in the primary tumors (r 2 = −0.09, P = 0.77) or in the metastases (r2= 0.12, P = 0.69). As for the proliferation index, as assessed by Ki-67, the mean value of percentage of positive cells in the primary tumors and metastases was 65.9 ± 8.7 and 55.9 ± 8.0, respectively (P = 0.07 by paired t-test or P = 1.00 by McNemar’s paired comparison test for binary data). Similarly, there was no correlation between p27 and Ki-67 in the primary tumors (r2= 0.11, P = 0.73) or the metastases (r2= 0.18, P = 0.55).
Ten patients received chemotherapy after resection of their primary tumors. There was no significant difference in the percentage of positive cells expressing p27 in the metachronous metastases of treated and untreated patients. (P = 0.9, Mann-Whitney rank sum test).
In the set of tumor/synchronous metastasis pairs, there was no significant difference in p27 protein expression between the primary tumors (mean = 8.3%) and metastases (mean = 9%) (P = 0.6582). As expected these patients were all low p27 expressors in the primary tumor (Table 2 ▶ : Figure 1, C and D ▶ ). Of note, we also identified distinct cytoplasmic expression in 7 pairs of primary tumors and their corresponding synchronous metastases. In the synchronous subgroup, the mean value of percentage of positive cells in the primary tumors and metastases for p53 was 79 ± 6.0 and 79.1 ± 5.9, respectively (P = 0.11). The mean value of percentage of positive cells in the primary tumors and metastases for Ki-67 in the synchronous subgroup was 64.2 ± 4.8 and 60.0 ± 5.0, respectively (P = 0.31). Similarly, comparison of p27, p53, and Ki-67 in the primary tumor/synchronous metastasis pairs was not significantly different when the McNemar’s comparison test for binary data was used (P = 1.00 for all three markers) (Table 3) ▶ .
Table 2.
Immunohistochemical Analysis of p27 Protein Expression in Primary Tumor-Synchronous Metastases Pairs
| Patient no. | Stage | Chemotherapy | p27 level (%) | |
|---|---|---|---|---|
| in primary | in metastases | |||
| 1 | IV | + | 0 | 0 |
| 2 | IV | + | 0 | 0 |
| 3 | IV | + | 0 | 0 |
| 4 | IV | + | 0 | 0 |
| 5 | IV | − | 0 | 0 |
| 6 | IV | + | 30 | 27 |
| 7 | IV | N/A | 0 | 0 |
| 8 | IV | + | 0 | 0 |
| 9 | IV | + | 0 | 0 |
| 10 | IV | − | 14 | 35 |
| 11 | IV | + | 11 | 2 |
| 12 | IV | + | 49 | 36 |
| 13 | IV | N/A | 0 | 0 |
| 14 | IV | + | 0 | 0 |
| 15 | IV | N/A | 0 | 0 |
| 16 | IV | + | 23 | 29 |
| 17 | IV | N/A | 0 | 0 |
| 18 | IV | + | 14 | 19 |
| 19 | IV | N/A | 9 | 30 |
| 20 | IV | N/A | 16 | 4 |
| 21 | IV | − | 12 | 17 |
| 22 | IV | + | 0 | 0 |
| 23 | IV | N/A | 12 | 8 |
| Mean± SEM | 8.3 ± 2.6 | 9.0± 2.8 (p = 0.6852) |
N/A, chemotherapy data not available.
Table 3.
Summary of p27, p53, and Ki-76 Percentage Positive Tumor Cells and P Values in the Metachronous and Synchronous Subgroups
| Subgroup | p27 | p53 | Ki-67 |
|---|---|---|---|
| Metachronous | |||
| Primary tumor | 41.7 ± 7.0 | 61.0 ± 10.6 | 65.9 ± 8.7 |
| Metastases | 14.5 ± 3.2 | 50.6 ± 10.7 | 55.9 ± 8.0 |
| P value (paired t-test) | 0.0023 | 0.41 | 0.07 |
| P value (McNemar’s paired test) | 0.03 | 0.75 | 1.00 |
| Synchronous | |||
| Primary tumor | 8.3 ± 2.6 | 79.5 ± 6.0 | 64.2 ± 4.8 |
| Metastases | 9.0 ± 2.8 | 79.1 ± 5.9 | 60.0 ± 5.0 |
| P value (paired t-test) | 0.69 | 0.11 | 0.31 |
| P value (McNemar’s paired test) | 1.00 | 1.00 | 1.00 |
The difference in p27, p53, and ki-67 expression was assessed by the paired t-test when comparing absolute numbers and the McNemar’s paired test for binary data when cutoffs were used (see Statistical Methods) for individual tumor/metastasis pairs in both the synchronous and metachronous subgroups.
Values are mean ± SEM.
Discussion
The CKI p27 plays a significant role in the regulation of the transition from G1 to S phase in both normal and neoplastic cells. 1,3 Defective regulation of this important checkpoint may result in uncontrolled cellular proliferation. 1 In fact, inactivation of several CKIs has been associated with neoplastic transformation in a variety of tissues. 1-3 The development of multiple organ hyperplasia and pituitary tumors in p27 knockout mice demonstrates that p27 plays an important role in repressing tumor growth in vivo. 28-30
We have previously demonstrated the role of p27 loss in determining prognosis in colorectal cancer. 16 Here we evaluated the potential role of p27 loss in the process of metastasis. All of the primary and subsequent metastatic tumors tested in the metachronous subgroup expressed high levels of p27 mRNA via in situ hybridization. In contrast, all metachronous metastases were low expressors of p27 protein by immunohistochemistry, suggesting that loss of p27 resulted from a posttranslational target-specific enhanced proteosome-mediated degradation as shown previously. 16,21,31 This may be a mechanism used by aggressive tumors to eliminate p27. Although the database is small, loss of p27 appears to be independent of the effects of chemotherapy.
There was no direct inverse or positive correlation between p27 and Ki-67 expression in the metachronous subgroup. Proliferative index has not been shown to have a prognostic significance in colorectal cancer. 32,33 Furthermore, we and others have previously demonstrated a lack of correlation between p27 and Ki-67 in colorectal adenocarcinoma 16,34 as well as in breast cancer. 13 It is becoming progressively clearer that CKIs have functions other than cell cycle inhibition. In fact, CKIs are expressed in postmitotic cells in the absence of any CDK activity. For instance, p27 has been shown to be involved in apoptosis, 35 whereas p21 appears to have a role in differentiation. 36 Both of these effects are independent of those involving cell cycle kinetics.
Our study indicates yet another potential mechanism of action of p27, namely its involvement in the process of cell adhesion. Cell adhesion has an essential role in regulating proliferation during the G1 phase and loss of this adhesion capacity is a feature of oncogenic transformation. Cell-cell contact such as that occurring at the confluence of epithelial or mesenchymal cells in culture and loss of cell adhesion, eg, when cells are grown in suspension, up-regulate p27 levels. 3,37-41 In cells growing in suspension, p27 up-regulation is due to an increase in protein stability. 39 Enforced expression of G1 cyclins allows cell proliferation in suspension. 40,41 Increased expression of cyclin D1 in both colon tumor cell lines and human colonic tumors has been shown to contribute to tumorigenecity. 42,43 Cyclin overexpression or loss of p27 protein via enhanced tumor-specific degradation may give tumor cells the ability to grow in the presence of altered extracellular matrix properties and altered intercellular adhesion, two conditions that might facilitate metastasis.
p53 mutations are a late event in colorectal carcinogenesis 44 and have been shown to be associated with the development of metastases in colorectal adenocarcinomas. 23,24 However, no difference in p53 overexpression (indicative of mutation in the gene) was found in the primary or metastatic tumors of either the metachronous or synchronous subgroups. In addition, there was no correlation between p27 and p53 expression, suggesting an independent pathway for p53 in the development of metastases.
Altered cellular compartmentalization may also play a role in the down-regulation of p27 in anchorage-independent cells. 45 Although p27 normally is localized to the nucleus, we noted distinct cytoplasmic localization of p27 in both the primary tumor and its corresponding synchronous metastasis in a small subset of patients. Previously we showed that confinement of p27 to the cytoplasm, confirmed by subcellular fractionation, was associated with a poor prognosis in Barrett’s associated esophageal adenocarcinoma. 20 The mechanism for nuclear exclusion is unclear at present. It remains to be determined whether the nuclear exclusion of p27 is involved in the metastatic process.
We have shown here that p27 protein expression is down-regulated or lost in metachronous metastases in patients with primary colorectal adenocarcinomas. This novel finding implies that p27 protein down-regulation in colorectal adenocarcinomas is associated with tumor progression and may be an important factor in the development of metastases.
Acknowledgments
We thank S. Galli for helpful discussion, P. Lavin for assistance with statistical computation, and P. Godschall and J. Hayward for assistance with photography.
Footnotes
Address reprint requests to Dr. Massimo Loda, Department of Adult Oncology, Dana Farber Cancer Institute and Department of Pathology, Brigham and Women’s Hospital, Harvard Medical School, 44 Binney Street, Boston, MA 02215. E-mail: massimo_loda@dfci.harvard.edu.
Supported by NIH Grants CA 44704–09 (to M. L.), CA 76584–01A1, and GM/CA 57587–01 (to M. P.).
The first two authors contributed equally to this study.
References
- 1.Sherr CJ: Cancer cell cycles. Science 1996, 274:1672-1677 [DOI] [PubMed] [Google Scholar]
- 2.Cordon-Cardo C: Mutations of cell cycle regulators: biological and clinical implications for human neoplasia. Am J Pathol 1995, 147:545-560 [PMC free article] [PubMed] [Google Scholar]
- 3.Del Sal G, Loda M, Pagano M: Cell cycle and cancer: critical events at the G1 restriction point. Crit Rev Oncog 1996, 7:127-142 [DOI] [PubMed] [Google Scholar]
- 4.Toyoshima T, Hunter T: p27, a novel inhibitor of G1-cyclin cdk protein kinase activity is related to p21. Cell 1994, 78:67-74 [DOI] [PubMed] [Google Scholar]
- 5.Polyak K, Lee MH, Erdjement-Bromage H, Koff A, Roberts JM, Tempst P, Massague J: Cloning of p27/kip1, a new cyclin kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell 1994, 78:59-66 [DOI] [PubMed] [Google Scholar]
- 6.Pagano M, Beer-Romano P, Glass S, Tam SW, Theodoras A, Rolfe M, Draetta G: Targeting ubiquitin-mediated degradation for proliferation inhibitors. Milhich E Housman D eds. Cancer Genes. 1997, :pp 255-268 Plenum Publishing Corp, New York [Google Scholar]
- 7.Hengst L, Reed S: Translation control of p27kip1 accumulation during the cell cycle. Science 1996, 271:1861-1864 [DOI] [PubMed] [Google Scholar]
- 8.Sandhu C, Garbe J, Daksis J, Pan C-H, Bhattacharya N, Yaswen P, Koh J, Slingerland J, Stampfer MR: TGF-β stabilizes p15 INK4B protein, increases p15 INK4B/cdk4 complexes, and inhibits cyclin D1/cdk4 association in human mammary epithelial cells. Mol Cell Biol 1997, 17:2458-2467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ponce-Castañeda V, Lee M, Latres E, Polyak K, Lacombe L, Montgomery K, Mathew K, Krauter K, Sheinfeld J, Massague J, Cordon-Cardo C: p27kip1: Chromosomal mapping to 12p12–12p13.1 and absence of mutations in human tumors. Cancer Res 1995, 55:1211-1214 [PubMed] [Google Scholar]
- 10.Kawamata N, Morosetti R, Miller CW, Park D, Spirin K, Nakamaki T, Takeuchi S, Hatta Y, Simpson J, Wilczynski S, Lee Y, Bartram C, Koeffler H: Molecular analysis of the cyclin dependent kinase inhibitor p27/kip1 in human malignancies. Cancer Res 1995, 55:2266-2269 [PubMed] [Google Scholar]
- 11.Spirin KS, Simpson JF, Takeuchi S, Kawamata N, Miller CW, Koeffler H: p27/kip1 mutation found in breast cancer. Cancer Res 1996, 56:2400-2404 [PubMed] [Google Scholar]
- 12.Pietenpol JS, Bohlander SK, Sato Y, Papadopoulos N, Liu B, Friedman C, Trask B: Assignment of the human p27kip1 gene to 12p13 and its analysis in leukemias. Cancer Res 1995, 55:1206-1210 [PubMed] [Google Scholar]
- 13.Tan P, Cady B, Wanner M, Worland P, Cukor B, Magi-Galluzzi C, Pagano M, Loda M: The cell cycle inhibitor p27 is an independent prognostic marker in small (T1a, b) invasive breast carcinomas. Cancer Res 1997, 57:1259-1263 [PubMed] [Google Scholar]
- 14.Porter PL, Malone KE, Heagerty PJ, Alexander GM, Gatti LA, Firpo EJ, Daling JR, Roberts JM: Expression of cell cycle regulators p27kip1 and cyclin E, alone and in combination, correlate with survival in young breast cancer patients. Nat Med 1997, 3:222-225 [DOI] [PubMed] [Google Scholar]
- 15.Catzavelos C, Bhattacharya N, Ung YC, Wilson JA, Roncari L, Sandhu C, Shaw P, Yeger H, Morava-Protzner I, Kapusta L, Franssen E, Pritchard KI, Slingerland JM: Decreased levels of the cell cycle inhibitor p27kip1 protein: prognostic implications in primary breast cancer. Nat Med 1997, 3:227-230 [DOI] [PubMed] [Google Scholar]
- 16.Loda M, Cukor B, Tam SW, Lavin P, Fiorentino M, Draetta GF, Jessup JM, Pagano M: Increased proteosome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med 1997, 3:231-234 [DOI] [PubMed] [Google Scholar]
- 17.Mori M, Mimori K, Shiraishi T, Tanaka S, Ueo H, Sugimachi K, Akiyoshi T: p27 expression and gastric carcinoma. Nat Med 1997, 3:593. [DOI] [PubMed] [Google Scholar]
- 18.Yang RM, Naitoh J, Philipson J, Wang H, deKernion JB, Loda M, Reiter RE: Low levels of p27 protein expression predict poor disease-free survival in patients with pathological T2a-T3b adenocarcinoma of the prostate. J Urol 1998, 159:941-945 [PubMed] [Google Scholar]
- 19.Tsihlias J, Kapusta LR, DeBoer G, Morava-Protzner I, Zbieranowski I, Bhattacharya N, Catzavelos GC, Klotz LH, Slingerland JM: Loss of cyclin-dependent kinase inhibitor p27kip1 is a novel prognostic factor in localized human prostate adenocarcinoma. Cancer Res 1998, 58:542-548 [PubMed] [Google Scholar]
- 20.Singh SP, Lipman J, Goldman H, Ellis FH, Aizenman L, Cangi MG, Signoretti S, Chiaur DS, Pagano M, Loda M: Loss or altered subcellular localization of p27 in Barrett’s associated adenocarcinoma. Cancer Res 1998, 58:1730-1735 [PubMed] [Google Scholar]
- 21.Esposito V, Baldi A, De Luca A, Groger AM, Loda M, Giordano GG, Caputi M, Baldi F, Pagano M, Giordano A: Prognostic role of the cyclin-dependent kinase inhibitor p27 in non-small cell lung cancer. Cancer Res 1997, 57:3381-3385 [PubMed] [Google Scholar]
- 22.Yatabe Y, Masuda A, Takashi K, Nakamura S, Kuroishi T, Osada H, Takahashi T, Mitsudomi T, Takahashi T: p27kip1 in human lung cancers: differential changes in small cell and non-small cell carcinomas. Cancer Res 1998, 58:1042-1047 [PubMed] [Google Scholar]
- 23.Kastrinakis WV, Ramchurren N, Reiger KM, Hess DT, Loda M, Steele G, Summerhayes IC: Increased incidence of p53 mutations is associated with hepatic metastases in colorectal neoplastic progression. Oncogene 1995, 11:647-652 [PubMed] [Google Scholar]
- 24.Bertorelle R, Esposito G, Del Mistro A, Belluco C, Nitti D, Lise M, Chieco-Bianchi L: Association of p53 gene and protein alterations with metastases in colorectal cancer. Am J Surg Pathol 1995, 19:463-471 [DOI] [PubMed] [Google Scholar]
- 25.Cady B, Stone M, McDermott WV, Jenkins RL, Bothe A, Lavin PT, Lovett EJ, Steele GD: Technical and biological factors in disease-free survival after hepatic resection for colorectal cancer metastases. Arch Surg 1992, 127:561-568 [DOI] [PubMed] [Google Scholar]
- 26.Ambiru S, Masaru M, Hiroshi I, Nakagawa K, Shimizu H, Kato A, Nakamura S, Omoto H, Nakajima N: Resection of hepatic and pulmonary metastases in patients with colorectal carcinoma. Cancer 1998, 82:274-278 [DOI] [PubMed] [Google Scholar]
- 27.Manual for staging of cancer: American Joint Committee on Cancer. Edited by Beahrs OH, Henson DE, Hutter RVP, Kennedy BJ. New York, J. B. Lippincott, 1992
- 28.Fero M, Rivkin M, Tasch M, Porter P, Carcow C, Firpo E, Polyak K: A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis and female sterility in p27kip1 deficient mice. Cell 1996, 85:733-744 [DOI] [PubMed] [Google Scholar]
- 29.Kiyokawa H, Kineman R, Manova-Todorova KO, Soares VC, Frohman LA, Koff A: Enhanced growth of mice lacking the cyclin dependent kinase inhibitor function of p27kip1. Cell 1996, 85:721-732 [DOI] [PubMed] [Google Scholar]
- 30.Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horri I, Loh D, Nakayama K: Mice lacking p27 display increased body size, multiple organ hyperplasia, retinal dysplasia and pituitary tumors. Cell 1996, 85:707-720 [DOI] [PubMed] [Google Scholar]
- 31.Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M: Role of the ubiqitin-proteosome pathway in regulating abundance of cyclin-dependant kinase inhibitor p27. Science 1995, 269:682-685 [DOI] [PubMed] [Google Scholar]
- 32.Sahin AA, Ro JY, Brown RW, Ordonez NG, Cleary KR, El-Naggar AK, Wilson P, Ayala AG: Assessment of Ki-67 derived tumor proliferative activity in colorectal adenocarcinomas. Mod Pathol 1994, 7:17-21 [PubMed] [Google Scholar]
- 33.Risio M, Coverlizza S, Ferrari A, Candelaresi G, Rossini S: Immunohistochemical study of epithelial cell proliferation in hyperplastic polyps, adenomas and adenocarcinomas of the large bowel. Gastroenterology 1988, 94:899-906 [DOI] [PubMed] [Google Scholar]
- 34.Ciaparrone M, Yamamoto H, Yao Y, Sgambato A, Cattoretti G, Tomita N, Monden T, Rotterdam H, Weinstein B: Localization and expression of p27 in multistage colorectal carcinogenesis. Cancer Res 1998, 58:114-122 [PubMed] [Google Scholar]
- 35.Katayose Y, Kim M, Rakkar ANS, Li Z, Cowan KH, Seth P: Promoting apoptosis: A novel activity associated with the cyclin dependent kinase inhibitor p27. Cancer Res 1997, 57:5441-5445 [PubMed] [Google Scholar]
- 36.Di Cunto F, Topley G, Calautti E, Hsiao J, Ong L, Seth PK, Dotto GP: Inhibitory function of p21 Cip1/WAF1 in differentiation of primary mouse keratinocytes independent of cell cycle control. Science 1998, 280:1069-1072 [DOI] [PubMed] [Google Scholar]
- 37.St. Croix B, Florenes V, Rak A, Flaagan JW, Bhattacharya N, Slingrland JM, Kerbel RS: Impact of the cyclin dependent kinase inhibitor p27 kip1 on adhesion-dependent resistance of tumor cells to anticancer agents. Nat Med 1996, 2:1204-1210 [DOI] [PubMed] [Google Scholar]
- 38.Polyak K, Kato M, Saloman MJ, Sherr CJ, Massague J, Roberts JM, Koff A: p27, a cyclin -cdk inhibitor, links TGF-β and contact inhibition to cell cycle arrest. Genes Dev 1994, 8:9-22 [DOI] [PubMed] [Google Scholar]
- 39.Schulze A, Zerfass-Thome J, Middendorp S, Jansen-Durr P, Henglein B: Anchorage-dependent transcription of the cyclin A gene. Mol Cell Biol 1996, 16:4632-4638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu X, Ohtsubo M, Roberts J, Assosian R: Adhesion-dependent cell cycle progression linked to the expression of cyclin D1, activation of cyclin E-cdk2 and phosphorylation of the retinoblastoma protein. J Cell Biol 1996, 133:391-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang F, Orend G, Watanabe N, Hunter T, Rouslathi E: Dependence of cyclin E-cdk2 kinase activity on cell anchorage. Science 1996, 271:499-502 [DOI] [PubMed] [Google Scholar]
- 42.Arber N, Doki Y, Han EK, Sgambato A, Zhou P, Kim NH, Delohery T, Klein MG, Holt PR, Weinstein IB: Antisense to cyclin D1 inhibits the growth and tumorigenecity of human colon cancer cells. Cancer Res 1997, 57:1569-1574 [PubMed] [Google Scholar]
- 43.Arber N, Hibshoosh H, Moss SF, Sutter T, Zhang Y, Begg M, Wang S, Weinstein IB, Holt PR: Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology 1996, 110:669-674 [DOI] [PubMed] [Google Scholar]
- 44.Fearon ER, Volgelstein B: A genetic model for colorectal carcinogenesis. Cell 1990, 61:759-767 [DOI] [PubMed] [Google Scholar]
- 45.Orend G, Hunter T, Ruoslathi E: Cytoplasmic displacement of cyclin E-cdk2 inhibitors p21cip1 and p27kip1 in anchorage-independent cells. Oncogene 1998, 16:2575-2583 [DOI] [PubMed] [Google Scholar]