

Abstract
Microsatellite instability (MSI) has been identified in various human cancers, particularly those associated with the hereditary nonpolyposis colorectal cancer syndrome. Although gliomas have been reported in a few hereditary nonpolyposis colorectal cancer syndrome kindred, data on the incidence of MSI in gliomas are conflicting, and the nature of the mismatch repair (MMR) defect is not known. We established the incidence of MSI and the underlying MMR gene mutation in 22 patients ages 45 years or less with sporadic high-grade gliomas (17 glioblastomas, 3 anaplastic astrocytomas, and 2 mixed gliomas, grade III). Using five microsatellite loci, four patients (18%) had high level MSI, with at least 40% unstable loci. Germline MMR gene mutation was detected in all four patients, with inactivation of the second allele of the corresponding MMR gene or loss of protein expression in the tumor tissue. Frameshift mutation in the mononucleotide tract of insulin-like growth factor type II receptor was found in one high-level MSI glioma, but none was found in the transforming growth factor β type II receptor and the Bax genes. There was no family history of cancer in three of the patients, and although one patient did have a family history of colorectal carcinoma, the case did not satisfy the Amsterdam criteria for hereditary nonpolyposis colorectal cancer syndrome. Three patients developed metachronous colorectal adenocarcinomas, fitting the criteria of Turcot’s syndrome. Thus, MSI and germline MMR gene mutation is present in a subset of young glioma patients, and these patients and their family members are at risk of developing other hereditary nonpolyposis colorectal cancer syndrome-related tumors, in particular colorectal carcinomas. These results have important implications in the genetic testing and management of young patients with glioma and their families.
Microsatellite instability (MSI) is characterized by the expansion and contraction of small repeat sequences during DNA replication and is present in the majority of tumors in the hereditary nonpolyposis colon cancer (HNPCC) syndrome. 1 HNPCC is characterized by familial occurrence of cancer in various sites, including the colon, endometrium, and urinary tract, at an early age. 2 The mechanism leading to MSI is related to a defect in the DNA mismatch repair (MMR) system, of which more than 5 DNA mismatch repair (MMR) genes are now known. 3-12 Mutation of the hMSH2 and hMLH1 genes accounts for more than 60% of individuals with the syndrome. 13
Many sporadic cancers have also been found to show MSI. 14 For gliomas, relatively few studies have been performed, and the results are conflicting. Izumoto et al 15 and Dams et al 16 demonstrated the presence of MSI in 20 to 45% of glioblastomas and anaplastic astrocytomas and in no low-grade astrocytomas. Zhu et al 17 found MSI in 17% of oligodendrogliomas and 3% of astrocytic tumors. Wooster et al 18 and Amariglio et al 19 found MSI in 1.9% of brain tumors and in no brain tumors, respectively. In most of these series, MSI was considered positive if there was an allelic shift in a single locus only. The status of the MMR gene, be it germline or somatic, is largely unknown for these MSI-positive gliomas. On the other hand, there have been reports of increased risk for brain tumors in HNPCC kindred, 20-22 and a few patients with Turcot’s syndrome, characterized by the development of both colorectal and brain cancers, have been shown to have MSI and to harbor germline mutation in the MMR genes. 23,24
We have previously reported an unusually high incidence of colorectal carcinoma in the young Hong Kong population. 25 Coincidentally, there have been several studies reporting an unexpected tendency for the occurrence of glioblastomas and anaplastic astrocytomas in young Chinese in Taiwan, the People’s Republic of China, and Hong Kong, 26-28 when compared with the West. 29,30 The incidence of MSI is high in cases of sporadic colorectal carcinoma in the young of Hong Kong 31 and elsewhere, 32 and germline mutation of the MMR genes has been found in a high proportion of these young patients with MSI, 31,32 but little is known of the MSI status or mutation of the MMR genes in the young patients with gliomas. Using stringent criteria for MSI, 33,34 we examined a series of local young patients with high-grade gliomas (grades III to IV by the World Health Organization system), 35 to look for the presence of MSI and examined for mutation, both somatic and germline, in the hMSH2 and hMLH1 genes.
Materials and Methods
Materials
Twenty-two patients, ages 45 years or less, with gliomas of grades III to IV, were included in this study. The mean age of the patients was 33 years (range, 13 to 44). The tumors included 17 glioblastomas, 3 anaplastic astrocytomas, 2 mixed gliomas (grade III), and the histological classification used was based on the World Health Organization system. 35 Either frozen or paraffin-embedded tumor tissue with more than 80% tumor cell content was used. For normal tissue, either blood leukocytes obtained by venipuncture with the patients’ consent or normal brain tissue adjacent to the tumor was used. For all tissue used for DNA extraction, frozen or paraffin sections were used to confirm the absence of tumor cell contamination in the normal tissues and to confirm the percentage of tumor in tumor blocks. DNA and RNA were extracted from blood leukocytes and frozen tumor blocks for germline and somatic MMR gene mutational analysis using standard methods.
MSI Analysis
Paired tumor and normal tissues were amplified by polymerase chain reaction (PCR) using 5 microsatellite loci. These included dinucleotide repeats (Tp53, D18S58, and D2S123) and polyadenine tracts (Bat40 and Bat26/A26). Tp53 was purchased from Research Genetics (Huntsville, AL). D18S58, D2S123, and Bat40 were synthesized according to the sequence published previously. 13 For the polyadenine tract in intron 5 of the hMSH2 gene, two pairs of primers were used, including Bat26 as previously published, 13 and another pair named A26, corresponding to nucleotides 123 to 143 (forward) and nucleotides 222 to 241 (reverse) of the hMSH2 exon 5 genomic sequence (GenBank accession no. U41210). In all cases, all five loci were analyzed.
The PCR was performed in a 10-μl reaction solution containing 50 ng of DNA, 10 mmol/L of Tris (ph 8.3), 50 mmol/L of KCl, 2 to 3 mmol/L of Mg2+, 200 μmol/L deoxynucleotide triphosphate, 1 μCi [α-32P]dCTP, 0.2 to 1 μmol/L of each primer, and 0.1 U Taq polymerase. A hot-start reaction was performed by preheating the mixture in the thermocycler at 95°C for 5 minutes, then cooling to 80°C before adding the Taq polymerase. An initial denaturation step of 95°C for 5 minutes and 25 to 40 cycles, including 95°C for 45 seconds (1 minute), 1 minute (1.5 minutes) in 52 to 64°C annealing temperature according to the specific primers, and 72°C for 1 minute (2 minutes) in frozen DNA (paraffin DNA), was performed, followed by a final extension of 5 minutes at 72°C.
The PCR products were diluted by loading buffer, heated at 95°C for 5 minutes, and loaded onto 6% vertical polyacrylamide gel. After electrophoresis, the gels were fixed, dried, and exposed to X-ray film for 12 hours to 7 days.
The results were interpreted independently by two observers. Results with discrepancy in interpretation were discussed and PCR was repeated if necessary. MSI was defined as the presence of allelic shift or additional bands in the tumor compared with normal tissue. All cases with MSI were repeated once. A case was defined as high-level MSI if there were more than 40% unstable loci, low-level MSI if less than 40%, and microsatellite stable if there were no unstable loci. 33,34
MSI in the (A)10 tract of type II transforming growth factor β receptor (TβRII), (G)8 tracts of Bax, and insulin-like growth factor type II receptor (IGFIIR) genes was also analyzed in the microsatellite-unstable cases. The primer sequences were as described previously. 36-38
hMSH2 and hMLH1 Mutational Analysis
Mutational analysis for hMSH2 and hMLH1 was performed for the high-level MSI cases using the following methods.
In Vitro Synthesized Protein Assay
In vitro-synthesized protein assays to screen for truncation mutations in the MMR genes hMSH2 and hMLH1 were performed with primer sequences as described. 5,13 In brief, 3 μg of total RNA was reverse transcribed using 20 to 200 ng of random hexamers or oligo(dT), 20 units of RNAsin, 20 pmol of deoxynucleotide triphosphates, and 200 units of Superscript II reverse transcriptase (Life Technologies, Inc., Grand Island, NY) in a 20-μl reaction volume, using the manufacturers’ suggested reaction conditions. Forty cycles of PCR were performed in 50 μl and included 2 to 4 μl of first-strand cDNA mix, 10 mmol/L of Tris-HCl (pH 8.3), 50 mmol/L of KCl, 3 to 5 mmol/L of MgCl2, 5 pmol of each primer, 200 μmol/L of each nucleotide, and 2.5 units Taq polymerase (Life Technologies). Both hMSH2 and hMLH1 were amplified in two overlapping segments ranging between 1.2 and 2.0 kb. The left-hand primers of each segment were tagged with a T7 promoter sequence and a translation initiation site. The products were then subjected to in vitro transcription/translation using the linked T7 transcription-translation system (Amersham Corp., Little Chalfont, UK).
DNA Sequencing Analysis
Individual exons of hMSH2 and hMLH1 genes, including intron-exon boundaries, were PCR amplified. The primers’ sequences are available on request. The PCR products were then purified by High Pure PCR Product Purification Kit (Boehringer Mannheim, Mannheim, Germany) and directly sequenced by Sequenase V2.0 (Amersham) using both forward and reverse primers following the manufacturer’s protocols. The sequencing products were denatured at 80°C for 5 minutes and electrophoresed through a 6% polyacrylamide/urea gel at 70 W for 2 to 3 hours. The gels were then fixed, dried, and exposed to autoradiographic films.
Immunohistochemistry
Immunostaining for hMSH2 and hMLH1 was performed in the cases showing allelic shift in one or more loci, using the standard streptavidin-biotin-peroxidase complex method with 3,3′-diaminobenzidine as chromogen. Sections 6 μm thick of 10% neutral buffered formalin-fixed, paraffin-embedded tumor tissue were incubated for 1 hour at 37°C with monoclonal antibodies against the amino-terminal fragment (clone GB12; dilution 1:20; Oncogene Research Products, Cambridge, MA) and carboxy-terminal fragment of hMSH2 (clone FE11; dilution 1:200; Oncogene Research Products, Cambridge, MA). For hMLH1, sections were incubated at 37°C for 1 hour using a monoclonal antibody (clone G168-15; dilution 1:10; PharMingen, San Diego, CA). Microwave pretreatment at 95°C for 30 minutes in citrate buffer, pH 6.0, was performed after deparaffinization. For negative control, the primary antibodies were replaced by mouse immunoglobulin G (Dakopatts, Glostrup, Denmark).
Results
MSI
Four of the 22 high-grade gliomas from patients ages 45 years or less (18%) showed high-level MSI (Table 1) ▶ . These included three glioblastomas and one malignant mixed glioma. In all four cases, there was gross MSI with 75 to 100% loci involved. One additional tumor showed MSI in one locus only, and this case was thus considered low-level MSI. None of the tumors showed mutation in the mononucleotide tracts in the TβRII and Bax genes. One tumor showed frameshift mutation in the (G)8 tract of the IGFIIR gene. Representative results of the microsatellite analysis are shown in Figure 1 and 2 ▶ ▶ .
Table 1.
Results of Microsatellite Analysis and Immunohistochemistry for Cases Showing MSI
| Patients | |||||
|---|---|---|---|---|---|
| A | B | C | D | E | |
| Sex/age | F /27 | M /23 | M /35 | M /37 | F /38 |
| Diagnosis | GBM | Mixed glioma (grade III)* | GBM | GBM | Astro III |
| Tp53 | + | + | + | + | + |
| D18S58 | − | + | + | + | − |
| D2S123 | + | + | + | + | − |
| Bat40 | + | + | + | + | − |
| Bat26/A26† | + | + | + | + | − |
| % loci with MSI | 75% | 100% | 100% | 100% | 20% |
| TβRII (A)10 | − | − | − | − | − |
| Bax (G)8 | − | − | − | − | − |
| IGFIIR (G)8 | − | + | − | − | − |
| hMSH2 protein (IHC) | + | − | − | − | + |
| hMLH1 protein (IHC) | − | + | + | + | + |
GBM, glioblastoma multiforme; Astro III, anaplastic astrocytoma; IHC, immunohistochemistry.
†Both Bat26 and A26 amplify a polyadenine tract in intron 5 of the hMSH2 gene.
*According to World Health Organization system.
Figure 1.

MSI at various loci in gliomas: A, Tp53; B, D2S123; C, D18S58; D, Bat26; E, A26; and F, Bat40. N, normal; T, tumor.
Figure 2.
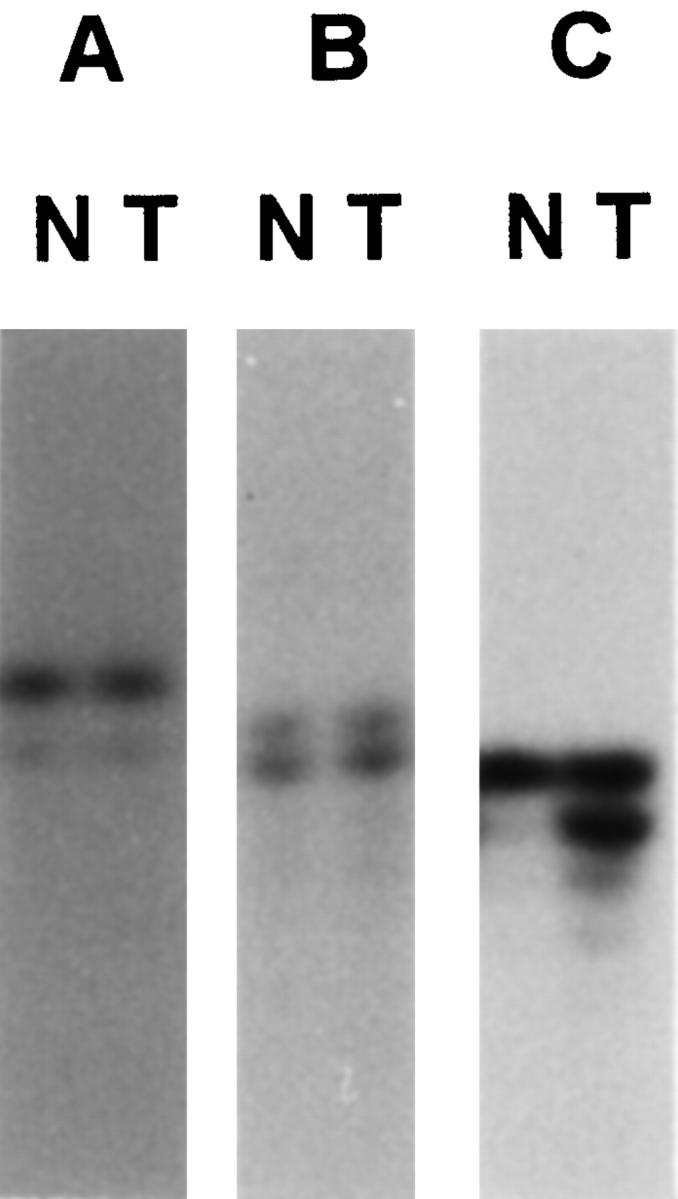
Frameshift mutation in the mononucleotide repeats of IGFIIR gene (C) but not in the TβRII (A) and Bax (B) genes in microsatellite-unstable gliomas. Lanes N, normal; lanes T, tumor.
Clinicopathological Data and Family History
The clinical data of the four patients with high-level MSI gliomas are shown in Table 2 ▶ . The patients were all relatively young; two of them were below 30 when they developed the glioblastoma. Histologically, the three glioblastomas showed primitive anaplastic cells in the background and the presence of many multinucleated tumor giant cells. Patient B had a malignant mixed glioma, with a prominent oligodendroglial element, although ependymal and astrocytic elements were noted in some areas. None except patient D had a family history of cancer. Patient D had a positive family history of colorectal carcinoma, but that did not satisfy the Amsterdam criteria for HNPCC syndrome. Interestingly, three of the patients had metachronous colorectal adenocarcinomas, thus satisfying the criteria of Turcot’s syndrome. 39 Patient A was only 27 years old when she developed a glioblastoma, and there was no previous tumor nor any family history of cancer.
Table 2.
Clinical Data and DNA MMR Gene Mutations in Four Patients with Microsatellite-Unstable Gliomas
| Patient | Sex/age | Histology | Family history | Other cancers (age, years) | Survival after craniotomy | MMR germline mutation | MMR somatic mutation in glioma |
|---|---|---|---|---|---|---|---|
| A | F /27 | GBM | None | None | Died of disease at 8 months | hMLH1 last nucleotide of exon 8 (CGgt→CAgt), resulting in splicing defect, skipping of exon 8, deletion of codon 197–226, and frameshift | hMLH1 exon 13, codon 487 CGA→TGA(stop) |
| B | M /23 | Malignant mixed glioma | None | Colon (25) | Died of disease (brain) at 4 years | hMSH2 exon 11 codon 580 GAA→TAA(stop) | Not determined; (absence of hMSH2 protein by immunostaining) |
| C | M /35 | GBM | Not available (adopted son) | Colon (29) | Alive with disease (brain) at 6 months | hMSH2 first nucleotide of exon 12 codon 587, deletion of G (agGCT→agCT), generating a stop codon 6 bp downstream | Wild-type allele loss in tumor by sequencing |
| D | M /37 | GBM | Two sisters had colorectal adenocarcinomas | Rectum (28) | Died of disease (brain) at 0.5 months | hMSH2 exon 3 codon 199 (TGT→CGT) resulting in change of an evolutionary conserved cysteine to arginine | Wild-type allele loss in tumor by sequencing |
GBM, glioblastoma multiforme.
Germline and Somatic Mutation of the MMR Genes
All four patients with high-level MSI gliomas showed germline mutation of the MMR genes, three of them in the hMSH2 and one in hMLH1 (Table 2) ▶ . Three of the mutations resulted in truncated protein products. One case (patient D) showed a missense mutation resulting in amino acid substitution in an evolutionary conserved residue. The wild-type allele was lost in the tumor in this patient.
In three cases, a second hit could be identified in the gliomas. Patient A showed two truncated protein products in the in vitro-synthesized protein assay of the tumor RNA (Figure 3) ▶ . The germline mutation was found in exon 8 of the hMLH1 gene, which resulted in skipping of the exon (Figure 4A and 5) ▶ ▶ . A second mutation, found only in the tumor DNA, resulted in a stop codon (Figure 4B) ▶ . For patients C and D, the normal allele was absent when tumor tissue was sequenced (Figure 6) ▶ . For patient B, the wild-type allele was retained in sequencing of exon 11 in the brain tumor. We did not screen for other somatic mutation in the hMSH2, because only paraffin blocks of the tumor were available.
Figure 3.

In vitro synthesized protein assay of 5′ (A) and 3′ (B) segments of the hMLH1 gene from the glioblastoma of patient A. The bands with highest molecular weight and strongest intensity correspond to the normal products. Truncated products of 21 and 18 kd (arrowheads) are noted in the 5′ and 3′ segments. Lanes C, blood leukocytes from a normal individual as control; lanes T, glioblastoma.
Figure 4.

A: Sequencing result of hMLH1 exon 8 from the blood leukocytes (lanes N) and glioblastoma (lanes T) of patient A. Both tumor and blood leukocytes demonstrate a mutation in the last nucleotide of exon 8, CGgt to CAgt (arrow), indicating that it is a germline mutation. B: Sequencing result of hMLH1 exon 13 from the blood leukocytes (lanes N) and glioblastoma (lanes T) of patient A. Somatic mutation in codon 487, CGA to TGA (arrow), generating a stop codon, is found in the tumor but not in blood leukocytes.
Figure 5.

Reverse transcription-PCR analysis using a pair of primers amplifying nucleotides 471 to 813 of hMLH1 cDNA. A wild-type band of 342 bp and an abnormal band of 253 bp (arrow), resulting from skipping of exon 8, are noted in the RNA extracted from the leukocytes of patient A (lane A) compared with a normal individual (lane C). Lane M: Bluescript-MSP1 marker.
Figure 6.

Sequencing result of hMSH2 exon 12 from the blood leukocytes (lanes L) and glioblastoma (lanes G) of patient C. Deletion of a guanosine in the first nucleotide of exon 12 (agGCT to agCT) is noted in both blood leukocytes and tumor tissue. The normal allele is lost in the tumor tissue.
Expression of the hMSH2 and hMLH1 Protein
Immunohistochemical staining revealed complete loss of hMSH2 protein when both antibodies on the tumor cells in patients B, C, and D were used, whereas the normal neurons and glial cells at the tumor borders were positive. Staining for hMLH1 was retained in the tumors in these three patients. The tumor cells in patient A were negative for hMLH1 but positive for hMSH2 proteins, whereas the normal cells were positive for both.
Discussion
Three important pieces of information resulted from this study. 1) A proportion of young patients (18%) with high-grade gliomas showed gross replication error, characteristic of defects in the DNA MMR system. 2) In all of these patients with gross replication errors, germline mutation in one of the MMR genes could be detected. 3) In three of the four cases, inactivation of the second allele was found in the tumor tissue. Although the second hit could not be detected in patient B, the tumor cells were immunohistochemically negative for hMSH2, supporting the presence of a second inactivating event. This double-hit phenomenon for MMR genes, identical to Knudson’s theory for a tumor suppressor gene, was also previously suggested in HNPCC-related and sporadic colorectal carcinomas. 4,6,7,40
To our knowledge, this is the first study documenting the presence of a germline MMR gene mutation in young patients with sporadic gliomas. A similar study in young patients with sporadic colorectal carcinoma revealed MSI in 58%, with germline MMR mutation detected in 42% of the MSI-positive cases. 32 Although germline MMR gene mutation has previously been found in four patients with Turcot’s syndrome, 21,23,24 patient A developed and died of the glioma without antecedent cancer. Patient B presented with the glioma first and only subsequently developed the colorectal carcinoma. Neither A nor B had a family history of cancer. Two other patients have antecedent colorectal carcinoma. In patient D, a family history of colorectal carcinomas was also obtained. This raises an important point concerning the management of young patients with microsatellite-unstable gliomas and their family members. From the information in our study, we conclude that screening for replication error is useful in young patients with high-grade gliomas. For high-level MSI patients, germline mutation of the MMR gene should be sought. Regular colonoscopic screening for colorectal carcinoma should be offered to the patient and the family members with demonstrable MMR gene mutation. Also, we should be alert to and check for the possibility of brain tumor by regular neurological examination. It is of note that, whereas the colorectal carcinoma could be successfully treated, three of the patients succumbed to the gliomas 0.5 months to 4 years after the craniotomy. This was in contrast to the prolonged survival noted in three patients with MSI-positive glioblastoma in a previous series. 23
Concerning the histological type of high-level MSI gliomas, three were glioblastomas. Interestingly, one case was a malignant mixed glioma with a prominent oligodendroglial component and also focal ependymal differentiation. In HNPCC kindred, the possible histological type of brain tumor includes not only astrocytomas but also oligodendrogliomas and rarely ependymomas. 21 Thus, mutation of the MMR gene may lead not only to glioblastomas, but to high-grade gliomas of oligodendroglial or even ependymal differentiation.
Mononucleotide tracts of various growth-regulatory genes are frequently the target of mutational inactivation in microsatellite-unstable tumors. The (A)10 tract in the TβRII gene is mutated in 70 to 90% of microsatellite-unstable colorectal and gastric cancers. 36,41 Frameshift mutation of the (G)8 tract in Bax is also reported in more than 50% of these cancers. 37,42,43 Apart from frameshift mutation in the (G)8 tract, somatic mutation of Bax genes is frequent in MSI-positive gastric and colorectal carcinomas, 43 but not in gliomas. 44 Interestingly, none of the MSI-positive gliomas in this study showed mutation in the TβRII and Bax genes. This may be the result of selection pressure, in which mutation of genes caused by MMR defects are selected for if they confer growth advantage in that organ.
We identified a frameshift mutation in the IGFIIR gene in the malignant mixed glioma from patient B, the first reported mutation in this gene in a glioma, although IGFIIR mutation has been reported in MSI colorectal, gastric, and endometrial carcinomas. 38,45 IGFIIR plays a role in activation of transforming growth factor β, 46 which is a potent growth inhibitor. Also, it antagonizes the growth-stimulatory effect of IGFII by internalizing and degrading the protein. 47 Given that enhanced expression of IGFII mRNA has been reported in gliomas, 48 inactivating mutation of IGFIIR may remove the growth-inhibitory signal and confer growth advantage.
The molecular genetic pathways of different subsets of glioblastoma have been increasingly clarified in recent years. 29,49 Those arising de novo are referred to as primary glioblastomas, and those developed from a pre-existing astrocytoma are referred to as secondary glioblastomas. Most primary glioblastomas develop in older patients (mean, 55 years) with epidermal growth factor receptor amplification or overexpression, loss of heterozygosity in chromosome 10, and p16 deletion. Secondary glioblastomas tend to occur in younger patients (mean, 39 years), and most of them harbor p53 mutations. 50-54 We have demonstrated that a proportion of primary glioblastomas in young patients can be caused by germline MMR gene mutations, and these patients and their family members are at risk of developing other HNPCC-related tumors, in particular colorectal carcinomas. Screening for MSI and MMR gene mutation is thus of importance in the management of these patients.
Acknowledgments
We thank Mr. Samson W. C. Shum for the technical assistance, Miss Kedo Kwan for collecting the family history, and Dr. R. J. Collins for his assistance with the manuscript.
Footnotes
Address reprint requests to Dr. Siu Tsan Yuen, Department of Pathology, Queen Mary Hospital, The University of Hong Kong, Pokfulam, Hong Kong. E-mail: styuen@hkucc.hku.hk.
Supported by Committee on Research and Conference Grant 337/046/0024 and University Research Committee Grant 344/046/0003 from the University of Hong Kong and by Croucher Foundation Research Grant 394/046/1238. TLC is a Ph.D. student of the University of Hong Kong.
References
- 1.Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, Petersen GM, Kinzler KW, Vogelstein B, de la Chapell A: Clues to the pathogenesis of familial colorectal cancer. Science 1993, 260:812-816 [DOI] [PubMed] [Google Scholar]
- 2.Lynch HT, Smyrk TC, Watson P, Lanspa SJ, Lynch JF, Lynch PM, Cavalieri RJ, Boland CR: Genetics, natural history, tumor spectrum, and pathology of hereditary nonpolyposis colorectal cancer: an updated review. Gastroenterology 1993, 104:1535-1549 [DOI] [PubMed] [Google Scholar]
- 3.Fishel R, Lescoe MK, Rao MR, Copeland NG, Jenkins NA, Garber J, Kane M, Kolodner R: The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell 1993, 75:1027-1038 [DOI] [PubMed] [Google Scholar]
- 4.Leach FS, Nicolaides NC, Papadopoulos N, Liu B, Jen J, Parsons R, Peltomaki P, Sistonen P, Aaltonen LA, Nystrom Lahti M, Guan XY, Zhang J, Meltzer PS, Yu JW, Kao FT, Chen DJ, Cerosaletti KM, Fournier RE, Todd S, Lewis T, Leach RJ, Naylor SL, Weissenbach J, Mecklin JP, Jarvinen H, Petersen GM, Hamilton SR, Green J, Jass J, Watson P, Lynch HT, Trent JM, de la Chapelle A, Kinzler KW, Vogelstein B: Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell 1993, 75:1215-1225 [DOI] [PubMed] [Google Scholar]
- 5.Liu B, Parsons RE, Hamilton SR, Petersen GM, Lynch HT, Watson P, Markowitz S, Willson JK, Green J, de la Chapelle A, Kinzler KW, Vogelstein B: hMSH2 mutations in hereditary nonpolyposis colorectal cancer kindreds. Cancer Res 1994, 54:4590-4594 [PubMed] [Google Scholar]
- 6.Papadopoulos N, Nicolaides NC, Wei YF, Ruben SM, Carter KC, Rosen C, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD, Venter JC, Hamilton SR, Petersen GM, Watson P, Lynch HT, Peltomaki P, Mecklin JP, de la Chapelle A, Kinzler KW, Vogelstein B: Mutation of a mutL homolog in hereditary colon cancer. Science 1994, 263:1625-1629 [DOI] [PubMed] [Google Scholar]
- 7.Nicolaides NC, Papadopoulos N, Liu B, Wei YF, Carter KC, Ruben SM, Rosen CA, Haseltine WA, Fleischmann RD, Fraser CM, Adams MD, Venter JC, Dunlop MG, Hamilton SR, Petersen GM, de la Chapelle A, Vogelstein B, Kinzler KW: Mutations of two PMS homologues in hereditary nonpolyposis colon cancer. Nature 1994, 371:75-80 [DOI] [PubMed] [Google Scholar]
- 8.Bronner CE, Baker SM, Morrison PT, Warren G, Smith LG, Lescoe MK, Kane M, Earabino C, Lipford J, Lindblom A, Tannergard P, Bollag RJ, Godwin AR, Ward DC, Nordenskjold M, Fishel R, Kolodner R, Liskay RM: Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature 1994, 368:258-261 [DOI] [PubMed] [Google Scholar]
- 9.Palombo F, Hughes M, Jiricny J, Truong O, Hsuan J: Mismatch repair and cancer. Nature 1994, 367:417. [DOI] [PubMed] [Google Scholar]
- 10.Palombo F, Gallinari P, Iaccarino I, Lettieri T, Hughes M, D’Arrigo A, Truong O, Hsuan JJ, Jiricny J: GTBP, a 160-kilodalton protein essential for mismatch-binding activity in human cells. Science 1995, 268:1912-1914 [DOI] [PubMed] [Google Scholar]
- 11.Drummond JT, Li GM, Longley MJ, Modrich P: Isolation of an hMSH2–p160 heterodimer that restores DNA mismatch repair to tumor cells. Science 1995, 268:1909-1912 [DOI] [PubMed] [Google Scholar]
- 12.Papadopoulos N, Nicolaides NC, Liu B, Parsons R, Lengauer C, Palombo F, D’Arrigo A, Markowitz S, Willson JK, Kinzler KW, Jiricny J, Vogelstein B: Mutations of GTBP in genetically unstable cells. Science 1995, 268:1915-1917 [DOI] [PubMed] [Google Scholar]
- 13.Liu B, Parsons R, Papadopoulos N, Nicolaides NC, Lynch HT, Watson P, Jass JR, Dunlop M, Wyllie A, Peltomaki P, de la Chapelle A, Hamilton SR, Vogelstein B, Kinzler KW: Analysis of mismatch repair genes in hereditary non-polyposis colorectal cancer patients. Nat Med 1996, 2:169-174 [DOI] [PubMed] [Google Scholar]
- 14.Eshleman JR, Markowitz SD: Microsatellite instability in inherited and sporadic neoplasms. Curr Opin Oncol 1995, 7:83-89 [PubMed] [Google Scholar]
- 15.Izumoto S, Arita N, Ohnishi T, Hiraga S, Taki T, Tomita N, Ohue M, Hayakawa T: Microsatellite instability and mutated type II transforming growth factor-β receptor gene in gliomas. Cancer Lett 1997, 112:251-256 [DOI] [PubMed] [Google Scholar]
- 16.Dams E, Van de Kelft EJ, Martin JJ, Verlooy J, Willems PJ: Instability of microsatellites in human gliomas. Cancer Res 1995, 55:1547-1549 [PubMed] [Google Scholar]
- 17.Zhu J, Guo SZ, Beggs AH, Maruyama T, Santarius T, Dashner K, Olsen N, Wu JK, Black P: Microsatellite instability analysis of primary human brain tumors. Oncogene 1996, 12:1417-1423 [PubMed] [Google Scholar]
- 18.Wooster R, Cleton Jansen AM, Collins N, Mangion J, Cornelis RS, Cooper CS, Gusterson BA, Ponder BA, von Deimling A, Wiestler OD, Cornelisse CJ, Devilee P, Stratton MR: Instability of short tandem repeats (microsatellites) in human cancers. Nat Genet 1994, 6:152-156 [DOI] [PubMed] [Google Scholar]
- 19.Amariglio N, Friedman E, Mor O, Stiebel H, Phelan C, Collins P, Nordenskjold M, Brok Simoni F, Rechavi G: Analysis of microsatellite repeats in pediatric brain tumors. Cancer Genet Cytogenet 1995, 84:56-59 [DOI] [PubMed] [Google Scholar]
- 20.Ponz de Leon M, Benatti P, Pedroni M, Sassatelli R, Roncucci L: Risk of cancer revealed by follow-up of families with hereditary non-polyposis colorectal cancer: a population-based study. Int J Cancer 1993, 55:202-207 [DOI] [PubMed] [Google Scholar]
- 21.Vasen HF, Sanders EA, Taal BG, Nagengast FM, Griffioen G, Menko FH, Kleibeuker JH, Houwing Duistermaat JJ, Meera Khan P: The risk of brain tumours in hereditary non-polyposis colorectal cancer (HNPCC). Int J Cancer 1996, 65:422-425 [DOI] [PubMed] [Google Scholar]
- 22.Heinimann K, Muller H, Weber W, Scott RJ: Disease expression in Swiss hereditary non-polyposis colorectal cancer (HNPCC) kindreds. Int J Cancer 1997, 74:281-285 [DOI] [PubMed] [Google Scholar]
- 23.Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, Krush AJ, Berk T, Cohen Z, Tetu B, Burger PC, Wood PA, Taqi F, Booker SV, Petersden GM, Offerhaus GJA, Tersmette AC, Giardiello FM, Vogelstein B, Kinzler KW: The molecular basis of Turcot’s syndrome. N Engl J Med 1995, 332:839-847 [DOI] [PubMed] [Google Scholar]
- 24.Miyaki M, Nishio J, Konishi M, Kikuchi-Yanoshita R, Tanaka K, Muraoka M, Nagato M, Chong JM, Koika M, Terada T, Yutaka K, Fukutome A, Tomiyama J, Chuganji Y, Momoi M, Utsunomiya J: Drastic genetic instability of tumours and normal tissues in Turcot syndrome. Oncogene 1997, 15:2877-2881 [DOI] [PubMed] [Google Scholar]
- 25.Yuen ST, Chung LP, Leung SY, Luk ISC, Chan SY, Ho JWC, Wyllie AH: Colorectal carcinoma in Hong Kong: epidemiology and genetic mutations. Br J Cancer 1997, 76:1610-1616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang WQ, Zheng SJ, Tian QS, Huang JQ, Li Yx, Xu QZ, Liu ZJ, Zhang WC: Statistical analysis of central nervous system tumors in China. J Neurosurg 1982, 56:555-564 [DOI] [PubMed] [Google Scholar]
- 27.Kepes JJ, Chen WY, Pang LC, Kepes M: Tumors of the central nervous system in Taiwan, Republic of China. Surg Neurol 1984, 22:149-156 [DOI] [PubMed] [Google Scholar]
- 28.Ng HK, Poon WS, South JR, Lee JC: Tumours of the central nervous system in Chinese in Hong Kong: a histological review. Aust N Z J Surg 1988, 58:573-578 [DOI] [PubMed] [Google Scholar]
- 29.Kleihues P, Burger PC, Plate KH, Ohgaki H, Cavenee WK: Glioblastoma: pathology and genetics of tumours of the nervous system. Edited by Kleihues P, Cavenee WK. Lyon: International Agency for Research on Cancer, 1997, pp 16–28
- 30.Zulch KJ: Brain Tumors: Their Biology and Pathology, ed 3. Berlin, Springer-Verlag, 1986
- 31.Chan TL, Yuen ST, Chung LP, Ho J, Leung SY, Chan AS, Chan LC: Replication errors (RER) and mismatch repair (MMR) gene mutations in young colorectal carcinoma patients in Hong Kong Chinese. Genetics of human cancer: pathogenesis and diagnosis, Proceedings of Keystone Symposium on Molecular and Cellular Biology, 1997, p 9
- 32.Liu B, Farrington SM, Petersen GM, Hamilton SR, Parsons R, Papadopoulos N, Fujiwara T, Jen J, Kinzler KW, Wyllie AH, Vogelstein B, Dunlop MG: Genetic instability occurs in the majority of young patients with colorectal cancer. Nat Med 1995, 1:348-352 [DOI] [PubMed] [Google Scholar]
- 33.Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J: Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res 1997, 57:4749-4756 [PubMed] [Google Scholar]
- 34.Bocker T, Diermann J, Friedl W, Gebert J, Holinski Feder E, Karner H, von Knebel Doeberitz M, Koelble K, Moeslein G, Schackert HK, Wirtz HC, Fishel R, Ruschoff J: Microsatellite instability analysis: a multicenter study for reliability and quality control. Cancer Res 1997, 57:4739-4743 [PubMed] [Google Scholar]
- 35.Kleihues P, Burger PC, Scheithauer BW: Histological Typing of Tumours of the Central Nervous System. 1993. Springer-Verlag, Berlin
- 36.Myeroff LL, Parsons R, Kim SJ, Hedrick L, Cho KR, Orth K, Mathis M, Kinzler KW, Lutterbaugh J, Park K, Bang YJ, Lee HY, Park JG, Lynch HT, Roberts AB, Vogelstein B, Markowitz SD: A transforming growth factor β receptor type II gene mutation common in colon and gastric but rare in endometrial cancers with microsatellite instability. Cancer Res 1995, 55:5545-5547 [PubMed] [Google Scholar]
- 37.Rampino N, Yamamoto H, Ionov Y, Li Y, Sawai H, Reed JC, Perucho M: Somatic frameshift mutations in the BAX gene in colon cancers of the microsatellite mutator phenotype. Science 1997, 275:967-969 [DOI] [PubMed] [Google Scholar]
- 38.Souza RF, Appel R, Yin J, Wang S, Smolinski KN, Abraham JM, Zou TT, Shi YQ, Lei J, Cottrell J, Cymes K, Biden K, Simms L, Leggett B, Lynch PM, Frazier M, Powell SM, Harpaz N, Sugimura H, Young J, Meltzer SJ: Microsatellite instability in the insulin-like growth factor II receptor gene in gastrointestinal tumours. Nat Genet 1996, 14:255-257 [DOI] [PubMed] [Google Scholar]
- 39.Turcot J, Despres JP, St. Pierre F: Malignant tumors of the central nervous system associated with familial polyposis of the colon: report of two cases. Dis Colon Rectum 1959, 2:465-468 [DOI] [PubMed] [Google Scholar]
- 40.Liu B, Nicolaides NC, Markowitz S, Willson JK, Parsons RE, Jen J, Papadopoulos N, Peltomaki P, de la Chapelle A, Hamilton SR, Kinzler KW, Vogelstein B: Mismatch repair gene defects in sporadic colorectal cancers with microsatellite instability. Nat Genet 1995, 9:48-55 [DOI] [PubMed] [Google Scholar]
- 41.Parsons R, Myeroff LL, Liu B, Willson JK, Markowitz SD, Kinzler KW, Vogelstein B: Microsatellite instability and mutations of the transforming growth factor β type II receptor gene in colorectal cancer. Cancer Res 1995, 55:5548-5550 [PubMed] [Google Scholar]
- 42.Chung YJ, Park SW, Song JM, Lee KY, Seo EJ, Choi SW, Rhyu MG: Evidence of genetic progression in human gastric carcinomas with microsatellite instability. Oncogene 1997, 15:1719-1726 [DOI] [PubMed] [Google Scholar]
- 43.Yamamoto H, Sawai H, Perucho M: Frameshift somatic mutations in gastrointestinal cancer of the microsatellite mutator phenotype. Cancer Res 1997, 57:4420-4426 [PubMed] [Google Scholar]
- 44.Chou D, Miyashita T, Mohrenweiser HW, Ueki K, Kastury K, Druck T, von Deimling A, Huebner K, Reed JC, Louis DN: The BAX gene maps to the glioma candidate region at 19q13.3, but is not altered in human gliomas. Cancer Genet Cytogenet 1996, 88:136-140 [DOI] [PubMed] [Google Scholar]
- 45.Ouyang H, Shiwaku HO, Hagiwara H, Miura K, Abe T, Kato Y, Ohtani H, Shiiba K, Souza RF, Meltzer SJ, Horii A: The insulin-like growth factor II receptor gene is mutated in genetically unstable cancers of the endometrium, stomach, and colorectum. Cancer Res 1997, 57:1851-1854 [PubMed] [Google Scholar]
- 46.Dennis PA, Rifkin DB: Cellular activation of latent transforming growth factor β requires binding to the cation-independent mannose 6-phosphate/insulin-like growth factor type II receptor. Proc Natl Acad Sci USA 1991, 88:580-584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kornfeld S: Structure and function of the mannose 6-phosphate/insulin-like growth factor II receptors. Annu Rev Biochem 1992, 61:307-330 [DOI] [PubMed] [Google Scholar]
- 48.Sandberg AC, Engberg C, Lake M, von Holst H, Sara VR: The expression of insulin-like growth factor I and insulin-like growth factor II genes in the human fetal and adult brain and in glioma. Neurosci Lett 1988, 93:114-119 [DOI] [PubMed] [Google Scholar]
- 49.Kleihues P, Ohgaki H: Genetics of glioma progression and the definition of primary and secondary glioblastoma. Brain Pathol 1997, 7:1131-1136 [Google Scholar]
- 50.Lang FF, Miller DC, Koslow M, Newcomb EW: Pathways leading to glioblastoma multiforme: a molecular analysis of genetic alterations in 65 astrocytic tumors. J Neurosurg 1994, 81:427-436 [DOI] [PubMed] [Google Scholar]
- 51.von Deimling A, von Ammon K, Schoenfeld D, Wiestler OD, Seizinger B, Louis DN: Subsets of glioblastoma multiforme defined by molecular genetic analysis. Brain Pathol 1993, 3:19-26 [DOI] [PubMed] [Google Scholar]
- 52.Watanabe K, Tachibana O, Sata K, Yonekawa Y, Kleihues P, Ohgaki H: Overexpression of the EGF receptor and p53 mutations are mutually exclusive in the evolution of primary and secondary glioblastomas. Brain Pathol 1996, 6:217-223 [DOI] [PubMed] [Google Scholar]
- 53.Hayashi Y, Ueki K, Waha A, Wiestler OD, Louis DN, von Deimling A: Association of EGFR gene amplification and CDKN2 (p16/MTS1) gene deletion in glioblastoma multiforme. Brain Pathol 1997, 7:871-875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Biernat W, Tohma Y, Yonekawa Y, Kleihues P, Ohgaki H: Alterations of cell cycle regulatory genes in primary (de novo) and secondary glioblastomas. Acta Neuropathol Berl 1997, 94:303-309 [DOI] [PubMed] [Google Scholar]