

Abstract
Monoclonal antibodies were raised to two regions of calpain 3 (muscle-specific calcium-activated neutral protease), which is the product of the gene that is defective in limb-girdle muscular dystrophy type 2A. The antibodies produced characteristic patterns of bands on Western blots: normal calpain 3 protein was represented by bands at 94 kd, plus additional fragments at ∼60 or 30 kd, according to the antibody used. Specificity was confirmed by the loss of all bands in patients with null gene mutations. The “normal” profile of bands was observed in muscle from 33 control subjects and 70 disease-control patients. Calpain 3 protein was found to be extremely stable in fresh human muscle, with full-size protein being detected 8 hours after the muscle had been removed. Blots of muscle from nine limb-girdle muscular dystrophy type 2A patients with defined mutations showed variation in protein expression, with seven showing a clear reduction in the abundance of protein detected. No simple relationship was found between the abundance and clinical severity. Two patients showed normal expression of the full-size 94 kd band accompanied by a clear reduction in the smaller fragments. This pattern was also observed in one patient with an undefined form of limb-girdle dystrophy. These results indicate that immunodiagnosis is feasible, but caution will need to be exercised with the interpretation of near-normal protein profiles.
To date, at least seven forms of autosomal recessive muscular dystrophy (MD) have appeared under the “umbrella” name of limb-girdle muscular dystrophy (LGMD). These forms are in two groups: those with abnormal expression of the dystrophin-glycoprotein complex, and those in which labeling of proteins in this complex is unaffected. Thus, the sarcoglycanopathies (sometimes known as LGMD types 2C, 2D, 2E, and 2F), are caused by defects in the genes for γ-, α-, β- or δ-sarcoglycan on chromosomes 13q12, 17q12, 4q12, and 5q33, respectively. 1-4 Among the dystrophies in which expression of the sarcoglycans is normal, the gene responsible for LGMD2A has been identified as the chromosome 15q15.1 to q21.1-encoded muscle-specific calcium-activated neutral protease (CAPN3), calpain 3. 5 The genes for LGMD2B and LGMD2G have been localized to 2p13 6 and 17q11, 7 and further recessive limb-girdle genes have been inferred. 7,8
The various forms of limb-girdle dystrophy are very difficult to differentiate on clinical grounds alone. 9 Thus, testing for defective protein expression is a very useful way of determining which gene or genes are at fault and therefore where to start the search for gene mutations. 10 Unlike the large multiexonic deletions that are the most common type of mutation in Duchenne and Becker dystrophy, the limb-girdle dystrophies typically have mutations that are smaller and more difficult to detect. The sarcoglycan group has very close interactions, and a mutation in one member protein may cause an absence of protein expression for all four. 4,11 As more patient biopsies are examined, however, a common observation (particularly in γ-sarcoglycanopathy) appears to be the deficiency or absence of labeling for one member accompanied by a more moderate reduction in the others. 12-15 In these cases the protein with the most severely altered expression has proved to be the one with the mutated gene, indicating that protein analysis may be useful even in the sarcoglycanopathies. 10,15
Unlike the other forms of LGMD, LGMD2A is not caused by defective expression of a structural protein, but by mutations in the gene for a soluble proteolytic enzyme. The calpain family of proteins contains three ubiquitous enzymes that have been studied extensively (see reviews in Refs. 16 and 17 ). They were named μ-calpain (calpain 1), m-calpain (calpain 2), and intermediate μ/m-calpain, after their distinct sensitivities to Ca2+ ions. All are dimers composed of distinct large catalytic subunits (around 80 kd) plus a shared regulatory small subunit of 30 kd. The large calpain subunits have four functional domains. 16 The N-terminal region of domain I is autocatalytically processed during activation by Ca2+ ions, suggesting that domain I is involved in the regulation of activity. Domain II is considered to be the cysteine protease module, and no function has yet been assigned to domain III. Domain IV contains structures for calcium binding, and thus this domain may be involved in the calcium activation of the calpains.
The founder members of this family have now been joined by an increasing number of tissue-specific calpains, but their precise roles remain uncertain. 17 The muscle-specific form, calpain 3, differs in structure from the ubiquitous forms by having three unique insertions (designated NS at the beginning of domain I, IS1 in protease domain II, and IS2 in domain III) which increase the molecular mass from 80 to 94 kd. Calpain 3 was originally named “p94” from this size. 18 The mRNA of a calpain 3 variant, predicted to code for an 82-kd protein, has recently been found in the lens of both rat 19 and mouse (M. Herasse et al, manuscript in preparation). This appears to differ in that the NS, IS1, and IS2 muscle-specific sequences are spliced out.
The use of calpain 3 protein expression to detect mutations in the CAPN3 gene for the diagnosis of LGMD2A has just started. Spencer et al 20 reported the use of polyclonal antibodies that recognized calpains 1, 2, and 3 in skeletal muscle to differentiate LGMD2A samples from others in a “blind” study. Here we report the first production of monoclonal antibodies to calpain 3, the characterization of their reactivity, and their use in analyzing protein expression in a group of LGMD2A patients with known mutations and clinical profiles.
Materials and Methods
Immunogens and Antibody Production
Two synthetic peptides from the published human CAPN3 sequence 5 were conjugated to keyhold limpet hemocyanin via an additional C residue and used to immunize CD1 mice. One peptide contained amino acids 1 to 19 at the N terminus (MPTVISASVAPRTAAEPRS-C) in the calpain 3-specific NS domain, and the other consisted of amino acids 355 to 370 (C-RLRNPWGQVEWNGSWS) in protease domain II, which is a region of sequence conservation between calpains 1, 2, and 3. This peptide corresponded to the human version of the chicken sequence used previously to raise polyclonal antibodies. 20 The mice were immunized over a period of 6 months, during which time several tail bleeds were taken, and mice were killed for unsuccessful fusion experiments. The experiments were conducted under a British Home Office license, and at the end of the specified 6-month time limit, the remaining mice had to be killed. The mice were therefore boosted before being killed, the splenocytes were frozen in medium containing 20% fetal calf serum + 10% dimethyl sulphoxide, and the cells were stored in liquid nitrogen. The final successful fusions, reported here, were performed on spleen cells that were rapidly thawed before immediate fusion with X63.Ag8.653 cells (assumed recovery of 5 × 10 7 viable splenocytes per spleen, with 5 × 10 6 myeloma cells). The antibody supernatants were screened on strips from Western blots of human muscle extracts. No significant labeling was obtained on unfixed frozen tissue sections with any of the antibodies. The cells in “positive” wells, which labeled bands of the correct size on Western blots, were cloned at least four times at 0.5 cells/well to ensure monoclonality. Specificity to calpain 3 was determined by loss of band reactivity in muscle from patients known to have null mutations in that gene.
Electrophoresis and Western Blotting
Standard buffers for electrophoresis and blotting were employed, 21 although we now routinely use a biphasic system that is optimized to permit resolution of all the known muscular dystrophy proteins on a pair of gels/blots. 22 Thus, the lower half of the gel contained 7% acrylamide (for resolving calpain 3, merosin, and the sarcoglycans, in the molecular mass range of 30 to 100 kd), whereas the upper half contained a gradient of 5.5 to 4% (for resolving myosin heavy chain and dystrophin in the range of 200 to 400 kd). A 3% stacking gel was used. The frozen tissue samples were weighed and kept frozen until homogenized with 19 volumes of electrophoresis treatment buffer containing 4% sodium dodecyl sulphate and 4 mol/L urea (no additional protease inhibitors). Lanes of control muscle (with no fat or fibrous connective tissue) typically contained 200 μg of protein. 21 After electrophoresis the gels were blotted, labeled with the antibodies (Calp3c/11B3 used undiluted, Calp3d/2C4, and Calp3c/12A2 diluted 1:10) followed by a peroxidase-conjugated secondary antibody, and visualized with hydrogen peroxide and diaminobenzidine. 21
Densitometric Analysis
Dried gels and blots were scanned at 400 dpi on an Epson GT8000 flatbed scanner using white light for gels and blue for blots. Each image was stored as a bit map where 8 bits = 1 byte = 1 pixel. Each pixel was graded from 0 (pure black) to 255 (pure white) on a 256 grayscale. Grayscale is not proportional to concentration, but optical density (OD) is according to Beer’s law, and is therefore the proper measure for use in quantitation. Software that was written by one of us (KD) for the Optimas v5.2 image analysis package was used for the densitometric analysis. In this, grayscale values were converted to OD units by scanning a Kodak SR-37 step wedge that has 37 steps of OD values from 0.05 to 1.85. The grayscale values measured for each step were then plotted against the true OD values, so that the scanner was calibrated as a true densitometer. Subsequently, the grayscale value for each pixel was automatically converted to an OD value. The outline of each band was defined by a software algorithm involving background measurements, to obtain a value for volume OD where:
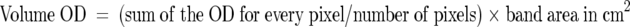 |
Pathological muscle samples contain a variable amount of fat and fibrous connective tissue. Therefore, myosin heavy chain staining on the postblotted gel was used as an indication of how much true muscle protein was loaded in each sample. Thus, the volume OD for each protein band in a sample was divided by the corresponding value for myosin heavy chain in that sample, to produce a density value that was “normalized volume OD.” 22
Human Muscle Samples from Normal and Disease Control Patients
Biopsies from patients with different neuromuscular diseases were obtained as part of routine diagnostic procedures at Newcastle General Hospital. These were stored in liquid nitrogen until required. More than 70 samples from patients with neuromuscular diseases have now been examined. Samples of rectus abdominis muscle were obtained, with informed consent, from normal control subjects undergoing a variety of abdominal operations. Control samples were also obtained from amputated legs. Altogether, samples from 33 different individuals without neuromuscular disease were examined.
LGMD2A Patients and Their Mutations
Nine patient biopsies from eight families with LGMD2A (from Réunion Island, Germany and Spain) were obtained for diagnostic procedures. Mutation analysis was undertaken at Généthon in Paris, 5,23 Rudolf Virchow Klinikum, Humboldt-Universität, Berlin, 24 and Unitat de Genètica Molecular, Hospital de la Santa Creu i Sant Pau, Barcelona. Clinical assessments were made by MF, CH, and JC.
Immunoreactivity of Calpain 3 Antibodies in Different Animals and Tissues
Because fresh human tissues are difficult to obtain, samples from a number of different animals were analyzed. Thus, skeletal muscle samples from human, mouse, rat, rabbit, dog, chicken, hamster, and pig were compared on the same blots labeled with calpain 3 antibodies. Subsequently, we examined samples of rabbit skeletal muscle, vas deferens, heart, brain, spinal cord, thymus, lung, kidney, spleen, and liver, and rat skeletal muscle, stomach, uterus, heart, brain, lung, kidney, spleen, and liver.
Stability of Calpain 3 Protein in Muscle
Three sets of experiments were conducted to investigate the stability of calpain 3 in normal human rectus abdominis muscle and to elucidate the nature of the lower molecular mass bands recognized by the antibodies.
Time before Freezing Fresh Tissue
A sample of fresh rectus abdominis muscle was taken and divided into eight portions (each ∼50 mg) in a lidded Petri dish lined with filter paper moistened with phosphate-buffered saline. The samples were left at room temperature to emulate conditions that might occur if a biopsy sample were taken in a distant hospital and sent to our laboratory for processing and freezing. The fresh muscle samples were snap frozen, by dropping them in liquid nitrogen, at time points designated T0 (the quickest achieved, actually about 10 minutes after leaving the body), 30 minutes, 1 hour, 1.5 hours, 2 hours, 3 hours, 4 hours, and 8 hours.
Time Thawed before Homogenization
A sample of frozen rectus abdominis muscle was broken with a brass pestle and mortar chilled in liquid nitrogen. Pieces of tissue, each weighing between 25 and 30 mg, were left in tubes to thaw at room temperature for different lengths of time before homogenization. The time points were designated T0 (homogenized frozen), 5 minutes, 10 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, and 2 hours.
Time as Homogenate in Saline
A sample of frozen rectus abdominis muscle was homogenized in an equal volume (1 g/1 ml) of sterile saline (Sterets Normasol, Seton Prebbles, Oldham, UK). The homogenate was left for different lengths of time at room temperature before 100-μl samples were taken and mixed with an equal volume of double-strength electrophoresis treatment buffer, before proceeding with boiling and centrifugation as normal. This resulted in a sample of 50% normal protein loading.
Results
Antibodies
Three different monoclonal antibodies were isolated. One, designated Calp3d/2C4, binds to an epitope within amino acids 1 to 19 in exon 1 of the human sequence and recognized a single band at ∼94 kd plus an additional clear band at about 30 kd in blots of human skeletal muscle (Figure 1) ▶ . The other two bind to epitopes within amino acids 355 to 370 in exon 8. One, designated Calp3c/11B3, recognized at least four bands from 94 to 80 kd (see expanded view in Figure 1 ▶ ) corresponding to calpains 1, 2, and 3, exactly like the polyclonal antiserum used in Spencer et al. 20 The other, designated Calp3c/12A2, only recognized the uppermost calpain 3 band. Both of the exon 8 antibodies also recognized a series of bands from a fairly clear 60 kd down to a more fuzzy 45 kd (Figure 1) ▶ . The immunoreactivity of each antibody was abolished by adsorption with the appropriate immunizing peptide, whereas adsorption with the other peptide had no effect. Analysis of the clinical samples was undertaken with the two calpain 3-specific antibodies only. The results with muscle from LGMD2A patients with null mutations (see below) demonstrated that the bands at 94 kd, ∼60 kd, and 30 kd all represent products of the CAPN3 gene.
Figure 1.

Three strips from a Western blot of human skeletal muscle labeled with different monoclonal antibodies (indirect peroxidase). On the right is an expanded view of the bands in the region of 78 to 94 kd. Approximate molecular masses for the principal bands of interest are indicated as a guide on the left.
Different Species and Tissues
With the N-terminal antibody Calp3d/2C4, protein bands of the same size as the 94-kd calpain 3 band in humans were observed in rabbit, dog, and hamster, but immunoreactivity with a larger peptide (approximately 110 kd) was also seen in hamster, and in rat. No reactivity was seen in mouse, chicken, or pig (Figure 2 ▶ , top). The 30-kd fragment was clearly visible in human muscle, but it was only weak in dog and rabbit. In the various rabbit tissues, the 94-kd calpain 3 band was only observed in skeletal muscle, although a variety of bands of other sizes were detected in thymus, brain, kidney, and spleen (results not shown). The ∼110-kd protein was only present in rat skeletal muscle and heart, with a slightly larger form in brain (results not shown).
Figure 2.

Samples of skeletal muscle from different animals analyzed on Western blots labeled with exon 1 antibody Calp3d/2C4 (top), exon 8 antibody Calp3c/11B3 (middle), or exon 8 antibody Calp3d/12A2 (bottom). The samples are as follows: Ms, mouse; Rt, rat; Rb, rabbit; Dg, dog; Ch, chicken; Ha, hamster; Pg, pig; and Hu, human. Approximate molecular masses are shown as a guide on the right.
The ubiquitous calpain antibody, Calp3c/11B3, recognized calpain 3 in all species tested, plus additional bands corresponding in size to calpain 1 and/or 2 in all species (Figure 2 ▶ , middle).
The exon 8 antibody Calp3c/12A2, recognized the 94-kd calpain 3 band in all species tested. It appeared to be specific in human, rabbit, dog, and pig skeletal muscle, but some additional bands corresponding in size to calpains 1 and/or 2 were detected in skeletal muscle from mouse, rat, chicken, and hamster (Figure 2 ▶ , bottom). As with antibody Calp3c/11B3, the group of calpain 3 breakdown bands starting at ∼60 kd appeared to be present in all the animal muscle samples tested.
In the various rabbit tissues, the 94-kd calpain 3 band was only observed in skeletal muscle, although bands of other sizes were detected in thymus and spleen. Some clear immunoreactivity with myosin heavy chain was observed in the smooth muscle of rabbit vas deferens and more weakly in the skeletal muscle of all species examined (results not shown). The identity of the myosin heavy chain band was suggested by its very characteristic size (200 kd) and shape on blots.
Stability in Human Muscle
Protein bands that are smaller than expected are usually degradation bands, and we undertook some deliberate muscle breakdown experiments to determine whether there was a reciprocal relationship between the abundance of the full-size and lower molecular mass bands. Some selected time points from three experiments are shown in Figure 3 ▶ . For the N-terminal antibody, Calp3d/2C4 (Figure 3 ▶ , top), there was little significant change in the band profiles, with muscle kept up to 8 hours before freezing and up to 2 hours postthawing, with the 94-kd band showing no obvious drop in abundance. In the saline homogenate experiment, the 94-kd and 30-kd bands disappeared in parallel. For the exon 8 antibody, Calp3c/12A2 (Figure 3 ▶ , bottom), there was little change in the 94-kd band intensity up to 8 hours before freezing and up to 2 hours postthawing. In both experiments, however, there was a clear shift in the ∼60-kd band pattern, with bands around 45 kd becoming more intense. In the saline homogenate experiment, the 94-kd band became progressively fainter, whereas the lower bands (intense even at the T0 time point) shifted from ∼60 kd down to ∼45 kd.
Figure 3.

Western blots of samples from selected time points in three muscle breakdown experiments, labeled with exon 1 antibody Calp3d/2C4 (top) or exon 8 antibody (bottom). A, time before freezing fresh muscle; B, time frozen muscle was left to thaw before homogenization; C, time as homogenate in saline before the addition of double-strength electrophoresis treatment buffer. In C, only half the amount of muscle protein was loaded compared with A and B (see Materials and Methods). For each experiment: T0 = immediately or at the earliest time point possible; h, hours; m, minutes. Approximate molecular masses are shown as a guide on the right.
These results indicate that 94-kd calpain 3 protein is extremely stable in intact human muscle. It takes homogenization to induce significant breakdown. The 30-kd and uppermost 60-kd fragments were present from the very earliest time it was possible to obtain human muscle from the operating theater and freeze it. Whereas the bands from ∼60 kd down to ∼45 kd clearly change like a ladder of degradation products, the abundance of the 30-kd band did not appear to increase as the full-size protein broke down.
Clinical Samples
Samples from 33 normal control subjects showed the described profile of bands with almost no variation (see Figure 4 ▶ , lanes 1, 3, 5, 17, and 20). A similar pattern of reactivity was found in 12 patients with Duchenne MD; 5 with Becker MD; 7 with facioscapulohumeral MD; 4 with sarcoglycanopathy; 1 with Emery-Dreifuss MD; 3 with merosin-associated congenital MD; 4 with myopathy accompanied by a secondary deficiency of laminin β1 chain expression (as in Refs. 25 and 26 ), 6 adult limb-girdle patients showing a secondary merosin deficiency on blots, 8 and 8 patients with one of the inflammatory muscle diseases, polymyositis or dermatomyositis. Some of these disease control samples are shown in Figure 4 ▶ , lanes 7, 9, 12, 14, and 16.
Figure 4.

Twenty lanes of control and patient biopsies analyzed on Western blots labeled with exon 1 antibody Calp3d/2C4 (top) or exon 8 antibody Calp3c/12A2 (bottom). Each panel contains lanes from three different blots, as indicated by the vertical lines. Lanes 1, 3, 5, 17, and 20: Different normal control samples. Lanes 2 , 4, 6, 8, 10, 11, 13, 18, and 19: patients with LGMD2A (see Table 2 ▶ ). The other lanes are from patients with different types of MD: lane 7, merosin-deficient congenital MD; lane 9, facioscapulohumeral MD; lane 12, γ-sarcoglycanopathy (LGMD2C); lane 14, Becker MD; lane 15, undefined limb-girdle MD (?LGMD); and lane 16, Duchenne MD. Approximate molecular masses are shown as a guide on the right.
One undefined LGMD (?LGMD) sample was sent from Paris along with the Réunion Island samples. This was the only patient, thought to be non-LGMD2A, whose sample showed any variation in protein expression, in this case a reduction in labeling intensity for the lower molecular mass bands, accompanying a 94-kd calpain 3 band of normal intensity (Figure 4 ▶ , lane 15). No mutation has been found to date in this patient, with all exons having been sequenced. No blood or tissue samples were available from other family members to test for linkage to chromosome 15, and there was no evidence of consanguinity.
A summary of the clinical details from the LGMD2A patients is shown in Table 1 ▶ . The results of immunoanalysis and gene mutation analysis are summarized in Table 2 ▶ , with Figure 4 ▶ illustrating the blot lanes. Labeling of the 94-kd band was undetectable on visual inspection in patients 1a, 1b, and 4 (Figure 4 ▶ , lanes 2, 13, and 11). In patients 2, 3, 6, and 8, full-size protein was represented by weakly labeled bands (Figure 4 ▶ , lanes 4, 10, 6, and 19). The lower molecular mass bands (30 kd and ∼60 kd) were greatly reduced or absent in all these patients, indicating that these bands represented calpain 3 polypeptides derived from the same CAPN3 gene and not distinct cross-reactive proteins. A very different pattern of protein expression was seen in patients 5 and 7 (Figure 4 ▶ , lanes 8 and 18). Here the 94-kd band was indistinguishable from normal, but the lower molecular mass bands were clearly reduced in intensity. Table 3 ▶ shows the densitometric analysis of the calpain 3 bands labeled with the exon 1 antibody Calp3d/2C4. The density of the 94-kd band in patient 5 (0.079) and patient 7 (0.155) was within the range seen in control subjects (0.042 to 0.133). In these controls, the values for 30-kd band density expressed as a percentage of 94-kd band density ranged from 45 to 95%. In patients 5 and 7 this was reduced to 18% and 14%, whereas in the ?LGMD patient (Figure 4 ▶ , lane 15), it was 9%. Thus, the protein profile of two LGMD2A patients could not be differentiated from a ?LGMD patient.
Table 1.
Clinical Data on Patients with LGMD2A
| Patient | Origin | Age at onset | Age at biopsy | Age in 1998 | Pattern of muscle involvement | Creatine kinase (times upper normal limit) | Functional stage* |
|---|---|---|---|---|---|---|---|
| 1a | Réunion Island | 10 | 14 | 24 | Typical† | 3× at age 14 years | VI at age 17 years |
| 1b | Réunion Island | 10 | 18 | 28 | Typical | 20× at age 18 years | VII at age 20 years |
| 2 | Réunion Island | 20 | 47 | 51 | Typical | 20× at age 47 years | III at age 44 years |
| 3 | Réunion Island | 23 | 29 | 39 | Typical | 25× at age 29 years | III at age 32 years |
| 4 | Réunion Island | 10 | 31 | 41 | Typical | 3× at age 31 years | VII at age 26 years |
| 5 | Réunion Island | 10 to 11 | 41 | 43 | Typical, but with calf hypertrophy | 10× at age 41 years | III at age 41 years |
| 6 | Germany | 3 | 11 | 15 | Frequent falls at age 3 to 4 years, calf contractures at 6 years, hyperlordosis, scapulae alatae | 41× at age 6 years | III at age 10 years |
| 7 | Spain | 12 | 16 | 17 | Weakness in legs first, biceps later | 17× at age 16 years | V at age 16 years |
| 8 | Spain | 6 | 14 | 19 | Mild proximal weakness in legs, calf hypertrophy, currently only slight difficulty rising from floor | 24× at age 14 years | II to III at age 19 years |
Patients 1a and 1b are related.
*Functional stages: I, unable to run freely or stand up without using hands; II, unsteady gait, climbs stairs without using banister; III, climbs stairs only with a banister; IV, unable to climb stairs; V, unable to rise from a chair without aid; VI, walks only with aid; VII, unable to walk or to stand erect.
†As described by Fardeau et al. 36
Table 2.
Protein Expression in Patients with LGMD2A: Correlation with Genotype and Clinical Phenotype
| Calpain 3 protein expression | Calpain 3 gene expression | Phenotype (Rate of disease progression) | ||||
|---|---|---|---|---|---|---|
| 94-kd full size | 30-kd fragment | ∼60-kd products | Mutations | Effect | ||
| Normal control | ++++ | ++++ | +++ | − | − | Normal control |
| Patient 1a (Figure 4 ▶ , lane 2) | − | − | − | Homozygous 946-1 AG → AA | Aberrant splicing; exon 7 skipped, frame shift | Severe |
| Patient 1b (Figure 4 ▶ , lane 13) | − | − | − | Homozygous 946-1 AG → AA | Aberrant splicing; exon 7 skipped, frame shift | Severe |
| Patient 2 (Figure 4 ▶ , lane 4) | + | +/− | − | 2362 AG → TCATCT | Frame shift in exon 22 | Mild |
| S744G | Missense S → G in exon 21 (EF loop) | |||||
| Patient 3 (Figure 4 ▶ , lane 10) | +/− | − | − | 946-1 AG → AA | Aberrant splicing; exon 7 skipped, frame shift | Intermediate |
| S744G | Missense S → G in exon 21 (EF loop) | |||||
| Patient 4 (Figure 4 ▶ , lane 11) | − | − | − | Homozygous 946-1 AG → AA | Aberrant splicing; exon 7 skipped, frame shift | Severe |
| Patient 5 (Figure 4 ▶ , lane 8) | ++++ | ++ | + | 946-1 AG → AA | Aberrant splicing; exon 7 skipped, frame shift | Mild |
| T184M | Missense T → M in exon 4 | |||||
| Patient 6 (Figure 4 ▶ , lane 6) | +/− | − | − | 801+ 1 G → A | Aberrant splicing; exon 5 skipped, frame shift | Severe |
| 598 to 612 deleted (amino acids 200 to 204) | In-frame deletion of 5 amino acids in exon 4 (within highly conserved motif) | |||||
| Patient 7 (Figure 4 ▶ , lane 18) | ++++ | +/− | +/− | Homozygous G222R | Missense G → R in exon 5 | Severe |
| Patient 8 (Figure 4 ▶ , lane 19) | + | − | − | 1785 to 1788 deleted | Deletion in exon 15, frame shift | Intermediate |
| R748Q | Missense R → Q in exon 21 |
Band labeling: ++++ = strong, +++ = moderate, ++ = clearly visible, + = faint, +/− = very faint, − = not visible
Table 3.
Densitometric Analysis of Calpain 3 Labeling with Exon 1 Antibody Calp3d/2C4
| Patient no. | 94-kd band | 30-kd band | 30 as a percentage of 94 | |
|---|---|---|---|---|
| Control subjects | ||||
| Range | 0.042 to 0.133 | 0.026 to 0.087 | 45 to 95 | |
| Mean | 0.071 | 0.048 | 72 | |
| SD | 0.030 | 0.015 | 19 | |
| SEM | 0.008 | 0.004 | 5.5 | |
| Patients (lane) | ||||
| 2 | 1a | 0 | 0 | |
| 4 | 2 | 0.009 | 0.003 | |
| 6 | 6 | 0.003 | 0 | |
| 10 | 3 | 0.015 | 0.009 | |
| 11 | 4 | 0 | 0 | |
| 13 | 1b | 0.001 | 0 | |
| 19 | 8 | 0.029 | 0 | |
| 8 | 5 | 0.079 | 0.014 | 18 |
| 18 | 7 | 0.115 | 0.017 | 14 |
| 15 | ?LGMD | 0.086 | 0.008 | 9 |
Density values are volume OD normalized for the myosin heavy chain content (see Materials and Methods).
Control subjects include the normal individuals and patients with defined dystrophies analyzed on the blots shown in Figure 4 ▶ .
Discussion
Characterization of Epitopes
This is the first time that potentially diagnostic monoclonal antibodies to human calpain 3 have been reported. This paper includes details of antibody characterization with respect to blot band patterns and species reactivity, because this is important for antibodies that will be available commercially for use with animal models as well as human diseases. There have been few reports of the immunological detection of calpain 3 in skeletal muscle, and early studies suggested that, although the mRNA was abundant in muscle, the protein had a very short half-life because of destruction by autolysis. 27 More recently, polyclonal antibodies that recognize calpains 1, 2, and 3, have shown that calpain 3 is detectable in mouse 28 and human 20 skeletal muscle.
Three different monoclonal antibodies have been produced. One is to an epitope within amino acids 1 to 19 in exon 1 in the calpain 3-specific NS domain. This sequence shows few differences between species, and the lack of immunoreactivity to rat, mouse, and pig calpain 3 suggests that the epitope may span amino acids 7 and 8 or 14, which are the only differences in the human sequence (Figure 5) ▶ . The other antibodies are to two different epitopes in exon 8 in protease domain II, a region of sequence homology common to all the calpains. The exon 8 immunogen contained a sequence of 16 amino acids (355 to 370), 11 of which are common to calpains 1, 2, and 3. Thus, the epitope recognized by antibody Calp3c/11B3 might span amino acids 357 to 361 (five contiguous residues common to human calpains 1, 2, and 3), whereas the epitope recognized by antibody Calp3c/12A2 might involve amino acids 366 to 370 (a group of five amino acids with two or three differences between calpain 3 and calpains 1 or 2) (Figure 5) ▶ . It is quite unusual to isolate common and specific antibodies from immunization with a single peptide. However, the degree of homology is similar to that in the C-terminal region of dystrophin and utrophin, and immunization with sequences from these proteins has also resulted in specific and cross-reactive antibodies. 29,30
Figure 5.

Comparison of the amino acids in the two human calpain 3 peptide sequences used to raise monoclonal antibodies. Amino acids 1 to 19 only occur in calpain 3, but amino acids 355 to 370 have corresponding sequences in calpains 2 and 1, as indicated in the (top). Only the amino acids that differ are indicated; a dash means that the amino acids are the same. Bottom: The human sequences in calpains 3, 2, and 1 are compared with the corresponding sequences in different animals. The first row depicts the human sequences, and subsequent rows show any differences in the corresponding amino acids in mouse, rat, pig, and chicken. Sequences are not currently available for pig calpains 2 and 1 or for calpains 3, 2, and 1 in the other species examined on blots.
The chosen regions of the human calpain 3 sequence seem to share homology with a number of other proteins in different species, judging from the range of bands detected in different tissues and different species with these antibodies. In particular, the exon 8 immunogen seems to share some homology with myosin heavy chain, because both polyclonal antibodies and these monoclonal antibodies showed some immunoreactivity toward this protein.
Band Patterns on Blots
Studies on mouse muscle 28 have shown that antibodies to an epitope shared by calpains 1, 2, and 3 in the common protease domain II produce a pattern of four or more related bands on blots. Apart from the 94-kd band of calpain 3, there are bands at 84 kd (representing intact calpain 1), 80 kd, and 78 kd. The latter two represent a mixture of the catalytic subunits of calpain 2 (both pre- and postautolysed) and autolysed calpain 1. 28 Adjacent strips from blots of human muscle labeled with these polyclonal antibodies and monoclonal antibody Calp3c/11B3 showed an identical pattern of grouped bands.
Two of the monoclonal antibodies produced recognized calpain 3 alone, but the protein was present as more than one band on blots of human skeletal muscle. In addition to the full-sized 94-kd band, a clean 30-kd fragment was detected with the exon 1 NS domain antibody Calp3d/2C4 (at 45 to 95% the density of the 94-kd band in control muscle; see Table 3 ▶ ), and a group of degradation bands running from about 60 kd down to 45 kd was detected with the exon 8 domain II antibody Calp3c/12A2. The total loss of immunoreactivity in LGMD2A patients demonstrated that these additional polypeptides were related to calpain 3 and the CAPN3 gene and not other unrelated proteins.
The identity of these bands is not really certain, but they are most likely to be proteolytic products of the full-size protein, possibly with the 30-kd and 60-kd fragments arising from a single proteolytic cut of the 94-kd protein. If this is the case, they may have been caused by ex vivo degradation after tissue was removed from the body or be normally present in muscle, having been generated as part of the in vivo calpain 3 physiological process. The 30-kd band was clearly present in human muscle but only weakly visible in muscle from dog and rabbit. This was not related to the time taken to freeze the muscle initially, because this was comparable between the species. In the degradation experiments we undertook there was no simple reciprocal relationship between the full-size and smaller polypeptides: the abundance of the 94-kd protein did not decrease in parallel to an increase in the smaller fragments. Both 30-kd and 60-kd bands were present from the earliest time points obtainable (Figure 3) ▶ , although they were slightly darker at the subsequent time points. In these experiments, the 60-kd band did appear to degrade further into a ladder of smaller bands down to about 45 kd, but there were no smaller bands than the 30-kd one. The N-terminal region of domain 1 is autocatalytically processed during Ca2+-dependent activation in calpains 1 and 2, 16 but the N terminus in calpain 3 (exemplified by the epitope recognized by antibody Calp3d/2C4) appears to be very stable in whole muscle, and it may be that the calpain 3-specific NS sequence insertion blocks the autolysis that occurs in calpains 1 and 2.
It is possible that the 30-kd fragment is a separate polypeptide product in its own right, expressed from exons 1 to 5 of the CAPN3 gene. Another tissue-specific calpain, which is expressed only in stomach, has two alternative gene products. The full-size stomach calpain is designated nCL-2, whereas the peptide corresponding to the N-terminal part is designated nCL-2′. 31 No function has yet been allocated to this half-size polypeptide, which is missing the calcium binding domain characteristically required by the calpain family for activation. It is interesting to note, however, that the tissue-specific calpains, unlike their ubiquitous relatives, do not interact with the small regulatory subunit, which is also 30 kd. 31,32 It may just be possible that the tissue-specific calpains use N-terminal products from their own genes to fulfill a regulatory role.
The exon 1 antibody detected a band at about 110 kd in skeletal muscle from rat and hamster. It was also observed in rat heart, with a slightly larger form in brain. These bands may represent reactivity with unrelated gene product(s) or may represent novel isoforms of calpain 3. There is increasing evidence for tissue-related alternative splicing in this gene. The mRNA of an isoform, predicted to be an 82-kd protein, has been described in the lens of both rat 19 and mouse (M. Herasse et al, manuscript in preparation). There is also evidence for alternative splicing of the CAPN3 gene in the digestive tract smooth muscle of human fetuses early in development. 33
Stability of Calpain 3 in Muscle
Calpain 3 is stable in intact human muscle, there being little decrease in the abundance of full-size protein detected in biopsy samples rested at room temperature for up to 8 hours after leaving the body. This is in contrast to early reports that seemed to indicate rapid autolysis. 27 It appears that it is critical for the muscle to be unfractionated and simply applied straight to gels. There may also be variation between samples, given that in these experiments we also found little change up to 2 hours postthaw, whereas a single freeze/thaw cycle had previously been shown to reduce the abundance of full-size protein within 10 to 15 minutes. 20 The difference may also be accentuated by the presence of moisture. Our samples were thawed “dry” in the homogenization tube, whereas previously the samples were left on a damp gauze in a Petri dish. 20 Here, homogenization in saline was the only way of producing degradation with a clear reduction in full-size protein within 30 minutes. The breakdown under these conditions was in agreement with short half-life reported by Sorimachi et al. 27
The stability of calpain 3 in whole muscle may be mediated by its binding to titin, a giant molecule that stretches between the M and Z lines, and may act as a template around which other myofibrillar proteins are assembled. 32,34 So far, titin is the only candidate protein that appears to bind calpain 3; other potential substrates and functions for calpain 3 remain elusive. The association with titin may indicate a function in maintaining myofibrillar integrity, and studies on rat myoblast cultures have shown that blocking calpain 3 expression with antisense oligonucleotides leads to muscle fiber instability and Z-line disorganization. 35 However, patients lacking calpain 3 are reported not to show dramatic ultrastructural changes, 35,36 and it does not seem likely that the pathogenesis of the human MD involves titin instability.
Protein Expression in Patients with LGMD2A
Three types of mutation are represented in this group of patients: homozygous null, homozygous missense, and heterozygous combinations of null/missense and null/in-frame deletion. As expected, the homozygous null mutation produced the most profound effect on protein expression, with undetectable calpain bands (see Figure 4 ▶ , lanes 2, 11, and 13). Evolution of the disease was considered to be severe in these patients.
Patients heterozygous for a null mutation and a missense change in exon 21 (S744G or R748, in an EF loop that might indicate calcium binding) had intermediate or mild disease progression but severely reduced calpain 3 expression (Figure 4 ▶ , lanes 4, 10, and 19). Thus, missense mutations in this region may affect either protein production or protein stability but presumably preserve some residual enzymatic activity. Thus, this result also indicates that milder clinical phenotypes can exist in the presence of greatly reduced enzyme levels. Calpain 3 protein was also severely reduced in one patient with a null mutation plus a small in-frame deletion within exon 4 in a highly conserved region (Figure 4 ▶ , lane 6). A small in-frame deletion might have resulted in translation of a slightly smaller protein in normal abundance, so again this result appears to confirm a region is important for protein stability and/or function. In this instance, the patient had a severe clinical phenotype. In summary, very low levels of calpain 3 were found in patients who varied in the classification of their disease severity from “mild” to “severe.”
One patient (no. 7) had a homozygous missense mutation situated in exon 5, in the protease domain II within a region of sequence conservation between species. In this mutation the 222nd amino acid was changed from glycine to arginine, thereby converting a nonpolar side chain to one that is very basic. Expression of the full-size protein appeared to be unaffected, but the band intensity (indicating abundance) of the 30-kd and ∼60-kd bands was very severely reduced (Figure 4 ▶ , lane 18; Table 3 ▶ ). Disease progression in this patient was classed as “severe” compared with the others. Thus, it appears that this mutation had affected calpain 3 function, and that the inactive protein was not removed from, or accumulated within, the muscle as a result of this dysfunction.
Exactly the same pattern of calpain 3 bands was observed in a patient (no. 5) in whom progression of the disease was mild (Figure 4 ▶ , lane 8). In this case the underlying mutations were a null mutation plus a missense mutation in exon 4 (the 184th amino acid changed from threonine to methionine, thereby converting a polar side chain to a nonpolar one). Again, the smaller fragments (30 kd and ∼60 kd) were clearly reduced in abundance (see Table 3 ▶ ). In summary, normal levels of the full-size calpain 3 band were found in patients who varied in disease severity and progression from “mild” to “severe.” In these cases, however, the normal full-size band was accompanied by obvious changes to the abundance of the smaller bands.
Given that the only apparent change in protein expression in patients 5 and 7 was the reduced abundance of the 30-kd and ∼60-kd bands, is the generation of these smaller bands part of normal calpain 3 function, and is their reduction a specific (ie, diagnostic) indication of loss of enzyme function? Possibly not: although this protein profile has not been seen in any other patients, it was observed in one ?LGMD sample in which mutations in the CAPN3 gene had not been found (Figure 4 ▶ , lane 15; Table 3 ▶ ). We need to discover a lot more about the physiology of calpain 3 and the circumstances under which the smaller bands are generated before any firm conclusions about the diagnostic usefulness of these minor changes in protein expression can be assessed.
Feasibility of Immunodiagnosis
This study has shown very clearly that the stability of calpain 3 protein in human muscle biopsies is not a problem for immunodetection. Very little change was observed in human muscle left on moist filter paper for up to 8 hours before freezing. Thus, biopsies taken in outlying hospitals and sent to a major center for analysis would yield useful results. During the course of this study, we found that samples prepared for electrophoresis showed no obvious loss of immunoreactivity when they sat for several hours before analysis, or even after four or five freeze/thaw cycles. It should be noted, however, that the experiments on protein stability were undertaken on “wild-type” calpain 3 in normal control muscle: mutated forms of the protein might behave differently.
A limitation on the widespread use of immunodiagnosis for calpain 3 is that the current antibodies only work on Western blots and not on tissue sections. Work is currently underway to obtain antibodies that will work for both techniques. Nevertheless, if the generation of the 30-kd and ∼60-kd bands has some functional significance, this could only be examined on blots. However, if the different fragments/isoforms have a different subcellular distribution, immunocytochemistry on sections would still be useful.
How accurate is immunodiagnosis likely to be? Absence of calpain 3 protein probably indicates the presence of null mutations (as the absence of protein expression does for other recessive diseases). Similarly, significantly reduced levels of protein are usually associated with pathological mutations. False-negative results might arise in circumstances in which calpain 3 is broken down (eg, after transporting a biopsy to a referral center in liquid, or prolonged thawing of a muscle sample in transit), but this would probably be detected by an increase in lower molecular mass bands on the gel and the loss of all the proteins to be analyzed. The use of another protein as an internal control for labeling would be useful in this context, and the multiplex blotting system is ideal. 22 From this study, false-positive results appear to be much more of a problem. Among nine patients, two were found to have normal levels of full-size calpain 3 protein. The diagnostic significance of the observed reduction in the 30-kd and ∼60-kd bands in these two patients is uncertain.
Prognosis based on dystrophin analysis was generally easy for patients with Duchenne and Becker dystrophy. It was much less easy in the sarcoglycanopathies, although there was a general rule that null mutations were associated with absence of detectable protein and, often, a severe phenotype. Most other mutations resulted in reduced protein expression, so that there was little correlation between clinical severity and figures for protein abundance. In LGMD2A, in which the gene product is a proteolytic enzyme, the situation is likely to be more complicated. Different mutations might result in enzyme levels that were reduced, normal, or even increased. 37 From these initial results, it seems very unlikely that calpain 3 protein expression alone will be of value for the prediction of clinical severity in LGMD2A.
In summary, these results indicate that immunodiagnosis for some patients is feasible, and this represents an important advance, because it has been estimated that 50% of patients with autosomal recessive LGMD actually have LGMD2A. 38 However, caution will need to be exercised with the interpretation of near-normal protein profiles. The confidence with which immunodiagnosis is used will only improve when many more patients with defined mutations have been analyzed. It is quite clear that this is an urgent priority for the future.
Acknowledgments
We wish to thank Drs. Melissa Spencer and James Tidball for providing polyclonal antibodies that recognized calpains 1, 2, and 3.
Footnotes
Address reprint requests to Dr. Louise V. B. Anderson, Neurobiology Department, University Medical School, Framlington Place, Newcastle upon Tyne, NE2 4HH, United Kingdom. E-mail: l.v.b.anderson@ncl.ac.uk.
Supported by the Muscular Dystrophy Group of Great Britain and the Association Française contre les Myopathies.
References
- 1.Noguchi S, McNally EM, Othmane KB, Hagiwara Y, Mizuno Y, Yoshida M, Yamanmoto H, Bönnemann CG, Gussoni E, Denton PH, Kyriakides T, Middleton L, Hentati F, Hamida MB, Nonaka I, Vance JM, Kunkel LM, Ozawa E: Mutations in the dystrophin-associated protein-γ-sarcoglycan in chromosome 13 muscular dystrophy. Science 1995, 270:819-822 [DOI] [PubMed] [Google Scholar]
- 2.Roberds SL, Leturcq F, Allamand V, Piccolo F, Jeanpierre M, Anderson RD, Lim LE, Lee JC, Tomé FMS, Romero NB, Fardeau M, Beckmann JS, Kaplan J-C, Campbell KP: Missense mutations in the adhalin gene linked to autosomal recessive muscular dystrophy. Cell 1994, 78:625-633 [DOI] [PubMed] [Google Scholar]
- 3.Lim LE, Duclos F, Broux O, Bourg N, Sanada Y, Allamand V, Meyer J, Richard I, Moomaw C, Slaughter C, Tomé FMS, Fardeau M, Jackson CE, Beckmann JS, Campbell KP: β-Sarcoglycan: characterization and role in limb-girdle muscular dystrophy linked to 4q12. Nat Genet 1995, 11:257-265 [DOI] [PubMed] [Google Scholar]
- 4.Nigro V, de Sá Moreira E, Piluso G, Vainzof M, Belsito A, Politano L, Puca AA, Passos-Bueno MR, Zatz M: Autosomal recessive limb-girdle muscular dystrophy, LGMD2F, is caused by a mutation in the δ-sarcoglycan gene. Nat Genet 1996, 14:195-198 [DOI] [PubMed] [Google Scholar]
- 5.Richard I, Broux O, Allamand V, Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C, Pasturaud P, Roudaut C, Hillaire D, Passos-Bueno MR, Zatz M, Tischfield JA, Fardeau M, Jackson CE, Cohen D, Beckmann JS: Mutations in the proteolytic enzyme calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell 1995, 81:27-40 [DOI] [PubMed] [Google Scholar]
- 6.Bushby K, Bashir R, Keers S, Britton S, Zatz M, Passos-Bueno MR, Lovett M, Mahjneh I, Marconi G, Strachan T: The molecular biology of LGMD2B: towards the identification of the LGMD gene on chromosome 2p13. Neuromusc Disord 1996, 6:491-492 [DOI] [PubMed] [Google Scholar]
- 7.Moreira ES, Vainzof M, Marie SK, Sertié AL, Zatz M, Passos-Bueno MR: The seventh form of autosomal recessive limb-girdle muscular dystrophy is mapped to 17q11–12. Am J Hum Genet 1997, 61:151-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bushby KMD, Anderson LVB, Pollitt C, Naom I, Muntoni F, Bindoff L: Abnormal merosin in adults: a new form of late onset muscular dystrophy not linked to chromosome 6q2. Brain 1998, 121:581-588 [DOI] [PubMed] [Google Scholar]
- 9.Bushby KMD: Diagnostic criteria for the limb-girdle muscular dystrophies: report of the ENMC consortium on limb-girdle dystrophies. Neuromusc Disord 1995, 5:71-74 [DOI] [PubMed] [Google Scholar]
- 10.Anderson LVB: Optimised protein diagnosis in the autosomal recessive limb-girdle muscular dystrophies. Neuromusc Disord 1996, 6:443-446 [DOI] [PubMed] [Google Scholar]
- 11.Bönnemann CG, Passos-Bueno MR, McNally EM, Vainzof M, de Sá Moreira E, Marie SK, Pavanello RCM, Noguchi S, Ozawa E, Zatz M, Kunkel LM: Genomic screening for β-sarcoglycan gene mutations: missense mutations may cause severe limb-girdle muscular dystrophy type 2E (LGMD 2E). Hum Mol Genet 1996, 5:1953-1961 [DOI] [PubMed] [Google Scholar]
- 12.Vainzof M, Passos-Bueno MR, Canovas M, de Sá Moreira E, Pavanello RCM, Marie SK, Anderson LVB, Bönnemann CG, McNally EM, Nigro V, Kunkel LM, Zatz M: The sarcoglycan complex in the six autosomal recessive limb-girdle muscular dystrophies. Hum Mol Genet 1996, 5:1963-1969 [DOI] [PubMed] [Google Scholar]
- 13.Sewry C, Taylor J, Anderson LVB, Ozawa E, Pogue R, Piccolo F, Bushby KMD, Dubovitz V, Muntoni F: Abnormalities in α-, β- and γ-sarcoglycan in patients with limb-girdle muscular dystrophy. Neuromusc Disord 1996, 6:467-474 [DOI] [PubMed] [Google Scholar]
- 14.Eymard B, Romero NB, Leturcq F, Piccolo F, Carrié A, Jeanpierre M, Collin H, Deburgrave N, Azibi K, Chaouch M, Merlini L, Thémar-Noël C, Penisson I, Mayer M, Tanguy O, Campbell KP, Kaplan JC, Tomé FMS, Fardeau M: Primary adhalinopathy (α-sarcoglycanopathy): clinical, pathologic, and genetic correlation in 20 patients with autosomal recessive muscular dystrophy. Neurology 1997, 48:1227-1234 [DOI] [PubMed] [Google Scholar]
- 15.Barresi R, Confalonieri V, Lanfossi M, Di Blasi C, Torchiana E, Mantegazza R, Jarre L, Nardocci N, Boffi P, Tezzon F, Pini A, Cornelio F, Mora M, Morandi L: Concomitant deficiency of β- and γ-sarcoglycans in 20 α-sarcoglycan (adhalin)-deficient patients: immunohistochemical analysis and clinical aspects. Acta Neuropathol 1997, 94:28-35 [DOI] [PubMed] [Google Scholar]
- 16.Sorimachi H, Kimura S, Kinbara K, Kazama J, Takahashi M, Yajima H, Ishiura S, Sasagawa N, Nonaka I, Sugita H, Maruyama K, Suzuki K: Structure and physiological functions of ubiquitous and tissue-specific calpain species: muscle-specific calpain, p94, interacts with connectin/titin. Adv Biophys 1996, 33:101-122 [DOI] [PubMed] [Google Scholar]
- 17.Sorimachi H, Ishiura S, Suzuki K: Structure and physiological function of Calpains. Biochem J 1998, 328:721-732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sorimachi H, Imajoh-Ohmi S, Emori Y, Kawasaki H, Ohno S, Minami Y, Suzuki K: Molecular cloning of a novel mammalian calcium-dependent protease distinct from both m- and n-types: specific expression of the mRNA in skeletal muscle. J Biol Chem 1989, 264:20106-20111 [PubMed] [Google Scholar]
- 19.Ma H, Fukiage C, Azuma M, Shearer TR: Cloning and expression of mRNA for calpain Lp82 from rat lens: splice variant of p94. Invest Ophthalmol Vis Sci 1998, 39:454-461 [PubMed] [Google Scholar]
- 20.Spencer MJ, Tidball JG, Anderson LVB, Bushby KMD, Harris JB, Passos-Bueno MR, Somer H, Vainzof M, Zatz M: Absence of calpain 3 in a form of limb-girdle muscular dystrophy (LGMD2A). J Neurol Sci 1997, 146:173-178 [DOI] [PubMed] [Google Scholar]
- 21.Nicholson LVB, Davison K, Falkous G, Harwood C, O’Donnell E, Slater CR, Harris JB: Dystrophin in skeletal muscle. I. Western blot analysis using a monoclonal antibody. J Neurol Sci 1989, 94:125-136 [DOI] [PubMed] [Google Scholar]
- 22.Anderson LVB. Multiplex Western blot analysis of the muscular dystrophy proteins. Muscular Dystrophy: Methods and Protocols. Methods in Molecular Medicine Series. Edited by KMD Bushby, LVB Anderson. New Jersey, Humana Press, 1998 (in press)
- 23.Richard I, Brenguier L, Dinçer P, Roudaut C, Bady B, Burgunder JM, Chemaly R, Garcia CA, Halaby G, Jackson CE, Kurnit DM, Lefranc G, Legum C, Loiselet J, Merlini L, Nivelon-Chevallier A, Ollagnon-Roman E, Restagno G, Topaloglu H, Beckmann JS: Multiple independent molecular etiology for limb-girdle muscular dystrophy type 2A patients from various geographical origins. Am J Hum Genet 1997, 60:1128-1138 [PMC free article] [PubMed] [Google Scholar]
- 24.Häffner K, Speer A, Hübner C, Voit T, Oexle K: A small in-frame deletion within the protease domain of muscle-specific calpain, p94 causes early-onset limb-girdle muscular dystrophy 2A. Hum Mutat 1998, (Suppl.) 1:S298–S300 [DOI] [PubMed]
- 25.Taylor J, Muntoni F, Robb S, Dubowitz V, Sewry C: Early onset autosomal dominant myopathy with rigidity of the spine: a possible role for laminin β1? Neuromusc Disord 1997, 7:211-216 [DOI] [PubMed] [Google Scholar]
- 26.Li M, Dickson DW, Spiro AJ: Abnormal expression of laminin β1 chain in skeletal muscle of adult-onset limb-girdle muscular dystrophy. Arch Neurol 1997, 54:1457-1461 [DOI] [PubMed] [Google Scholar]
- 27.Sorimachi H, Toyama-Sorimachi N, Saido TC, Kawasaki H, Sugita H, Miyasaka M, Arahata K, Ishiura S, Suzuki K: Muscle-specific calpain, p94, is degraded by autolysis immediately after translation, resulting in disappearance from muscle. J Biol Chem 1993, 268:10593-10605 [PubMed] [Google Scholar]
- 28.Spencer MJ, Croall DE, Tidball JG: Calpains are activated in necrotic fibers from mdx dystrophic mice. J Biol Chem 1995, 270:10909-10914 [DOI] [PubMed] [Google Scholar]
- 29.Pons F, Nicholson LVB, Robert A, Voit T, Léger JJ: Dystrophin and dystrophin-related protein (utrophin) distribution in normal and dystrophin-deficient skeletal muscles. Neuromusc Disord 1993, 3:507-514 [DOI] [PubMed] [Google Scholar]
- 30.Rivier F, Robert A, Latouche J, Hugon G, Mornet D: Presence of long and short dystrophin and/or utrophin products in Torpedo marmorata peripheral nerves. FEBS Lett 1996, 378:272-276 [DOI] [PubMed] [Google Scholar]
- 31.Sorimachi H, Ishiura S, Suzuki K: A novel tissue-specific calpain species expressed predominantly in the stomach comprises two alternative splicing products with and without Ca2+-binding domain. J Biol Chem 1993, 268:19476-19482 [PubMed] [Google Scholar]
- 32.Sorimachi H, Kinbara K, Kimura S, Takahashi M, Ishiura S, Sasagawa N, Sorimachi N, Shimada H, Tagawa K, Maruyama K, Suzuki K: Muscle-specific calpain, p94, responsible for limb girdle muscular dystrophy type 2A, associates with connectin through IS2, a p94-specific sequence. J Biol Chem 1995, 270:31158-31162 [DOI] [PubMed] [Google Scholar]
- 33.Fougerousse F, Durand M, Suel L, Pourquié O, Delezoide A-L, Romero NB, Abitbol M, Beckmann JS: Expression of genes (CAPN3, SGCA, SGCB, and TTN) involved in progressive muscular dystrophies during early human development. Genomics 1998, 48:145-156 [DOI] [PubMed] [Google Scholar]
- 34.Kinbara K, Sorimachi H, Ishiura S, Suzuki K: Muscle-specific calpain, p94, interacts with the extreme C-terminal region of connectin, a unique region flanked by two immunoglobulin C2 motifs. Arch Biochem Biophys 1997, 342:99-107 [DOI] [PubMed] [Google Scholar]
- 35.Poussard S, Duvert M, Balcerzak D, Ramassamy S, Brustis JJ, Cottin P, Ducastaing A: Evidence for implication of muscle-specific calpain (p94) in myofibrillar integrity. Cell Growth Differ 1996, 7:1461-1469 [PubMed] [Google Scholar]
- 36.Fardeau M, Hillaire D, Mignard C, Feingold N, Feingold J, Mignard D, De Ubeda B, Collin H, Tomé FMS, Richard I, Beckmann J: Juvenile limb-girdle muscular dystrophy: clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain 1996, 119:295-308 [DOI] [PubMed] [Google Scholar]
- 37.Beckmann JS, Fardeau M: Limb-girdle muscular dystrophies. Emery AEH eds. Neuromuscular Disorders: Clinical and Molecular Genetics, ch 6. 1998, :pp 123-156 John Wiley, New York [Google Scholar]
- 38.Topaloglu H, Dinçer P, Richard I, Akcoren Z, Alehan D, Ozme S, Caglar M, Karaduman A, Urtizberea JA, Beckmann JS: Calpain-3 deficiency causes a mild muscular dystrophy in childhood. Neuropediatrics 1997, 28:212-216 [DOI] [PubMed] [Google Scholar]