

Abstract
Cerebral amyloid angiopathy in Alzheimer’s disease is characterized by deposition of amyloid β (Aβ) in cortical and leptomeningeal vessel walls. Although it has been suggested that Aβ is derived from vascular smooth muscle, deposition of Aβ is not seen in larger cerebral vessel walls nor in extracranial vessels. In the present study, we examine evidence for the hypothesis that Aβ is deposited in periarterial interstitial fluid drainage pathways of the brain in Alzheimer’s disease and that this contributes significantly to cerebral amyloid angiopathy. There is firm evidence in animals for drainage of interstitial fluid from the brain to cervical lymph nodes along periarterial spaces; similar periarterial channels exist in humans. Biochemical study of 6 brains without Alzheimer’s disease revealed a pool of soluble Aβ in the cortex. Histology and immunocytochemistry of 17 brains with Alzheimer’s disease showed that Aβ accumulates five times more frequently around arteries than around veins, with selective involvement of smaller arteries. Initial deposits of Aβ occur at the periphery of arteries at the site of the putative interstitial fluid drainage pathways. These observations support the hypothesis that Aβ is deposited in periarterial interstitial fluid drainage pathways of the brain and contributes significantly to cerebral amyloid angiopathy in Alzheimer’s disease.
Amyloid β (Aβ) peptides accumulate in the brains of patients with Alzheimer’s disease (AD) in senile plaques and in vessel walls as cerebral amyloid angiopathy (CAA). Neither the exact origin of Aβ in plaques and vessel walls nor the mechanism by which it is deposited has, as yet, been resolved. One widely accepted view with regard to CAA is that Aβ in vessel walls is derived from vascular smooth muscle cells, 1,2 but this does not explain why CAA rarely involves larger intracranial arteries or why Aβ does not accumulate in the walls of extracranial vessels. 3 In the present study, we challenge the view that Aβ in CAA is derived solely from smooth muscle cells; we then examine the evidence that Aβ is deposited in the periarterial pathways along which interstitial fluid (ISF) drains from the cerebral cortex, and that this contributes significantly to CAA in AD.
There is now firm evidence that ISF from the brain drains to regional cervical lymph nodes in a range of species from rodents to sheep. 4-6 Injections of tracers into the rat brain show that ISF drains along perivascular spaces around intracortical and leptomeningeal arteries; these channels eventually connect with nasal lymphatics at the cribriform plate of the ethmoid bone. 4,7-9 The immunological significance of ISF drainage to cervical lymph nodes is emphasized by experiments in the rat showing that cervical lymphadenectomy reduces both B-cell immune reactions to antigens draining from the brain 10,11 and T cell-mediated immune responses in the brain. 12-14
Periarterial pathways, homologous with ISF drainage pathways in the rat, have been identified by histological and ultrastructural studies in the human brain. 15-18 In the human cerebral cortex, periarterial spaces are encompassed by an outer sheath of leptomeningeal cells derived from the pia mater, and these spaces are in direct continuity with perivascular spaces around arteries in the subarachnoid space. 15,17 In the basal ganglia, the arrangement is slightly different, as periarterial spaces are lined both on the inner and the outer aspects by layers of leptomeninges; however, the periarterial spaces within the brain are still in continuity with the perivascular spaces of leptomeningeal arteries. 18 No such well defined continuity of intracortical and leptomeningeal perivascular spaces has been observed in relation to veins. 15 Reports of glioblastoma metastasizing from the brain to cervical lymph nodes 19 suggest that they may act as regional lymph nodes for the brain in humans as in other mammals.
Evidence that smooth muscle cells are a source of vascular amyloid 1,20 derives mainly from the observations that they contain β-amyloid precursor protein (APP) fragments 21 and that they lie immediately adjacent to vascular Aβ deposits. However, there are a number of problems with accepting that smooth muscle cells are the sole source of Aβ in CAA. First, CAA is strictly confined to the central nervous system in AD, and extracranial vessels are free of Aβ deposits, 3 but there is little evidence that smooth muscle cells in extracranial arteries differ from intracranial arteries in terms of APP metabolism. 3 Second, if Aβ were produced by smooth muscle cells, early deposits of amyloid would be located throughout the tunica media, rather than in the adventitia and outer media. 20-22 Third, if vascular smooth muscle cells were the only or major source of Aβ, it would be expected that the larger arteries would be more prone to CAA, as they contain more smooth muscle cells, but, in fact, they are less severely involved than the smaller arteries. 3
In the present study to test the hypothesis that Aβ is deposited in periarterial ISF drainage pathways of the brain in AD, we use both biochemical and histological methods. Biochemical techniques are used to show that there is a pool of soluble Aβ in non-AD cerebral cortex, which is presumably eliminated continuously from the normal brain. Qualitative and quantitative histological and immunohistochemical techniques are used to demonstrate 1) that there is a correlation between the presence of Aβ senile plaques in the brain and amyloid angiopathy in AD; 2) that insoluble Aβ accumulates selectively around small arteries in the brain and leptomeninges with relative sparing of large arteries and veins, suggesting that deposition of Aβ occurs in drainage pathways nearest the site of origin of Aβ from the brain; and 3) that small and possibly the earliest deposits of Aβ in the walls of leptomeningeal arteries occur at the site of putative ISF fluid drainage pathways in the adventitia, and that only later does Aβ accumulate in the tunica media.
Materials and Methods
Tissue Samples and Materials
Biochemical quantitation of soluble Aβ was performed on cerebral cortex from six brains of clinically nondemented control cases without a history of neurological disease (average age, 79 years; range, 69–92 years) selected from the Sun Health Research Institute brain bank. All six cases had a ε3/ε3 apolipoprotein E (ApoE) genotype. On histological (thioflavin S and Campbell-Zwitzer stains) and immunocytochemical (4G8, 10D5, and ubiquitin antibodies) examination, these brains showed no visible deposits of amyloid in the parenchyma or in the vasculature and no neurofibrillary tangles. Quantitative and qualitative studies of senile plaques and CAA were performed on 17 formalin-fixed AD brains; 7 were from the Sun Health Research Institute brain bank, and 10 were from the Neuropathology Laboratory, University of Southampton.
Tris-hydroxymethyl aminomethane (Tris), ethylenediaminetetraacetic acid, leupeptin, pepstatin, phenylmethylsulfonyl fluoride, phenanthroline, benzamidine, and thioflavin S were from Sigma Chemical Co. (St. Louis, MO). Gentamicin sulfate, amphotericin B, hydrochloric acid, and formic acid were obtained from Fluka Chemie AG (Buchs, Switzerland). On all occasions, formic acid was glass distilled in our laboratory before use. Antibody 4G8 (against Aβ17–24) was obtained from Senetek (Maryland Heights, MO) and was labeled with europium according to the manufacturer’s procedure (Wallac Inc., Gaithersburg, MD). Eu and enhancement solution were from Delfia (Wallac, Inc.). Polyclonal antibodies R163 and R165 specifically raised against Aβ40 and Aβ42, respectively, were obtained from Dr. P. Mehta of the New York State Institute for Brain Research (Staten Island, NY). These antibodies do not exhibit cross-reactivity between each other up to 50 ng/ml. For immunocytochemistry on paraffin sections, an antibody raised against Aβ1-39/42 was kindly donated by Dr. C. Wischik of the MRC Laboratory for Molecular Biology (Cambridge, UK).
Europium Immunoassay of Aβ Peptides
Cerebral cortex (0.8 g) from the superior frontal gyrus was carefully separated from the underlying white matter. The tissue was minced into fine pieces and, to remove unwanted blood, rinsed three times with 5 ml of protease inhibitor buffer (20 mmol/L Tris-HCl, pH 8.5, 3 mmol/L ethylenediaminetetraacetic acid, 500 μg/L leupeptin, 700 μg/L pepstatin, 35 mg/L phenylmethylsulfonyl fluoride, 100 mg/L 1,10 phenanthroline, 100 mg/L benzamidine, 50 mg/L gentamicin sulfate, and 250 μg/L amphotericin B). The tissue was thoroughly homogenized with a motorized Duall tissue grinder (100 μm clearance) in the presence of 3 ml of the above buffer. The brain homogenates, 4 ml per sample, were spun at 100,000 × g for 1 hour at 4°C. The supernatants and pellets were collected and stored separately. For Aβ quantitation, polyclonal antibodies R163 and R165 were coated to the wells of microtiter plates and used as the capture antibodies. Bovine serum albumin (1%) in TTBS (0.05% Tween in 20 mmol/L Tris-HCl and 0.5 mol/L NaCl, pH 7.4) was used as blocking solution. One hundred μl of the supernatant or of Aβ standards was applied to the wells and allowed to stand at room temperature for 2 hours on a rocking platform. The unbound materials were removed by washing the plate three times with TTBS. Europium-labeled 4G8 was added to the wells and incubated for 2 hours followed by four washes with TTBS and two washes with deionized water. Finally, the Eu-enhancement solution was added to each well and the plates were read in a fluorimeter using excitation and emission wavelengths of 320 and 615 nm, respectively. The values, obtained from triplicated wells, were calculated based on standard curves generated on each plate.
Quantitation of Amyloid Angiopathy in Relation to Amyloid Plaque Density
A total of 24 blocks of cerebral cortex were taken from 3 AD brains. From each brain, two blocks were taken from frontal, temporal, parietal, and occipital lobes. The leptomeningeal vessels were carefully stripped from the surface of the blocks of brain, stained free floating with thioflavin S, and spread onto glass slides for observation in either ultraviolet light (395 nm) or visible light (450 nm). Aβ deposition in leptomeningeal vessels was recorded on a semiquantitative scale in these preparations: −, no evidence of amyloid; +, moderate amyloid deposition and no aneurysm formation on the vessels; ++, clear evidence of amyloid angiopathy and some vessels with occasional aneurysm formation; +++, obvious amyloid angiopathy in the majority of vessels; and ++++, virtually all vessels showing evidence of amyloid angiopathy and multiple aneurysms.
Blocks of cerebral cortex from which the leptomeninges had been stripped were dehydrated and embedded in paraffin, and 5-μm sections were stained with thioflavin S. The total number of thioflavin S-positive amyloid plaques in the gray matter on each slide (mean area, 30 mm2) was counted and the number of plaques per unit area of cortex (plaque density) was estimated using Color Vision version 1.7.4.A on an Apple Macintosh Quadra 700 computer. The semiquantitative estimation of thioflavin S-stained amyloid deposition in the leptomeningeal vessels was compared with the plaque density in the underlying cortex.
Quantitation of Amyloid According to Vessel Type
A total of 56 blocks of cerebral cortex were taken from seven AD brains. Two blocks were taken from the frontal, temporal, parietal, and occipital cortex of each brain. Leptomeningeal vessels were carefully stripped from the brain’s surface and histologically processed, sectioned (5 μm), and stained with thioflavin S. A total of 681 arteries and 352 veins were investigated. For immunocytochemical studies, 5-μm sections were stained with an antibody against Aβ1-39/42. The number of diffuse and fibrillar plaques in the entire cortical area of each section was counted using a Leitz Laborlux K light microscope with a ×20 objective lens. The area of gray matter was measured in serial sections stained with the hematoxylin van Gieson technique using enhanced image analysis. Cortical plaque densities were calculated for each plaque type by dividing the relevant plaque count by the area of gray matter analyzed.
Assessment of amyloid angiopathy was performed on paraffin sections of leptomeningeal vessels stained with thioflavin S and examined in ultraviolet light under a fluorescence microscope. All vessels that had been sectioned transversely (ie, at no point was the maximum diameter more than twice the minimum diameter) were assessed. The diameter of each vessel was recorded using an eyepiece graticule. Identification of vessels as arteries and veins depended on the ratio of wall thickness to lumen diameter and the presence or not of an elastic lamina.
Qualitative Histology
Five brains from patients with AD were used for the isolation of intracortical and leptomeningeal vessels for qualitative assessment of the deposition of amyloid. Cerebral cortex from all four cerebral lobes were cut into approximately 1-cm 3 pieces and lysed in 20 volumes of 15% sodium dodecyl sulfate, prepared in 50 mmol/L Tris-HCl (pH 7.5) buffer containing 5 mmol/L ethylenediaminetetraacetic acid. After 72 hours of continuous stirring and two changes of sodium dodecyl sulfate, the remaining tufts of blood vessels were filtered out, washed with buffer, and stained with thioflavin S. Leptomeningeal vessels, stripped from the surface of cerebral cortex, were spread on glass slides, air dried, stained with thioflavin S, and examined in a fluorescence microscope by phase contrast and by Nomarski optics. Selected vessels were examined with a Leica TCS4D Krypton-Argon confocal laser scanning microscope; the image of thioflavin S fluorescence under light of 490-nm wavelength was digitally captured in 64 planes, allowing three-dimensional reconstruction.
Electron Microscopy
Cerebral cortex from three AD brains was taken fresh postmortem, fixed in glutaraldehyde, postfixed in osmium, stained with uranyl acetate, and embedded in Epon Araldite resin for electron microscopy. Thin sections of cerebral cortex were stained with lead citrate and examined in a Phillips 400 transmission electron microscope.
Results
Water-Soluble Aβ in Control and AD Brains
In the six brains from clinically nondemented control cases, Europium immunoassay demonstrated an average value of water-soluble Aβ of 2.75 ng/g of cerebral cortex (range, 0.2 to 8.6 ng/g). In only one instance was the level of water-soluble Aβ below the detection limit of 0.1 ng/g. Although this level is in contrast to the average of 36.8 ng/g of water-soluble Aβ in AD brains that we have previously demonstrated, 23 it nevertheless shows that Aβ is present in a water-soluble pool of non-AD brain tissue without visible deposits of Aβ.
Correlation between CAA and Plaque Density
Detailed examination of thioflavin S-stained sections of brain and overlying meninges stripped from the cerebral cortex showed that amyloid angiopathy did not occur in the absence of amyloid deposition in senile plaques in the underlying cortex. A positive correlation between the degree of amyloid angiopathy and the deposits of fibrillar amyloid in the subjacent cortex was observed up to the +++ levels of CAA (P < 0.05; Table 1 ▶ ). However, this increase was not maintained with the grade ++++ CAA.
Table 1.
Degree of Amyloid Angiopathy in Leptomeningeal Vessels Compared with Density of Fibrillar Amyloid Plaques in the Underlying Cortex: Thioflavin S.
| Severity of amyloid angiopathy | Plaques per mm2 cortex ±SD†† |
|---|---|
| −* | 0.00 |
| + | 0.23 ± 0.27 |
| ++ | 0.7 ± 0.4 |
| +++ | 1.04 ± 0.5† |
| ++++ | 0.33 ± 0.34 |
*No amyloid angiopathy.
†The mean density of plaques in the cortex underlying vessels with grade +++ amyloid angiopathy is significantly higher than plaque density with no (−), +, and ++ grades of amyloid angiopathy (P = 0.05).
††Thioflavin S-stained plaques were up to 100 times less numerous than those identified by Aβ immunocytochemistry.
In a parallel study in which senile plaques were immunocytochemically stained, a significant correlation between the number of plaques in the cerebral cortex and the extent of CAA in the overlying meninges, detected by thioflavin S, was observed in four of seven cases (P < 0.05). The r values in these cases ranged from 0.6697 to 0.9139 with a mean of 0.7817. In the other three cases examined, no such correlation was observed, except that CAA did not occur in the absence of amyloid plaques in the associated cortex.
Accumulation of Amyloid in Walls of Arteries and Veins of Different Calibre
In paraffin sections of leptomeninges stained with thioflavin S, amyloid deposition was detected in the walls of 20.2% of 681 arteries and in 4.5% of 352 veins (Table 2) ▶ . This represents a ratio of 5:1 for amyloid deposition in the walls of arteries compared with veins. Deposition of amyloid in arterial walls was observed in 24.7% of small leptomeningeal arteries (less than 60 μm in diameter) and in 11.2% of larger arteries (60 to 300 μm in diameter), giving a ratio of 2:1 between the two groups. No difference in the degree of amyloid deposition between veins of large and small diameter was seen. Amyloid deposition in intracortical vessels followed a similar pattern to that observed in the leptomeninges with arteries predominating over veins.
Table 2.
Amyloid Deposition in Leptomeningeal Vessels: Thioflavin S Stain
| Nature of vessels | Number counted | Proportion of vessels positive for amyloid |
|---|---|---|
| Total arteries | 681 | 20.3% |
| Total veins | 352 | 4.4% |
| Arteries <60 μm in diameter | 458 | 24.7% |
| Arteries >60 μm in diameter | 223 | 11.2% |
| Veins <60 μm in diameter | 193 | 4.5% |
| Veins >60 μm in diameter | 159 | 4.2% |
Distribution of Aβ in Intracortical Blood Vessels
Treatment of cerebral cortex from brains of patients with AD with 10% SDS resulted in isolation of the blood vessels; vessel basement membranes, perivascular connective tissue, and amyloid deposits were preserved, because these structures are insoluble in the detergent. Thioflavin S-stained preparations revealed globular deposits of amyloid arranged like beads along small blood vessels (Figure 1A) ▶ with linear deposits of amyloid outlining the intervening blood vessel walls (Figure 1B) ▶ . Arteries of larger diameter within the cortex contained band-like deposits of amyloid distributed transversely, or circumferentially around the vessel wall (Figure 1C) ▶ . Immunocytochemistry for Aβ in paraffin sections showed that the band-like disposition of the amyloid was due to the interposition of smooth muscle cells between the Aβ deposits (Figure 1D) ▶ ; preserved smooth muscle cells were clearly seen surrounded by Aβ. At the surface of the brain, Aβ was observed in the walls of arteries as they penetrated the cortex, in leptomeningeal arteries, and deposited at the glia limitans (Figure 1D) ▶ .
Figure 1.

CAA of cortical vessels in AD. Aβ deposits associated with intracortical vessels, isolated by 10% sodium dodecyl sulfate treatment, stained by thioflavin S, and viewed in a fluorescence microscope (A to C). A: Globular deposits of amyloid are arranged along a small intracortical blood vessel. B: Linear deposits of amyloid (arrow) outline the wall of a small intracortical blood vessel between the globular deposits. C: Intracortical arteries showing transverse bands of amyloid deposition in the vessel walls. D: Paraffin section stained by immunocytochemistry for Aβ. Two leptomeningeal arteries can be seen on the right of the picture. In the larger vessel (top right), Aβ is deposited in the adventitial region and the media is intact. The smaller vessel sends a branch into the cortex; smooth muscle cells in the media of this artery are surrounded by amyloid deposits producing a band-like appearance similar to that seen in thioflavin S preparations. Amyloid is also present at the glia limitans on the surface of the cortex (arrow).
Electron microscopy of intracortical arteries showed deposition of amyloid within dilated perivascular spaces separating the glia limitans from the apparently intact vessel wall (Figure 2A) ▶ . In other arteries, amyloid was deposited between smooth muscle cells with considerable disruption of the vessel wall (Figure 2B) ▶ . Thin profiles of smooth muscle were observed within the amyloid, and smooth muscle cells in the subintimal region appeared to be better preserved than those in the outer aspects of the wall (Figure 2B) ▶ .
Figure 2.
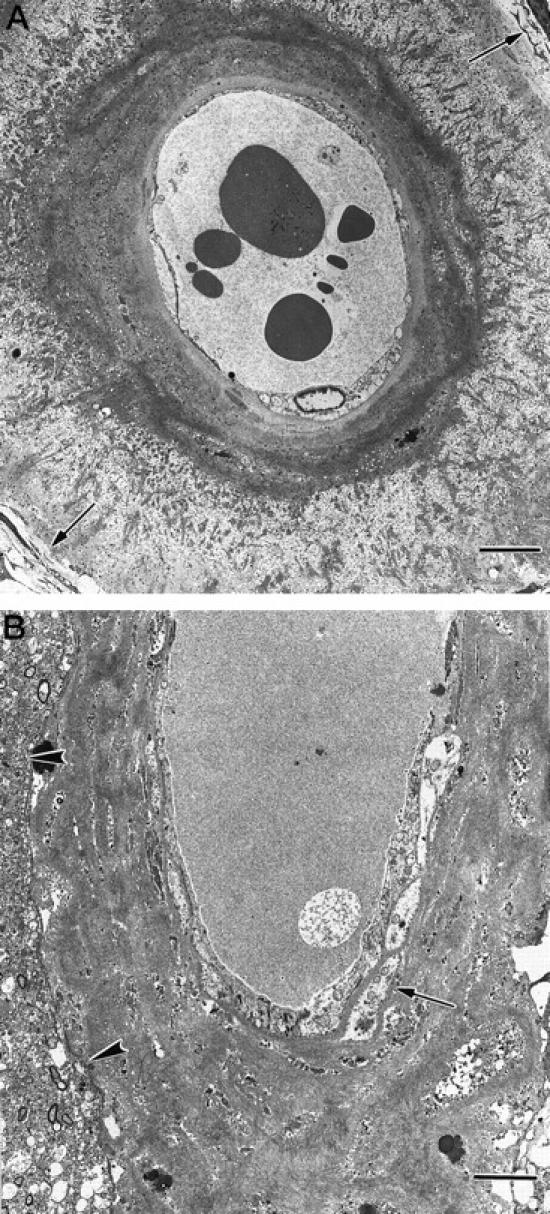
CAA of cortical vessels in AD. Transmission electron micrographs of intracortical arteries showing the pattern of deposition of amyloid in perivascular spaces and in the media. A: The perivascular space of this small intracortical artery is expanded by darkly stained wisps of amyloid, separating the glia limitans (arrows, top right, and bottom left) from the smooth muscle cells of the intact arterial media. Bar = 5 μm. B: Artery in which amyloid has been deposited within the perivascular space and between smooth muscle cells in the media. Glia limitans (arrowheads). Deposition of amyloid is heavier in the outer aspect of the media than in the inner aspects, where the smooth muscle cells remain better preserved (arrow). Bar = 5 μm.
Distribution of Aβ in Leptomeningeal Blood Vessels
The pattern of Aβ deposits in the walls of leptomeningeal arteries was similar to that in the larger intracortical arteries. Bands of amyloid were disposed intermittently along the length of arteries, often concentrated distal to bifurcations (Figure 3A) ▶ . In areas where amyloid deposition was heavy, aneurysm formation occurred either as fusiform expansion of the vessel (Figure 3A) ▶ or as more saccular extrusions (Figure 3B) ▶ . In some instances, the microaneurysms were surrounded by fine deposits of hemosiderin, suggesting past hemorrhage.
Figure 3.
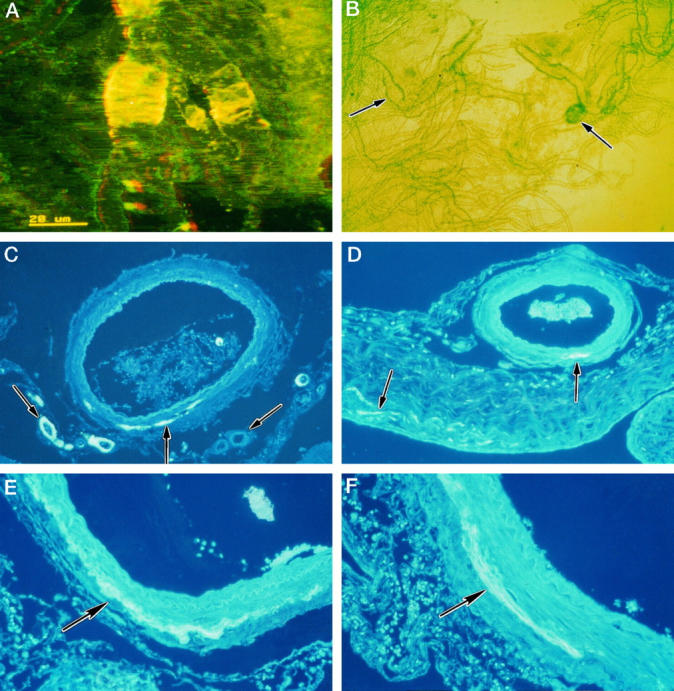
CAA of leptomeningeal arteries in AD. A: Isolated leptomeningeal artery showing heavy deposition of amyloid as transverse bands within its walls, associated with aneurysmal dilatation. Confocal microscopy: three-dimensional reconstruction, thioflavin S stain. B: Isolated leptomeningeal vessels viewed by phase-contrast microscopy showing a fusiform microaneurysm (left arrow) and a saccular microaneurysm (right arrow). C to F: Patterns of amyloid deposition in paraffin sections of leptomeningeal vessels and arachnoid stained with thioflavin S. C: Some smaller arteries show amyloid deposition throughout the thickness of their walls (left arrow), whereas other small vessels show complete absence of amyloid (right arrow). In the larger vessel, amyloid is deposited as a linear streak (middle arrow) in the adventitia and perivascular space. D: Leptomeningeal artery with a small streak of amyloid deposited at the junction of the media and adventitia (right arrow). The associated arachnoid mater also contains small streaks of amyloid (left arrow). E: Part of the wall of a leptomeningeal artery showing a linear circumferential deposit of amyloid at the junction of the adventitia and the media (arrow). F: Linear deposit of amyloid (arrow) in the wall of a leptomeningeal artery; the deposit is mostly in the adventitia near the perivascular space but also extends into the outer media.
Paraffin sections stained with thioflavin S revealed patchy deposition of amyloid in the small leptomeningeal arteries; either the whole thickness of the artery wall was involved or amyloid was completely absent (Figure 3C) ▶ . Small deposits of Aβ in the larger leptomeningeal arteries were always at the periphery of the tunica media where it meets the adventitia close to the periarterial space (Figure 3, C and D) ▶ . Early deposits of amyloid were never observed in the vicinity of the endothelium or the internal elastic lamina. As the amount of amyloid increased, deposits involved the outer part of the media in addition to the more peripheral regions of the artery wall (Figure 3, E and F) ▶ . Occasionally, small deposits of amyloid were present in the connective tissue of the arachnoid mater (Figure 3D) ▶ , but they were always extremely small in comparison with deposits in arterial walls. Deposition of amyloid in leptomeningeal veins was much less common than in arteries (Table 2) ▶ and very small in amount.
Discussion
The results of the present study suggest that Aβ peptides are produced by neural tissue and drain with ISF along periarterial pathways which, by analogy with other species, 4,24 are the lymphatic drainage pathways of the human brain. 15 Deposition of Aβ peptides in such drainage pathways may contribute significantly to CAA in AD.
Figure 4 ▶ summarizes diagrammatically the distribution of Aβ in CAA emphasized in the present study. Aβ forms globular and linear deposits in association with the walls of small intracortical vessels and band-like deposits in the walls of larger arteries in the cortex. In the leptomeninges, it is the small arteries that are most severely affected by Aβ deposition; larger arteries are relatively or completely spared, and veins show only minor Aβ deposition. The small, and probably early, deposits of Aβ in leptomeningeal artery walls are in the adventitia and thus at the site of the putative periarterial drainage pathways for ISF from the brain. It is unclear, as yet, whether such drainage pathways in humans have the same immunological significance as those in other species 4,13,14 or to what extent ISF leaks into the cerebrospinal fluid (CSF) of the subarachnoid space. 15 The high concentration of Aβ in perivascular compartments around arteries and the relatively small amounts of Aβ in the arachnoid do suggest, however, that substantial amounts of Aβ remain within perivascular ISF drainage pathways.
Figure 4.

Diagram summarizing the pattern of the distribution of amyloid in CAA associated with AD. a: Aβ accumulates as globules or linear deposits in the perivascular spaces of small intracortical blood vessels or as transverse bands in the walls of larger intracortical arteries and in smaller leptomeningeal arteries. The severity of amyloid angiopathy decreases with increasing size of the artery, suggesting that Aβ is precipitated to a greater extent in the initial portions of the pathways draining ISF from the brain. b: With increasing deposition, Aβ surrounds smooth muscle cells in the media. c: Eventually, smooth muscle cells are lost and aneurysms may form.
The existence of water-soluble Aβ in the brains of control individuals, those with Down’s syndrome, and AD subjects has been demonstrated previously. 23,25-27 In this study, we carefully selected six control cases without any apparent sign of Aβ deposition. Despite the absence of Aβ deposits, soluble Aβ could still be detected in the range of low ng/g of gray matter. These results suggest that, in the normal brain, there is a pool of soluble Aβ that has the ability to diffuse along the narrow extracellular spaces and to be eliminated along ISF drainage pathways. The pool of soluble Aβ is substantially increased in AD. 23 Both soluble and fibrillar Aβ accumulate in the glia limitans in the subpial region of the cortex and around vessels, reflecting the bulk flow of Aβ through the extracellular spaces.
In relation to CAA, the present study has shown that Aβ in AD brains accumulates around arteries five times more commonly than around veins, and that small arteries (<60μm diameter) in the leptomeninges are more severely involved than the larger arteries (60 to 300 μm in diameter). There is no selectivity of large or small veins in relation to the very small amount of Aβ deposited within their walls. Stainable fibrillar amyloid does not appear to accumulate within the walls of cerebral or leptomeningeal arteries larger than 500 μm in diameter. In general, therefore, there is an inverse gradient in the accumulation of Aβ related to the caliber of the artery; the larger the size of the artery, the smaller the amount of stainable amyloid. However, Aβ1-40 is detectable biochemically in the walls of the middle cerebral and basilar arteries even in young adults, 3 which suggests that soluble amyloid is in the perivascular spaces of even the largest arteries. Amyloid angiopathy was not observed in the absence of amyloid plaques in the underlying cortex, and a linear correlation between the severity of the CAA and the number of Aβ plaques in the underlying cortex was seen in four out of seven of the cases examined. All of these observations support the hypothesis that Aβ is eliminated from the brain along periarterial drainage pathways and accumulates in such pathways in CAA. As yet, it is not clear how ISF and Aβ in perivascular drainage compartments finally leave the cranial cavity and to which lymph nodes they drain.
Various hypotheses have been advanced to explain the origin and distribution of Aβ in blood vessel walls, and a number of mechanisms may contribute to CAA. 28
Hematogenous Origin
Blood-borne amyloid is known to be associated with angiopathy, as with the immunoglobulin amyloid of multiple myeloma. As the APP is normally expressed in platelets 29 and the blood levels of APP are increased in Down’s syndrome patients, 30 it is possible that APP in the blood may be subsequently deposited as Aβ in blood vessel walls. However, the present study and previous reports show that Aβ deposits in arterial walls are initially confined to the adventitia and outer media of the vessel wall rather than to the vicinity of the endothelium. 1,20-22 This abluminal site of deposition, in addition to the restriction of CAA to central nervous system vessels, 3 argues against a hematogenous origin for the amyloid. In blood-borne amyloidoses, such as multiple myeloma, arteries are affected throughout the body, whereas there is only one report of trace amounts of Aβ outside the central nervous system in AD. 31 In other amyloidoses, in which the amyloid precursor is known to be blood borne, amyloid is found in the walls of both arteries and veins. 32
Origin from CSF
The CSF could be a source of amyloid in CAA, particularly as Aβ is detected in CSF of normal and AD individuals, 33 and it has been suggested that amyloid is deposited in leptomeningeal vessels via the CSF in the transthyretin amyloidosis. 34 However, amyloid is much more frequently deposited in arteries than in veins, as has been shown in this and other studies, 22,28 and yet both types of vessel are exposed to the CSF. In addition, small arteries in the subarachnoid space are affected more commonly and more severely than larger arteries in the same location, yet they are all surrounded by CSF. Small intracortical arteries and capillaries are major sites for Aβ deposition, but they are at some distance from the CSF and separated from it by the pia mater. 35-37 These features argue strongly against the CSF in the subarachnoid space being the main source of Aβ in CAA.
Smooth Muscle Cells as a Source of Aβ
The relationships between the drainage of Aβ from brain tissue into the perivascular spaces in AD and the local production or uptake of amyloid by smooth muscle cells are a matter for conjecture. Aβ1-40 and Aβ1-42 are rapidly internalized by cultured human and canine smooth muscle cells derived from leptomeningeal vessels. Internalization appears to be in association with ApoE. 38 It is possible that the deposition of parenchymal amyloid in blood vessel walls may also stimulate the local production of Aβ, as the major species of Aβ that accumulate within the brain differ from those in vessel walls. Aβ in senile plaques consists mainly of the 1-42 species 39,40 whereas vascular amyloid is mostly composed of 1-40 species. 40,41 Aβ1-40 draining from the brain may be supplemented from Aβ1-40 produced by smooth muscle cells as part of a stress response to the accumulation of Aβ1-42, particularly as it has been shown that smooth muscle cells, when stimulated by exogenous Aβ, are capable of producing APP and Aβ. 42
Cerebral Cortex as a Source of Amyloid
The relative proportions of Aβ1-42 to Aβ1-40 are higher in cortical vessels than in leptomeningeal vessels, 40 which is consistent with neurons being the source of Aβ1-42 and the vasculature producing Aβ1-40 peptide in response. Cortical vessels are closer to the source of Aβ1-42 than leptomeningeal vessels and hence show deposition of proportionately more Aβ1-42. More convincing, however, is the observation that, whereas some vascular amyloid deposits in vessels are solely composed of Aβ1-42, deposits consisting of Aβ1-40 alone have not been recorded. 3 In hereditary cerebral hemorrhage with amyloidosis, Dutch type, 43 fibrillar Aβ1-40 is deposited in cortical and leptomeningeal vessel walls. In this disease, the Aβ in the neuropil is mainly of the diffuse, nonfibrillar, amorphous type, whereas fibrillar Aβ is only present in the glia limitans around the cortical vessels. 44 This suggests that Aβ1-42, derived from the brain, may be the initial amyloid to be deposited in vessel walls and that it entraps the more soluble Aβ1-40, which normally drains from the brain along perivascular spaces more easily than Aβ1-42. Soluble Aβ could be sequestered by the basal laminae of vascular endothelial cells, pericytes, and/or smooth muscle cells; such basal laminae are rich in proteoglycans for which Aβ has a high affinity. 45 Finally, the solubility properties of Aβ1-40 may permit this molecule to diffuse for larger distances than the less soluble Aβ1-42, resulting in an increased proportion of Aβ1-40 deposited around leptomeningeal vessels.
In conclusion, evidence from the pattern of distribution of Aβ in AD suggests that accumulation of Aβ in ISF drainage pathways contributes significantly to CAA 46 (Figure 4) ▶ . Similar criteria may be applicable to the amyloid angiopathy observed in other pathologies such as prion diseases 47 and cystatin C amyloidosis. 48 Amyloid, particularly Aβ1-40 peptide, is present in non-AD brains and has been detected in the walls of the large arteries of the circle of Willis in young adults. 3 These findings support the concept that, under normal circumstances, the brain eliminates Aβ peptides via periarterial ISF fluid drainage pathways. Aβ may be deposited within vessel walls, either through increased production of insoluble Aβ1-42, through the obstruction of ISF drainage pathways, or because of failure of solubility factors. Such deposition in turn may induce Aβ production by smooth muscle cells with further obstruction of drainage pathways and further accumulation of Aβ within the cerebral cortex and its vasculature in AD.
Consideration of possible solubility factors involved in the transport of Aβ along periarterial fluid drainage pathways may yield opportunities for therapeutically enhancing the elimination of Aβ from the brain. ApoE, for example, may act as a transport molecule for mobilization of Aβ1-40 in the ISF. 49 Because the predominant species of vascular amyloid is Aβ1-40, transport of Aβ from the brain along perivascular ISF drainage pathways may be defective in individuals with particular ApoE alleles, resulting in the accumulation of amyloid peptides in senile plaques and arterial walls. Compensating for ApoE or other solubility defects may reduce the deposition of amyloid in the brain.
Footnotes
Address reprint requests to RO Weller, Professor of Neuropathology, Pathology (Neuropathology), University of Southampton Medical School, Mailpoint 813, Southampton General Hospital, Southampton SO16 6YD, UK. E-mail: row@soton.ac.uk.
Supported in part by NIH grant AG-11925 (to AER) and by the Wessex Medical Trust Brain Fund and the David Gibson Fund (to ROW). MH is a recipient of VJ Chapman Research Fellowship from the New Zealand Neurological Foundation.
References
- 1.Wisniewski HM, Wegiel J, Kotula L: Some neuropathological aspects of Alzheimer disease and its relevance to other disciplines. Neuropathol Appl Neurobiol 1996, 22:3-11 [DOI] [PubMed] [Google Scholar]
- 2.Vinters HV, Wang ZZ, Secor DL: Brain parenchymal and microvascular amyloid in Alzheimer’s disease. Brain Pathol 1996, 6:179-195 [DOI] [PubMed] [Google Scholar]
- 3.Shinkai Y: Amyloid β-proteins 1-40, and 1-42(43) in the soluble fraction of extra-, and intracranial blood vessels. Ann Neurol 1995, 38:421-428 [DOI] [PubMed] [Google Scholar]
- 4.Cserr HF, Harling-Berg CJ, Knopf PM: Drainage of brain extracellular fluid into blood and deep cervical lymph and its immunological significance. Brain Pathol 1992, 2:269-276 [DOI] [PubMed] [Google Scholar]
- 5.Boulton M, Young A, Hay J, Armstrong D, Flessner M, Schwartz M, Johnston M: Drainage of CSF through lymphatic pathways and arachnoid villi in sheep: measurement of 125I-albumin clearance. Neuropathol Appl Neurobiol 1996, 22:325-333 [DOI] [PubMed] [Google Scholar]
- 6.Brinker T, Lüdemann W, Berens von Rautenfeld D, Samii M: Dynamic properties of lymphatic pathways for the absorption of cerebrospinal fluid. Acta Neuropathol 1997, 94:493-498 [DOI] [PubMed] [Google Scholar]
- 7.Szentistvanyi I, Patlak CS, Ellis RA, Cserr HF: Drainage of interstitial fluid from different regions of the rat brain. Am J Physiol 1984, 246:F835-F844 [DOI] [PubMed] [Google Scholar]
- 8.Zhang ET, Richards HK, Kida S, Weller RO: Directional and compartmentalised drainage of interstitial fluid and cerebrospinal fluid from the rat brain. Acta Neuropathol 1992, 83:233-239 [DOI] [PubMed] [Google Scholar]
- 9.Kida S, Pantazis A, Weller RO: CSF drains directly from the subarachnoid space into nasal lymphatics in the rat: anatomy, histology and immunological significance. Neuropathol Appl Neurobiol 1993, 19:480-488 [DOI] [PubMed] [Google Scholar]
- 10.Harling-Berg CJ, Knopf PM, Merriam J, Cserr HF: Role of the cervical lymph nodes in the systemic humoral immune response to human serum albumin microinfused into rat cerebrospinal fluid. J Neuroimmunol 1989, 25:185-193 [DOI] [PubMed] [Google Scholar]
- 11.Knopf PM, Cserr HF, Nolan SC, Wu T-Y, Harling-Berg CJ: Physiology and immunology of lymphatic drainage of interstitial and cerebrospinal fluid from the brain. Neuropathol Appl Neurobiol 1995, 21:175-180 [DOI] [PubMed] [Google Scholar]
- 12.Phillips MJ, Weller RO, Kida S, Iannotti F: Focal brain damage enhances experimental allergic encephalomyelitis in brain and spinal cord. Neuropathol Appl Neurobiol 1995, 21:189-200 [DOI] [PubMed] [Google Scholar]
- 13.Weller RO, Engelhardt B, Phillips MJ: Lymphocyte targeting of the central nervous system: a review of afferent and efferent CNS-immune pathways. Brain Pathol 1996, 6:275-288 [DOI] [PubMed] [Google Scholar]
- 14.Phillips MJ, Needham M, Weller RO: Role of cervical lymph nodes in autoimmune encephalomyelitis in the Lewis rat. J Pathol 1997, 182:457-464 [DOI] [PubMed] [Google Scholar]
- 15.Zhang ET, Inman CBE, Weller RO: Interrelationships of the pia mater and perivascular (Virchow-Robin) spaces in the human cerebrum. J Anat 1990, 170:111-123 [PMC free article] [PubMed] [Google Scholar]
- 16.Kida S, Ellison DW, Steart PV, Weller RO: Characterisation of perivascular cells in astrocytic tumours and oedematous brain. Neuropathol Appl Neurobiol 1995, 21:121-129 [DOI] [PubMed] [Google Scholar]
- 17.Weller RO: Fluid compartments and fluid balance in the central nervous system. ed 38 Williams PL eds. Gray’s Anatomy. 1995, :pp 1202-1224 Churchill Livingstone, Edinburgh [Google Scholar]
- 18.Pollock H, Hutchings M, Weller RO, Zhang E-T: Perivascular spaces in the basal ganglia of the human brain: their relationship to lacunes. J Anat 1997, 337–346 [DOI] [PMC free article] [PubMed]
- 19.Campora RG, Salaverri CO, Ramirez FV, Villaddiego MS, Davidson HG: Metastatic glioblastoma multiforme in cervical lymph nodes: report of a case with diagnosis by fine needle aspiration. Acta Cytologica 1993, 37:938-942 [PubMed] [Google Scholar]
- 20.Wisniewski HM, Wegiel J: β-Amyloid formation by myocytes of leptomeningeal vessels. Acta Neuropathol 1994, 87:233-241 [DOI] [PubMed] [Google Scholar]
- 21.Kawai M: Degeneration of vascular muscle cells in cerebral amyloid angiopathy of Alzheimer’s disease. Brain Res 1993, 623:142-146 [DOI] [PubMed] [Google Scholar]
- 22.Yamaguchi H: Beta amyloid is focally deposited within the outer basement membrane in the amyloid angiopathy of Alzheimer’s disease: an immunoelectron microscopic study. Am J Pathol 1992, 141:249-259 [PMC free article] [PubMed] [Google Scholar]
- 23.Kuo Y-M, Emmerling MR, Vigo-Pelfrey C, Kasunic TC, Kirkpatrick JB, Murdoch GH, Ball MJ, Roher AE: Water-soluble Aβ (N-40, N-42) oligomers in normal, and Alzheimer disease brains. J Biol Chem 1996, 271:4077-4081 [DOI] [PubMed] [Google Scholar]
- 24.Weller RO, Kida S, Zhang E-T: Pathways of fluid drainage from the brain: morphological aspects and immunological significance. Brain Pathol 1992, 2:227-284 [DOI] [PubMed] [Google Scholar]
- 25.Tabaton M, Nunzi MG, Xue R, Usiak M, Autilio-Gambetti L, Gambetti P: Soluble amyloid β-protein is a marker of Alzheimer amyloid in brain but not in cerebrospinal fluid. Biochem Biophys Res Commun 1994, 200:1598-1603 [DOI] [PubMed] [Google Scholar]
- 26.Tamaoka A, Kondo T, Odaka A, Sahara N, Sawamura N, Ozawa K, Suzuki N, Shoji S, Mori H: Biochemical evidence for the long-tail form (Aβ1-42/43) of amyloid β protein as a seed molecule in cerebral deposits of Alzheimer’s disease. Biochem Biophys Res Commun 1994, 205:834-842 [DOI] [PubMed] [Google Scholar]
- 27.Harigaya Y, Shoji M, Kawarabayashi T, Kanai M, Nakamura T, Iizuka T, Igeta T, Saido TC, Sahara N, Mori H, Hirai S: Modified amyloid β protein ending at 42 or 40 with different solubility accumulates in the brain of Alzheimer’s disease. Biochem Biophys Res Commun 1995, 211:1015-1022 [DOI] [PubMed] [Google Scholar]
- 28.Frautschy SA, Cole GM, Baird A: Phagocytosis and deposition of vascular β-amyloid in rat brains injected with Alzheimer β-amyloid. Am J Pathol 1992, 140:1389-1399 [PMC free article] [PubMed] [Google Scholar]
- 29.Bush A: The amyloid precursor protein of Alzheimer’s disease is released by human platelets. J Biol Chem 1990, 265:15977-15983 [PubMed] [Google Scholar]
- 30.Rumble B: Amyloid A4 protein, and its precursor in Down’s syndrome, and Alzheimer’s disease. N Engl J Med 1989, 320:1446-1452 [DOI] [PubMed] [Google Scholar]
- 31.Joachim CL, Mori H, Selkoe DJ: Amyloid β-protein deposition in tissues other than the brain in Alzheimer’s disease. Nature 1989, 341:226-230 [DOI] [PubMed] [Google Scholar]
- 32.Pepys MB: Amyloid, familial Mediterranean fever, and acute phase response. Weatherall DJ Ledingham JGG Warrell DA eds. The Oxford Textbook of Medicine. 1996, :pp 1512-1524 Oxford University Press, Oxford, UK, [Google Scholar]
- 33.Selkoe DJ: Physiological production of the β-amyloid protein and the mechanism of Alzheimer’s disease. Trends Neurosci 1993, 16:403-409 [DOI] [PubMed] [Google Scholar]
- 34.Vidal R: Meningocerebrovascular amyloidosis associated with a novel transthyretin missense mutation at codon 18 (TTRD18G). Am J Pathol 1966, 148:361-366 [PMC free article] [PubMed] [Google Scholar]
- 35.Hutchings M, Weller RO: Anatomical relationships of the pia mater to cerebral blood vessels in man. J Neurosurg 1986, 63:316-325 [DOI] [PubMed] [Google Scholar]
- 36.Alcolado R, Weller RO, Parrish EP, Garrod D: The cranial arachnoid and pia mater in man: anatomical and ultrastructural observations. Neuropathol Appl Neurobiol 1988, 14:1-17 [DOI] [PubMed] [Google Scholar]
- 37.Feurer DJ, Weller RO: Barrier functions of leptomeninges: a study of normal meninges and meningiomas in tissue culture. Neuropathol Appl Neurobiol 1991, 17:391-405 [DOI] [PubMed] [Google Scholar]
- 38.Urmoneit B, Prikulis I, Wihl G, D’Urso D, Frank R, Heeren J, Beisiegel U, Prior R: Cerebrovascular smooth muscle cells internalize Alzheimer amyloid β protein via a lipoprotein pathway: implications for cerebral amyloid angiopathy. Lab Invest 1997, 77:157-166 [PubMed] [Google Scholar]
- 39.Iwatsubo T: Visualisation of Aβ42(43) and Aβ(40) in senile plaques with end-specific Aβ monoclonals: evidence that an initially deposited species is Aβ42(43). Neuron 1994, 13:45-53 [DOI] [PubMed] [Google Scholar]
- 40.Roher AE: β-amyloid (1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer’s disease. Proc Natl Acad Sci USA 1993, 90:10836-10840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joachim CL: Protein chemical and immunocytochemical studies of meningovascular β-amyloid protein in Alzheimer’s disease and normal aging. Nature 1988, 474:100-111 [DOI] [PubMed] [Google Scholar]
- 42.Davis-Salinas J, Saporito-Irwin SM, Cotman CW, Van Nostrand WE: Amyloid β-protein induces its own production in cultured degenerating cerebrovascular smooth muscle cells. J Neurochem 1995, 65:931-934 [DOI] [PubMed] [Google Scholar]
- 43.Levy E: Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248:1124-1126 [DOI] [PubMed] [Google Scholar]
- 44.Tagliavini F, Giaccone G, Bugiani O, Frangione B: Ubiquinated neurites are associated with preamyloid and cerebral amyloid β deposits in patients with hereditary cerebral hemorrhage with amyloidosis Dutch type. Acta Neuropathol 1993, 85:267-271 [DOI] [PubMed] [Google Scholar]
- 45.Snow AD, Kinsella MG, Parks E, Sekiguchi RT, Miller JD, Kimata K, Wight TN: Differential binding of vascular cell-derived proteoglycans (perlecan, biglycan, decorin, and versican) to the β-amyloid protein of Alzheimer’s disease. Arch Biochem Biophys 1995, 320:84-95 [DOI] [PubMed] [Google Scholar]
- 46.Massey AP, Newman TA, Roher A, Weller RO: β-Amyloid accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Neuropathol Appl Neurobiol 1997, 23:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghetti B, Piccardo P, Frangione B, Bugiani O, Giaccone G, Young K, Prelli F, Farlow MR, Dlouhy SR, Tagliavini F: Prion protein amyloidosis. Brain Pathol 1996, 6:127-145 [DOI] [PubMed] [Google Scholar]
- 48.Olafsson I, Thorsteinsson L, Jensson O: The molecular pathology of hereditary cystatin C amyloid angiopathy causing brain hemorrhage. Brain Pathol 1996, 6:121-126 [DOI] [PubMed] [Google Scholar]
- 49.Rebeck GW, Reiter JS, Strickland DK, Hyman BT: Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron 1993, 11:575-580 [DOI] [PubMed] [Google Scholar]