

Abstract
Recent studies indicate that p21ras proteins mediate their multiple cell functions through interactions with multiple effectors and that the number of new effectors is growing. We recently reported that K-ras2 mutations in human colorectal adenomas were associated with chromosome instability and proliferation changes. In the present study, we extend these previous observations. Hereditary and multiple (n ≥ 5) adenomas and adenomas with early cancer were excluded. Dysplasia was moderate in 91 cases and high in 25, and the median adenoma size was 1.5 cm. K-ras2 spectrum analysis was done by sequence-specific oligonucleotide hybridization using nuclear suspensions provided by analysis and sorting of multiparameter flow cytometry. In particular, tissue inflammatory cells were separated for DNA diploid tumors, whereas DNA aneuploid epithelial subclones were analyzed separately. K-ras2 mutations and DNA aneuploidy were both detected in 29 of 116 (25%) cases. DNA aneuploid index was in the near-diploid region in the majority of cases. DNA aneuploidy was strongly associated with G→C/T transversions. An association was also found between low S-phase values and G→A transitions. These findings were confirmed using multivariate logistic regression analysis to account for the effects of size, dysplasia, site, type, age, and sex. These data suggest that specific K-ras2 mutations in a subgroup of human sporadic colorectal adenomas play a role in chromosome instability and, contrary to expectations, are associated with inhibition of proliferation.
Ras proteins participate at the plasma membrane level in transduction of diverse extracellular physiological signals that are thought to induce appropriate gene expression toward proliferation and differentiation. Ras proteins possess an intrinsic GTPase activity and alternate between activated and inactivated forms. The best-known Ras transduction mechanism is a pathway from receptor tyrosine kinases to transcription factors phosphorylated by mitogen-activated protein kinases. Mutations of the ras oncogenes result in constitutive signaling to downstream elements and are detected at high frequency in many types of human cancer. 1-3 Recently, a link of Ras proteins to cell cycle machinery and proliferation via regulation of cyclin D1 and cyclin-dependent kinases was reported. 4-6 The existence of other important cell functions of Ras is supported by a number of reports. For example, Ras proteins were reported to play a role in apoptosis by interacting with protein kinase C and Bcl-2 or by leading to the activation of stress-activating protein kinase (SAPK, also called jun kinase or JNK) and of the p38 subfamily. 7-11 Ras proteins were also reported to regulate the formation of stress fibers, focal cell adhesion, and cytokinesis. 12,13 Transfection of human mutated K-ras2 in mouse NIH-3T3 cells has been shown to induce destabilization of the chromosomes in mitosis 14,15 and generation of DNA aneuploid subclones as detected by flow cytometry (FCM). 15 Chromosome losses and chromatin textural changes by image cytometry were also shown to occur in H-ras-transformed human breast epithelial cells. 16 Dependence of aneuploidy from H-ras mutations was also shown in chemically induced mouse skin papillomas from a very early stage. 17,18
All these cell systems clearly lack the complexity of human systems and, to our knowledge, the role of K-ras2 activation on aneuploidy and proliferation for human sporadic colorectal adenomas is not yet well established. The incidence of K-ras2 mutations, mainly in codons 12 and 13, in human colorectal adenomas was reported to be up to 60%. 19-23 A similar incidence was detected for DNA aneuploidy using FCM. 24-27 K-ras2 mutations in colorectal adenomas were detected in both DNA diploid and aneuploid cells and in some cases in regions of histologically normal mucosa, suggesting that K-ras2 mutations occur before change in DNA ploidy. 22,23 Also, human colorectal aberrant crypt foci, suggested to be early precursor lesions of adenomas, were found to be mutated in K-ras2 up to the 85% level. 28-30 The present study addresses the possible relationship of specific K-ras2 mutations with DNA aneuploidy and proliferation in human sporadic colorectal adenomas.
Materials and Methods
Study Population
The study was performed on 116 polyps (size range, 0.3 to 5 cm: 15 were <1 cm, 77 were 1 to 2 cm, and 24 were >2 cm; median, 1.5 cm) with a histological diagnosis of adenomas. Sixty-two polyps were located in the sigmoid, 16 in the rectum, 11 in the ascending colon, 17 in the descending colon, 5 in the transverse colon, and 5 in the cecum. Patients (43 females and 60 males) age 33 to 86 years (median, 63 years) did not have history of familial adenomatous polyposis (FAP) or suspected attenuated FAP. Controls were taken in the vast majority of cases during endoscopy from the normal mucosa at the rectal-sigmoid junction 25 and in 19 cases from mucosa of healthy donors. 22
Histological Analysis and Topographic Selection
Histological diagnosis and grading were according to the World Health Organization criteria. 31 Two dysplasia grades were considered, ie, a low-grade class including mild and moderate dysplasia (n = 91) and high-grade class of severe dysplasia (n = 25). Adenomas with early cancer were excluded from this study. Polyps were divided into two specular parts by a central midsaggital section. One specimen was fixed in 10% buffered formalin for 24 hours, handled according to customary and histopathological diagnosis protocols, and embedded in paraffin. The other specimen was immediately frozen in liquid nitrogen and stored at −80°C for not more than a week. Using a hematoxylin and eosin-stained cryostatic section as histotopographic reference, samples were taken by hand with a scalpel blade from selected prevalent areas with homogeneous dysplasia grade. Areas with prevalent connective tissue and normal epithelial cells were also partly discarded. Multiple cryostatic sections were then taken from all of the sides of each sample, providing roughly cubic blocks with linear size ranging from about 4 to 10 mm. This procedure was repeated until the same histological features were observed on the whole surface of the block.
DNA FCM and Sorting
The specimens for FCM were treated as previously detailed. 25 In brief, the tissue fragments were minced with scalpels for 1 to 2 minutes directly in the staining solution composed of 10 mmol/L phosphate buffer in isotonic saline, 1 mmol/L CaCl2, 0.5 mmol/L MgSO4, 0.6% Nonidet P40 (v/v), 0.2% bovine serum albumin (w/v), and 10 mg/l of 4,6-diamidino-2-phenilindole-2-hydrochloride (DAPI; Sigma Chemical Co., St. Louis, MO). Nuclear suspensions were syringed, filtered through a 50 μm nylon filter, and immediately measured. The measurements were taken with a FACS 440 dual laser flow sorter system (Becton-Dickinson, Sunnyvale, CA). Three parameters were simultaneously measured in list mode acquisition for every individual nucleus, ie, blue emission (from DAPI), 0° forward scatter, and 90° perpendicular scatter. Excitation was provided by the ultraviolet 351 to 364-nm lines (100 mW) of an argon ion laser (model 2025, Spectra Physics, Mountain View, CA). DAPI emission fluorescence signals (obtained with suitable filters in the 450 to 490-nm range) and scatter signals were input to signal processing electronics using 1024 channels for subsequent storage, graphics, and analysis on a 486 personal computer equipped with dedicated software (Phoenix Flow Systems, San Diego, CA). Mixed samples of tissue nuclei and individual specific lymphocytes showed that infiltrating and external lymphocytes superimposed in all cases. Trout erythrocytes and individual-specific normal mucosa were also used as reference DNA standards. The degree of DNA aneuploidy (also known as DNA index (DI)) was calculated as the ratio of mean channel number of epithelial aneuploid G0-G1 peak to mean channel number of peak corresponding to tissue-infiltrating G0-G1 lymphocytes. DNA aneuploidy was taken only when lymphocyte and epithelial nuclei showed two clear-cut separated peaks. One region of abnormal DI values of special interest was defined as near-diploid aneuploidy (DI ≠ 1 and DI ≤ 1.4) in comparison with high aneuploidy (DI>1.4 and DI ≠ 2). DNA tetraploidy (DI = 2) was defined at the threshold value of the mean G2+M peak size among the controls after adding to it 3 standard deviation values. No DNA tetraploidy was detected in the present series of adenomas. Coefficient of variation values of the G0-G1 peaks and S-phase fraction values were evaluated, after gating out the tissue-infiltrating lymphocytes, using dedicated software (Phoenix Flow Systems). The mean coefficient of variation was 3.36 ± 0.68. Specifically selected FCM “sorting windows” were activated to enrich for the epithelial cell component. In the case of DNA diploid adenomas, the enrichment of the epithelial cell component was done by discarding lymphocytes. In the case of DNA aneuploid adenomas, the sorting was performed only for the DNA aneuploid epithelial nuclei.
K-ras2 Analysis
Peripheral blood lymphocytes from healthy donors were used as wild-type K-ras2 codon 12 GGT-gly and codon 13 GGC-gly controls. Additionally, six cell lines were used as controls for known K-ras2 mutations, ie, murine NIH3T3 with a transfected human K-ras2 CGT in codon 12, 32 human SW480 cells (American Type Culture Collection (ATCC), Manassas, VA) that are GTT homozygously mutated in codon 12; 33 human DLD-1 cells (ATCC) heterozygously GAC mutated in codon 13; human SW837 cells (ATCC) heterozygously TGT mutated in codon 12; and two human lung cancer cell lines, A549 and SKLU-1, respectively heterozygously AGT and GAT mutated in codon 12. High molecular weight genomic DNA was extracted by a standard method. 34
FCM-sorted nuclei from both control mucosa and adenomas were stored at −80°C and then treated as follows: they were first washed 30 minutes at 1500 × g in phosphate-buffered saline, then resuspended in 1× polymerase chain reaction (PCR) buffer (Perkin-Elmer Corp., Norwalk, CT), and finally heated to 100°C for 10 minutes before PCR. 35 PCR amplification of the K-ras2 fragment in exon 1-containing codons 12 and 13 was done in a total volume of 50 μl of PCR reaction mixture (50 mmol/L KCl; 10 mmol/L Tris-HCl, pH 8.3; 1.5 mmol/L MgCl2; 0.01% gelatin; 200 μmol/L each dATP, dGTP, dCTP, and dTTP; and 0.8 to 1.2 μmol/L for each oligonucleotide primer (Beckman, Fullerton, CA)) and 2.0 U of Taq polymerase (Perkin-Elmer). In each cycle, samples were denatured for 20 seconds at 98°C, followed by 30 seconds at 53°C (annealing) and 30 seconds at 72°C (polymerization). Using automatic gene-amplification PCR (System 9600, Perkin-Elmer), 35 cycles were performed. A PCR product of 69 bp was obtained after successive amplifications using both outside and inside (nested) primers flanking codons 12 and 13 of the K-ras2 gene, as follows: outside primers: 5′ primer, 5′-TAA GGC CTG CTG AAA ATG ACT GAA T-3′, and 3′ primer, 5′-CTC TAT TGT TGG ATC ATA TTC GTC-3′; inside primers (nested): 5′ primer, 5′-ACT GAA TAT AAA CTT GTG GTA GTT-3′, and 3′ primer, 5′-AAT TAG CTG TAT CGT CAA GGC-3′.
PCR products were routinely checked for amplified DNA on 1.5% agarose gel containing 0.5 μg/ml ethidium bromide. The oligonucleotide 20-mer panel (synthesized by TIB MOL, National Cancer Institute Advanced Biotechnology Center, Genoa, Italy) included K-ras2 codon 12 and 13 wild-type sequences, all possible mutations of codon 12, and the AGC and GAC mutations of codon 13. The probes were 5′-end labeled by phosphorylation with [γ-32P]ATP, according to the standard method. 34
Dot-blot and sequence-specific oligonucleotide probe hybridization was done with 10 μl of PCR products denatured and spotted onto Hybond-N-nylon filters (Amersham International, Buckinghamshire, United Kingdom) using a Microfiltration Bio Dot apparatus (Bio-Rad, Richmond, CA). The filters were then prehybridized for 1 hour at 65°C in 5× sodium saline phosphate ethylenediaminetetraacetic acid, 5× Denhardt’s solution, 0.5% sodium dodecyl sulfate, and 100 μg/ml DNA from herring sperm and hybridized (Hybridiser HB-1D; Techne, Cambridge, United Kingdom), in the same solution containing each 32P-end-labeled oligonucleotide probe, overnight at 58°C.
Filters were washed briefly twice in 6× standard saline citrate at room temperature, and, additionally, once under stringent conditions at 60°C for 40 minutes in 3 mol/L tetramethylammonium chloride, 50 mmol/L Tris-HCl pH 8.0, 2.5 mmol/L ethylenediaminetetraacetic acid, and 0.1% sodium dodecyl sulfate. Autoradiography was performed using Amersham MP-Hyperfilm at −80°C for 2 to 4 hours.
Statistical Analysis
Associations among the variables measured in the study, ie, DI, S-phase, K-ras2, adenoma size, dysplasia, site and type, and patient age and sex were investigated by using the Pearson correlation coefficient and the χ 2 test. 36 To this aim, variables were categorized as follows: DI (DNA diploid and aneuploid), S-phase (lower than/equal to and higher than the median value of 7.3%), K-ras2 (wild type, G→A transitions, and G→C/T transversions), size (lower than/equal to and higher than the median value of 1.5 cm), dysplasia (low-moderate and high), site (ascending, descending, or transverse colon and cecum versus sigmoid colon and versus rectum), type (tubular versus tubulovillous and villous), age (lower than/equal to and higher than the median value of 63 years), and sex (females and males). Statistically significant associations were indicated for P values of 0.05 and 0.01 (two tailed).
The associations of DI and S phase, as dependent variables, with K-ras2 were also investigated by using logistic regression analysis 37 to account for the effect of the other variables. The statistical analyses were performed using the SPSS statistical software, version 7.5. 38
Results
Figure 1 ▶ shows two examples of multiparameter FCM of DAPI-stained nuclei. Fluorescence emission from DAPI is proportional to nuclear DNA content, whereas forward and side scatter signals are associated with nuclear size and structure. A1 and B1 regions, as shown in the computer drawing (Figure 1) ▶ , mainly contained epithelial nuclei, whereas A2 and B2 regions comprised tissue-infiltrating inflammatory nuclei. When, in fact, external control mucosa and lymphocytes obtained from the same patients were added to these samples, the corresponding regions were increased by the same proportion of nuclei (not shown). When the samples contained only DNA diploid nuclei, the GO-G1 peaks corresponding to epithelial and inflammatory nuclei superimposed (Figure 1 ▶ , top). In these cases, the S-phase fraction could be corrected for the diluting effect of the presence of tissue-infiltrating inflammatory cells (corresponding to the broken line in the gated DNA histograms). Figure 1 ▶ , bottom, shows an example of a DNA near-diploid aneuploid peak overlapping with a DNA diploid peak that was resolved as a result of forward-side scatter gating.
Figure 1.
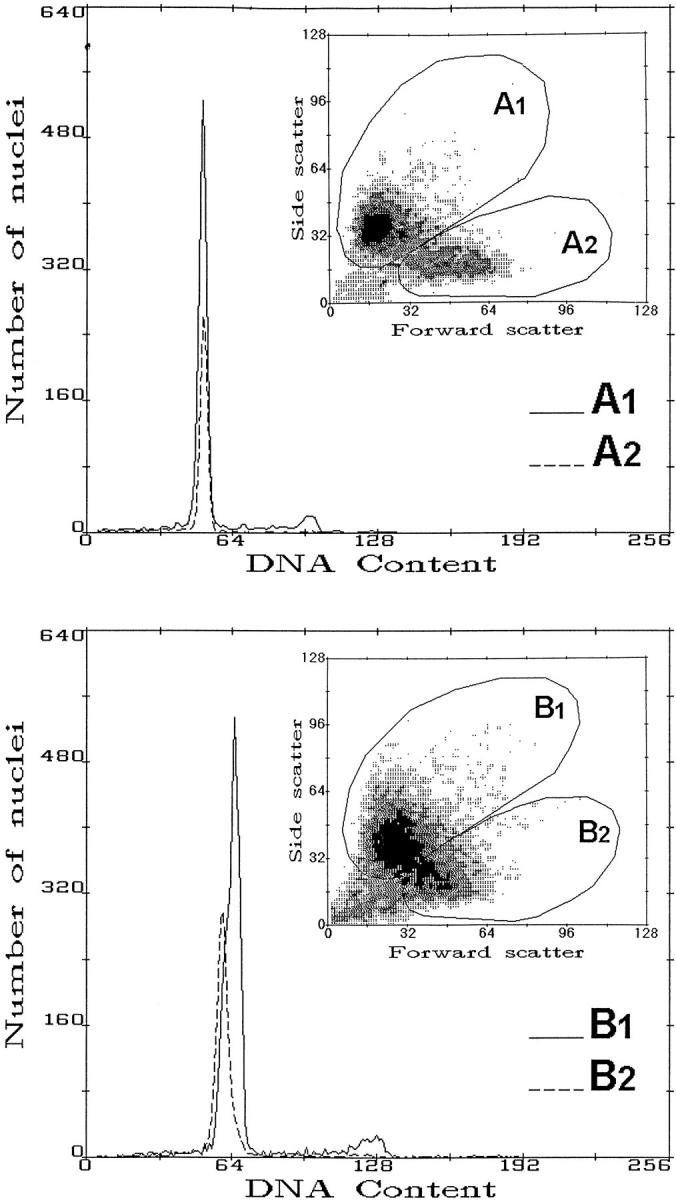
Examples of multiparameter FCM of DAPI-stained nuclear suspensions obtained from dysplastic areas of human sporadic colorectal adenomas. Top: Forward and side scatter parameters were used to gate out inflammatory cells (corresponding to A2 and B2 subregions) and correct S-phase fraction values among DNA diploid cases. Bottom: Detection of a DNA aneuploid near-diploid peak overlapping with the DNA peak of diploid inflammatory cells. The FCM sorting of an enriched epithelial cell component was done to improve the sensitivity of PCR and sequence-specific oligonucleotide analysis of the K-ras2 spectrum.
DNA aneuploid subpopulations were detected in 29 of 116 adenomas (25%). Figure 2 ▶ , top, shows that the vast majority of DI aneuploid values (DI ≠ 1) fell in the near-diploid region within 0.9 and 1.4 (24 out of the 29 cases, ie, 83%). S-phase fraction values were obtained for 109 adenomas giving a median population value of 7.3% and range from 2.5 to 31% (Figure 2 ▶ , center). Figure 2 ▶ , bottom, shows all DI values in relationship with S-phase values. No correlation was present between the two variables.
Figure 2.
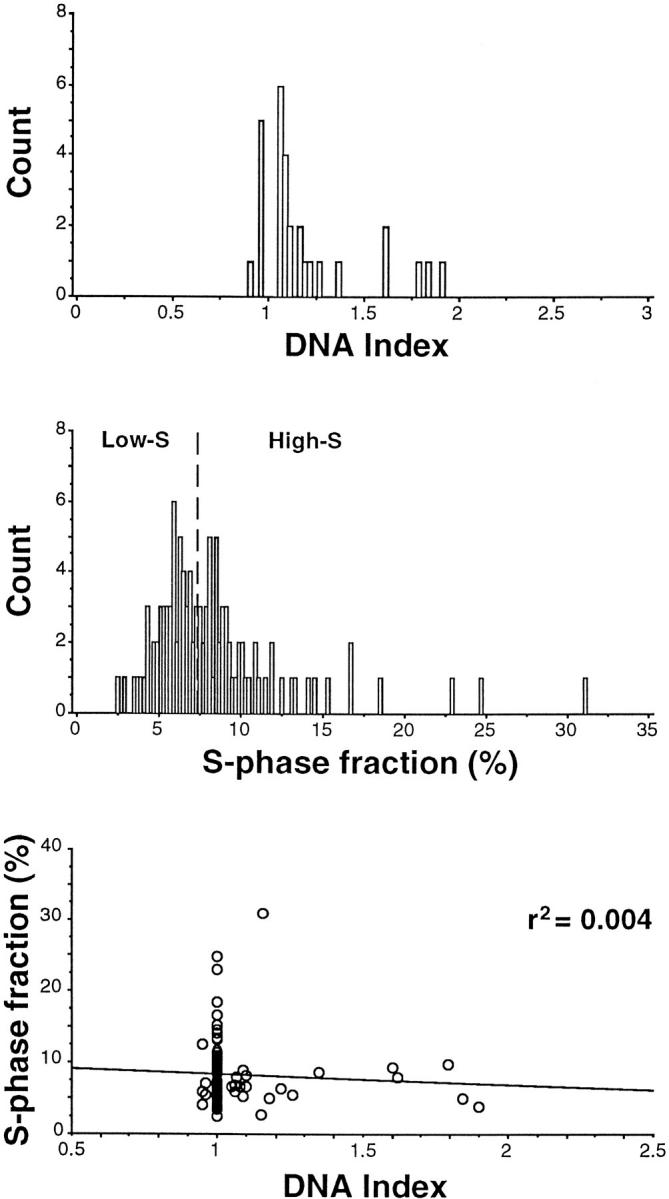
Top: DI aneuploid values (DI ≠ 1) detected by multiparameter FCM. Center: S-phase fraction values, respectively, below and above the median value of 7.3%. S-phase values in DNA diploid cases were corrected by subtracting the inflammatory cell component. Bottom: S-phase values versus all DIs.
K-ras2 analysis was performed using gated sorting of enriched epithelial nuclei (see also Materials and Methods).
Figure 3 ▶ shows an example of K-ras2 spectrum analysis. Human lymphocytes from healthy donors were used as wild-type K-ras2 codon 12 GGT-glycine and codon 13 GGC-glycine controls. Additionally, six cell lines were used as specific K-ras2-mutated controls (see Materials and Methods). K-ras2 mutations in codons 12 and 13 were detected in 29 of 116 adenomas (25%). There were 17 G→A transitions, ie, 11 in codon 12 as GAT (corresponding to aspartate), 1 in codon 13 AGC (serine), and 5 in codon 13 GAC (aspartate). G→C/T transversions were 12, ie, codon 12 CGT in 6 cases (arginine), GTT in 4 (valine), and TGT in 2 (cystein). When considering only codon 12, 65% of the mutations were at position 2 of the base triplet.
Figure 3.

Examples of K-ras2 mutation spectrum analysis. K-ras2 mutation analysis was for wild type in codons 12 and 13 (wild-type codon 12 GGT-gly and codon 13 GGC-gly), six possible mutations in codon 12 (AGT, TGT, CGT, GAT, GCT, and GTT), and two mutations in codon 13 (AGC and GAC). K-ras2 wild-type controls were the following: peripheral blood lymphocytes of healthy donors (g6), SW837 cell line with heterozygous TGT mutation (h2), NIH3T3 cell line with transfected CGT human mutated K-ras2 (h3), SW480 cell line with homozygous GTT mutation (h4), and DLD-1 cell line with heterozygous GAC mutation (h5). All other spots correspond to different wild-type or mutated adenoma samples. Of particular interest are adenomas with TGT mutation in both diploid and aneuploid subclones (e2 and e3) and adenomas with GTT (a6 and b1) and GAT (c2 and c3) mutations in both low-grade and high-grade dysplasia components.
The associations among the variables evaluated in the study were investigated by using the Pearson coefficient of correlation (Table 1) ▶ . A statistically significant correlation at the 0.01 level was detected between DI and K-ras2. S phase and type, age and site, and sex and type were also associated at the 0.05 level.
Table 1.
Pearson Correlation Coefficients
| DI | S-phase | K-ras2 | Size | Dysplasia* | Site | Type | Age | Sex | |
|---|---|---|---|---|---|---|---|---|---|
| DI | 1 | ||||||||
| S-phase | −0.158 | ||||||||
| K-ras2 | 0.264† | −0.114 | 1 | ||||||
| Size | 0.164 | −0.226 | 0.066 | 1 | |||||
| Dysplasia | 0.036 | 0.108 | 0.005 | 0.066 | 1 | ||||
| Site | 0.076 | 0.143 | 0.135 | −0.072 | −0.072 | 1 | |||
| Type | −0.119 | −0.226‡ | 0.103 | 0.144 | −0.025 | −0.010 | 1 | ||
| Age | 0.090 | 0.119 | 0.030 | −0.049 | −0.030 | 0.215‡ | 0.158 | 1 | |
| Sex | 0.112 | 0.120 | 0.043 | −0.079 | 0.006 | 0.029 | −0.223‡ | −0.043 | 1 |
*Categorized in low-moderate versus high. See Materials and Methods for other categories.
†Statistically significant at the 0.01 level (two-tailed).
‡Statistically is significant at the 0.05 level (two-tailed).
Table 2 ▶ shows the associations between K-ras2 G→A transitions and G→C/T transversions versus DNA ploidy (DNA diploidy with DI = 1 and aneuploidy with DI ≠ 1) and proliferation (low and high S-phase values respectively, below and above the 7.3% median) in addition to wild-type and mutated K-ras2. A positive association was found between K-ras2 (wild type, G→C/T transversions) and DNA ploidy (P = 0.003). A negative association was found between K-ras2 (wild type, G→A transitions) and S phase (P = 0.02).
Table 2.
DNA Ploidy, Proliferation, and K-ras2
| K-ras2 classes | DI = 1 | DI = 1* | P | Low-S | High-S† | P |
|---|---|---|---|---|---|---|
| 1. Wild type | 70 (80%) | 17 (20%) | 36 (44%) | 45 (56%) | ||
| 2. Mutated | 17 (59%) | 12 (41%) | P12 = 0.02‡ | 18 (64%) | 10 (36%) | P12 = 0.07 |
| 3. G→A transitions | 12 (70%) | 5 (30%) | 12 (75%) | 4 (25%) | P13 = 0.02 | |
| 4. G→C/T tranversions | 5 (42%) | 7 (58%) | P134 = 0.01 | 6 (50%) | 6 (50%) | P134 = 0.08 |
| P14 = 0.003 |
*DI indicates the degree of DNA aneuploidy (DI = 1 for DNA diploidy) evaluated according to multiparametric flow cytometry. Flow cytometry sorting was used to separate epithelial nuclei to be submitted to K-ras2 analysis (see Materials and Methods).
†High-S indicates the values of the S-phase fraction above the median value of 7.3% evaluated among 109 adenomas. S-phase values were corrected by the subtraction of the tissue-infiltrating inflammatory cells (see Materials and Methods). For DNA aneuploid cases, we have evaluated the S-phase fraction of the aneuploid component that mainly corresponds to the epithelial cell compartment.
‡χ2 test-associated probability.
The associations of K-ras2 (including G→A transitions and G→C/T transversions) with DI and S phase as dependent variables were also investigated by logistic regression analysis to take into consideration the effects of the other covariates investigated, ie, size, dysplasia, site, type, age, and sex (see also Materials and Methods). The results obtained for DI and S phase are shown in Tables 3 and 4 ▶ ▶ , respectively. One may notice that the effect of K-ras2 on DI was dominated by G→C/T transversions with a positive coefficient of correlation β = 1.8 and an odds ratio of 6.1 (ranging from lower and upper 95% confidence limits of 1.57 and 23.7). Increased adenoma size showed an association with DI (odds ratio = 2.7; 95% confidence limits (CL) = 1.04, 6.99).
Table 3.
Associations between DI and Independent Covariates: Odds Ratios Point Estimates (OR) and Their 95% CL Computed by Logistic Regression Analysis
| Covariates | β (SE) | OR | 95% CL | P | |
|---|---|---|---|---|---|
| Lower | Upper | ||||
| K-ras2 (G→A) | 0.6288 (0.6483) | 1.8753 | 0.5263 | 0.6820 | 0.3321 |
| K-ras2 (G→C/T) | 1.8096 (0.6918) | 6.1081 | 1.5742 | 23.7006 | 0.0089 |
| Size | 0.9938 (0.4854) | 2.7014 | 1.0433 | 6.9945 | 0.0406 |
| Dysplasia | 0.1346 (0.5619) | 1.1440 | 0.3804 | 3.4411 | 0.8107 |
| Site (sigma) | 0.3279 (0.5588) | 1.3880 | 0.4642 | 4.1502 | 0.5574 |
| Site (rectum) | 0.2373 (0.7894) | 1.2678 | 0.2698 | 5.9569 | 0.7637 |
| Type | −0.9878 (0.5898) | 0.3724 | 0.1172 | 1.1831 | 0.0940 |
| Age | 0.6079 (0.4904) | 1.8365 | 0.7023 | 4.8024 | 0.2152 |
| Sex | 0.3999 (0.5073) | 1.4916 | 0.5519 | 4.0314 | 0.4305 |
Table 4.
Associations between S-Phase and Independent Covariates: Odds Ratios Point Estimates (OR) and Their 95% CL Computed by Logistic Regressoin Analysis
| Covariates | β(SE) | OR | 95% CL | P | |
|---|---|---|---|---|---|
| Lower | Upper | ||||
| K-ras (G-A) | −1.3299 (0.6841) | 0.2645 | 0.0692 | 1.0109 | 0.0519 |
| K-ras (G-C/T) | −0.3540 (0.6659) | 0.7019 | 0.1903 | 2.5884 | 0.5949 |
| Size | 0.1073 (0.4400) | 1.1133 | 0.4700 | 2.6374 | 0.8073 |
| Dysplasia | 0.7775 (0.5388) | 2.1760 | 0.7569 | 6.2554 | 0.1490 |
| Site (sigma) | 0.8045 (0.4741) | 2.2355 | 0.8828 | 5.6609 | 0.0897 |
| Site (rectum) | 0.8968 (0.7161) | 2.4516 | 0.6025 | 9.9763 | 0.2104 |
| Type | −1.0170 (0.4822) | 0.3617 | 0.1405 | 0.9307 | 0.0349 |
| Age | 0.6733 (0.4383) | 1.9606 | 0.8304 | 4.6290 | 0.1245 |
| Sex | 0.3270 (0.4374) | 1.3868 | 0.5885 | 3.2682 | 0.4546 |
S-phase was, instead, found to be inversely associated with G→A transitions and tubular type. The odds ratios were, respectively, 0.26 (95% CL = 0.07, 1.01) and 0.36 (95% CL = 0.14, 0.93).
Discussion
Functions of the Ras mutated proteins associated with chromosome instability and proliferation changes have been suggested using in vitro systems, 4-6,14,15 as well as human colorectal adenomas. 22 In the present study, we extended these last observations. Adenomas investigated were overall 116 and were characterized for the majority by low-moderate dysplasia (78%) with a relatively small size (50% with size ≤1.5 cm) and absence of early cancer. FAP patients as well as patients with multiple polyps (suspected attenuated FAP) were excluded.
A method of enrichment of dysplastic epithelial adenoma regions was applied by means of cryocutting, microdissection, and morphological guided criteria using fresh-frozen material (see details in Materials and Methods). In addition, enrichment of epithelial cells was obtained by multiparameter DNA FCM. In particular, the use of nuclear suspensions and two scattering signals (forward and perpendicular to the incident laser beam associated, respectively, with area and internal structure of nuclear chromatin) made it possible to separate the infiltrating inflammatory cell component. This procedure was useful for improving detection of DNA aneuploidy in the near-diploid region and for correcting the S-phase fraction values in about two-thirds of the adenomas characterized by only DNA diploid cells. Moreover, FCM-based sorting of an enriched epithelial cell component (up to almost 100% for the DNA aneuploid subclones) was used for evaluating the K-ras2 mutation spectrum.
In the present study, we found that K-ras2 G→C/T transversions were strongly associated with DNA near-diploid aneuploidy. DNA aneuploidy was found, in fact, in 58% of the cases with K-ras2 G→C/T transversions with respect to 20% with K-ras2 wild type, whereas DNA diploidy was associated with wild-type K-ras2 in 80% of the cases (P = 0.01). This association was confirmed by logistic regression analysis taking into account the effects of the other variables investigated in the study. Only increased adenoma size was found to exert an additional influence.
This finding, which appears in agreement with other literature data and hypotheses, 14,15,22,27 suggests that G→C/T transversions disrupt chromosome stability. Although the exact mechanisms are not known, they may be related to mitotic checkpoints that maintain chromosome stability. 39 Although it clearly remains to be proven, we favor the hypothesis that a defect in chromosome segregation is linked with K-ras2 G→C/T transversions. The alternative interpretation is that we are in presence of a carcinogenic process that has produced concomitantly both K-ras2 G→C/T transversions and DNA aneuploidy.
The first hypothesis appears to be in agreement with K-ras2 oncogene transfection experiments using NIH3T3 cells, in which an increased rate of abnormal mitoses was correlated with a high expression of the mutated p21ras protein 14 and, in particular, with the codon 12 G→C mutation. 15 This specific mutation was also associated with generation of DNA aneuploid subclones. 15 That abnormal mitoses are massively present in human colorectal adenomas 40 and that there is a relatively high incidence of K-ras2 transversions in these lesions 19-23 also constitute indirect evidence of a link of the two variables.
We have previously postulated that K-ras2-activated proteins may participate in a mechanism of “loss of symmetry” in chromosome segregation during cell division 22,26,27,41 and that it appears important to understand those regulatory events that integrate chromosome motor activity into the signal transduction cascades of the cell cycle. 42 Perhaps ras oncogene functions that regulate the formation of stress fibers, focal cell adhesion, and cytokinesis 12,13 might also play a role in disrupting the symmetry of cell division.
The concept of “loss of symmetry” in cell division was also associated with recent results that indicated that tumor colorectal cell lines without instability at the nucleotide level 43 were characterized by chromosome gains or losses in excess of 10−2 per chromosome per generation. 44
The second main finding of the present study was that K-ras2 mutations and, in particular, G→A transitions correlated with decreased proliferation. Low S-phase (with values less than/equal to the median value of 7.3% as evaluated for the whole population) among G→A-mutated adenomas were found to represent 75% of the cases, compared with 44% in the K-ras2 wild-type group (P = 0.02). Logistic regression has confirmed this association together with an influence of the tubular type. This finding reinforces our previous observation from a study of only 54 cases. 22
K-ras2 activation (up to 85%) was reported in human aberrant crypt foci of the colon that may be considered in some cases to be early precursors of adenomas. 28-30 Moreover, K-ras2 mutation incidence in human sporadic colorectal adenomas (up to 60%) was reported to be higher than in adenocarcinomas. 1,2,20 Thus, K-ras2 mutations appear on one hand to be an initiating event of the sporadic colorectal aberrant crypt foci-adenoma-carcinoma sequence and on the other hand, for the specific G→A transitions, to be linked with a mechanism of regression.
So far, the regulatory events that integrate exit from G1 and entry into the S phase of the cell cycle via the Ras signal transduction cascades are not well understood. Recently, Ras proteins were reported to link growth factor signaling to cell cycle machinery via regulation of cyclin D1 and cyclin-dependent kinases. 4-6 These pathways might represent possible mechanisms to interpret the association between G→A transitions and decreased proliferation. On the other hand, because S-phase fraction and DNA aneuploidy were not correlated in the present series of cases, a mechanism of inhibition of proliferation caused by a new abnormal chromosomal setup was excluded.
We suggest that a specific carcinogenic process in the colon of human patients might favor specific mutations of the K-ras2 oncogene in precursor lesions that simultaneously may interfere with the cell cycle machinery and inhibit proliferation.
Overall, we suggest that specific K-ras2 mutations, as an early genetic event of the colorectal aberrant crypt foci-adenoma-carcinoma sequence, may potentially be responsible for aneuploidy and proliferation changes. In addition, decrease or inhibition of apoptosis caused by the ras oncogene clearly may play a concomitant important role. 7-11
K-ras2 mutations are not, however, the only genetic event potentially involved. A possible role of 1p deletions in association with chromosome instability in colorectal adenomas has been recently suggested using interphase cytogenetics. 45,46 A more complex situation, in which other new tumor suppressor genes might be involved, was recently suggested by data obtained by comparative genomic hybridization in colorectal adenomas. 47,48
DNA aneuploidy in colorectal adenomas obtained by various techniques including DNA FCM, classical cytogenetics, and interphase cytogenetics was found to be approximately as frequent as or higher than in the present series. 24-27,49-52 On the other hand, it is known that the incidence of DNA aneuploidy for colorectal adenocarcinomas is about three times higher than in adenomas. 53 Thus, it appears likely that later genetic alterations of the colorectal tumorigenesis, like the p53 tumor oncosuppressor gene, may be associated with (or may possibly cause) aneuploidy and that complex relationships govern the interaction of aneuploidy, proliferation, and apoptosis.
In other human tumor model systems of tumorigenesis, specifically the Barrett’s esophagus, p53 mutations were reported to represent an early event and were associated with the generation of aneuploidy. 54-56 The role of p53 in causing aneuploidy was shown, in particular, with cultured fibroblasts from p53-deficient mouse embryos. These experiments have shown, in fact, the formation of tetraploid and octaploid cells, as predicted by a model of aneuploidization, 57 suggesting that murine p53 inactivation is a component of a spindle G2 checkpoint that ensures the maintenance of diploidy. 58,59
The fact that we have not observed DNA tetraploidy in the present series of adenomas but near-diploid aneuploidy in the vast majority of cases (83%) suggests different types and mechanisms of aneuploidization. In this respect, we also note that in the present series of colorectal adenomas only a minority of cases is expected to be p53 mutated, according to literature data, because the majority of adenomas were characterized by low-moderate dysplasia. 19,20,60 Because DNA aneuploidy incidence was about 70% in adenomas with early cancer 26,27 and up to 90% in adenocarcinomas, 53,41 it appears that aneuploidy may be associated with p53 mutations in these late lesions and that tetraploidization of near-diploid aneuploid subclones may be a possible mechanism. 27
Specific mutations of the ras oncogene are known to induce a change of the three-dimensional structure of the p21ras protein and an alteration of its functioning in signal transduction. 2,61-63 The exogenous and endogenous factors that cause ras mutations have been investigated. Fidelity in DNA replication and repair presents endogenous factors that may be responsible for G→A transitions and G→T transversions. 1,64 Literature data, in addition, support the hypothesis that G→A transitions and G→C transversions are due to alkylating agents 2,65-67 and that G→T transversions are ascribed to the presence of polycyclic aromatic hydrocarbons and heterocyclic amines. 68-70
To our knowledge, the correlation of aneuploidy and proliferation during the human colorectal adenoma-carcinoma sequence with mutations of the APC gene and of the DNA mismatch repair genes 71,72 has not yet been investigated. DNA mismatch repair gene mutations, known to be infrequent in the sporadic colorectal adenomas, were recently shown to induce instability at DNA microsatellite levels in hereditary nonpolyposis colorectal cancer. 71 Interestingly, DNA microsatellite instability in these lesions is not accompanied by instability at chromosome level. 71,73
In conclusion, the present study has demonstrated that human sporadic colorectal adenomas without early cancer are characterized by a strong correlation between specific K-ras2 mutations, DNA near-diploid aneuploidy, and proliferation inhibition. Further work remains to be done with use of cell and animal model systems to understand cause and effect relationships and the relative importance of these changes for the possible progression and/or regression of these common human preneoplastic lesions. It is likely that this knowledge in conjunction with specific chemopreventive treatments, which have already been attempted in FAP patients, 74 may be useful to reduce the incidence of advanced colorectal cancer.
Acknowledgments
We thank N. Pujic for scientific and technical collaboration. The technical assistance of E. Infusini and R. Orecchia is also kindly acknowledged.
Footnotes
Address reprint requests to Dr. Walter Giaretti, Laboratory of Biophysics-Cytometry, National Cancer Institute, Viale R. Benzi, no. 10, 16132 Genoa, Italy. E-mail: giaretti@hp380.ist.unige.it.
This study was financially supported by A.I.R.C. (Associazione Italiana per la Ricerca sul Cancro), Ministero della Sanità and Fondazione Novello.
References
- 1.Bos JL, Fearon EL, Hamilton SR, Verlaan de Vries M, van Boom JH, Van der Eb AJ, Vogelstein B: Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327:293-297 [DOI] [PubMed] [Google Scholar]
- 2.Barbacid M: ras genes. Annu Rev Biochem 1987, 56:779-827 [DOI] [PubMed] [Google Scholar]
- 3.Bollag G, McCormick F: Regulators and effectors of ras proteins. Annu Rev Cell Biol 1991, 7:601-632 [DOI] [PubMed] [Google Scholar]
- 4.Aktas H, Cai H, Cooper GM: Ras links growth factor signaling to cell cycle machinery via regulation of cyclin D1 and the CdK inhibitor p27 KIP1. Mol Cell Biol 1997, 17:3850-3857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arber N, Hibshoosh H, Moss SF, Sutter T, Zhang Y, Begg M, Wang S, Weinstein B, Holt PR: Increased expression of cyclin D1 is an early event in multistage colorectal carcinogenesis. Gastroenterology 1996, 110:669-674 [DOI] [PubMed] [Google Scholar]
- 6.Serrano M, Gomez-Lahoz E, DePinho RA, Beach D, Bar-Sagi D: Inhibition of Ras-induced proliferation and cellular transformation by p16INK4. Science 1995, 267:249-252 [DOI] [PubMed] [Google Scholar]
- 7.Khosravi-Far R, Campbell S, Rossman KL, Der CJ: Increasing complexity of Ras signal transduction: involvement of Rho family proteins. Adv Cancer Res 1998, 72:57-107 [DOI] [PubMed] [Google Scholar]
- 8.Bedi A, Pasricha PJ, Akhtar AJ, Barber JP, Bedi GC, Giardiello FM, Zehnbauer BA, Hamilton SR, Jones RJ: Inhibition of apoptosis during development of colorectal cancer. Cancer Res 1995, 55:1811-1816 [PubMed] [Google Scholar]
- 9.Jimenez B, Arends M, Esteve P, Perona R, Sanchez R, Ramon y Cajal S, Wyllie A, Lacal JC: Induction of apoptosis in NIH3T3 cells after serum deprivation by overexpression of rho-p21, a GTPase protein of the ras superfamily. Oncogene 1995, 10:811-816 [PubMed] [Google Scholar]
- 10.Ward RL, Todd AV, Santiago F, O’Connor T, Hawkins N: Activation of the K-ras oncogene in colorectal neoplasms is associated with decreased apoptosis. Cancer 1997, 79:1106-1113 [PubMed] [Google Scholar]
- 11.Chen CY, Faller DV: Direction of p21ras-generated signals towards cell growth or apoptosis is determined by protein kinase C and Bcl-2. Oncogene 1995, 11:1487-1498 [PubMed] [Google Scholar]
- 12.Hall A: Small GTP-binding proteins, and the regulation of the actin cytoskeleton. Annu Rev Cell Biol 1994, 10:31-54 [DOI] [PubMed] [Google Scholar]
- 13.Nobes CD, Hall A: Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81:53-62 [DOI] [PubMed] [Google Scholar]
- 14.Hagag N, Diamond L, Palermo R, Lyubsky S: High expression of ras p21 correlates with increased rate of abnormal mitosis in NIH3T3 cells. Oncogene 1990, 5:1481-1489 [PubMed] [Google Scholar]
- 15.Nigro S, Geido E, Infusini E, Orecchia R, Giaretti W: Transfection of human mutated K-ras in mouse NIH-3T3 cells is associated with increased cloning efficiency and DNA aneuploidization. Int J Cancer 1996, 67:871-875 [DOI] [PubMed] [Google Scholar]
- 16.Mello MLS, Lin TY, Russo J: Scanning microphotometry image analysis of Ha-ras-transformed human breast epithelial cells. Anal Cell Pathol 1994, 7:301-319 [PubMed] [Google Scholar]
- 17.Aldaz CM, Conti CJ, Klein-Szanto AJP, Slaga TJ: Progressive dysplasia and aneuploidy are hallmarks of mouse skin papillomas: relevance to malignancy. Proc Natl Acad Sci USA 1987, 84:2029-2032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Quintanilla M, Brown K, Ramsden M, Balmain A: Carcinogen specific mutation and amplification of Ha-ras during mouse skin carcinogenesis. Nature 1986, 322:78-80 [DOI] [PubMed] [Google Scholar]
- 19.Fearon ER, Vogelstein B: A genetic model for colorectal tumorigenesis. Cell 1990, 61:759-767 [DOI] [PubMed] [Google Scholar]
- 20.Vogelstein B, Fearon ER, Hamilton SR, Kern, Preisinger AC, Leppert BAM, Nakamura Y, White R, Smits AMM, Bos JL: Genetic alterations during colorectal-tumor development. New Engl J Med 1988, 319:525–532 [DOI] [PubMed]
- 21.Hasegawa H, Ueda M, Watanabe M, Teramoto T, Mukai M, Kitajima M: K-ras gene mutations in early colorectal cancer: flat elevated vs. polyp-forming cancer. Oncogene 1995, 10:1413-1416 [PubMed] [Google Scholar]
- 22.Giaretti W, Pujic N, Rapallo A, Nigro S, Di Vinci A, Geido E, Risio M: K-ras2 G-C, and G-T transversions correlate with DNA aneuploidy in colorectal adenomas. Gastroenterology 1995, 108:1040-1047 [DOI] [PubMed] [Google Scholar]
- 23.Burmer GC, Loeb LA: Mutations in the K-ras2 oncogene during progressive stages of human colon carcinoma. Proc Natl Acad Sci USA 1989, 86:2403-2407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van den Ingh HF, Griffioen G, Cornelisse CJ: Flow cytometric detection of aneuploidy in colorectal adenomas. Cancer Res 1985, 45:3392-3397 [PubMed] [Google Scholar]
- 25.Giaretti W, Sciallero S, Bruno S, Geido E, Aste H, Di Vinci A: DNA flow cytometry of endoscopically examined colorectal adenomas and adenocarcinomas. Cytometry 1988, 9:238-244 [DOI] [PubMed] [Google Scholar]
- 26.Giaretti W, Santi L: Tumor progression by DNA flow cytometry in human colorectal cancer. Int J Cancer 1990, 45:597-603 [DOI] [PubMed] [Google Scholar]
- 27.Giaretti W: A model of DNA aneuploidization and evolution in colorectal cancer. Lab Invest 1994, 71:904-910 [PubMed] [Google Scholar]
- 28.Yamashita N, Minamoto T, Ochiai A, Onda M, Esumi H: Frequent and characteristic K-ras activation and absence of p53 protein accumulation in aberrant crypt foci of the colon. Gastroenterology 1995, 108:434-440 [DOI] [PubMed] [Google Scholar]
- 29.Otori K, Sugijama K, Hasebe T, Fukushima S, Esumi H: Emergence of adenomatous aberrant crypt foci (ACF) from hyperplastic ACF with concomitant increase in cell proliferation. Cancer Res 1995, 55:4743-4746 [PubMed] [Google Scholar]
- 30.Pretlow PT: Aberrant crypt foci and K-ras mutations: early recognized players or innocent bystanders in colorectal carcinogenesis? Gastroenterology 1995, 108:600-603 [DOI] [PubMed] [Google Scholar]
- 31.Jass JR, Sobin LH: Histological typing of intestinal tumors. WHO International Histological Classification of Tumors. Berlin, Springer-Verlag, 1989
- 32.Santos E, Martin-Zanca D, Reddy EP, Pierotti MA, Della Porta G, Barbacid M: Malignant activation of a K-ras oncogene in lung carcinoma but not in normal tissue of the same patient. Science 1984, 223:661-664 [DOI] [PubMed] [Google Scholar]
- 33.Verlaan-de-Vries M, Bogaard ME, van del Elst H, van Boom JH, van der Eb AJ, Bos JL: A dot-blot screening procedure for mutated ras oncogenes using synthetic oligodeoxynucleotides. Gene 1986, 50:313-320 [DOI] [PubMed] [Google Scholar]
- 34.Current Protocols in Molecular Biology. New York, John Wiley & Sons, 1988
- 35.Burmer GC, Rabinovitch PS, Haggit RC, Crispin DA, Brentnall TA, Kolli VR, Stevens AC, Rubin CE: Neoplastic progression in ulcerative colitis: histology, DNA content, and loss of a p53 allele. Gastroenterology 1992, 103:1602-1610 [DOI] [PubMed] [Google Scholar]
- 36.Fleiss JL: Statistical Methods for Rates and Proportions, ed 2. New York, John Wiley & Sons, 1981
- 37.Kleinbaum DG: Logistic Regression. 1994. Springer-Verlag, New York
- 38.SPSS Professional Statistics, version 7.5. Chicago, SPSS Inc., 1997
- 39.Hartwell L, Kastan MB: Cell cycle control and cancer. Science 1994, 266:1821-1828 [DOI] [PubMed] [Google Scholar]
- 40.Rubio CA: Atypical mitosis in colorectal adenomas. Pathol Res Pract 1991, 187:508-513 [DOI] [PubMed] [Google Scholar]
- 41.Giaretti W, Monaco R, Pujic N, Rapallo A, Nigro S, Geido E: Intratumor heterogeneity of K-ras2 mutations in colorectal adenocarcinomas. Am J Pathol 1996, 149:237-245 [PMC free article] [PubMed] [Google Scholar]
- 42.Barton NR, Goldstein LSB: Going mobile: microtubule motors and chromosome segregation. Proc Natl Acad Sci USA 1996, 93:1735-1742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marra G, Boland CR: Hereditary nonpolyposis colorectal cancer: the syndrome, the genes, and historical perspectives. J Natl Cancer Inst 1995, 87:114-1125 [DOI] [PubMed] [Google Scholar]
- 44.Lengauer C, Kinzler KW, Vogelstein B: Genetic instability in colorectal cancer. Nature 1992, 386:623-626 [DOI] [PubMed] [Google Scholar]
- 45.Di Vinci A, Infusini E, Peveri C, Risio M, Rossini FP, Giaretti W: Deletions at chromosome 1p by fluorescence in situ hybridization are an early event in human colorectal tumorigenesis. Gastroenterology 1996, 111:102-107 [DOI] [PubMed] [Google Scholar]
- 46.Di Vinci A, Infusini E, Peveri C, Sciutto A, Geido E, Risio M, Rossini FP, Giaretti W: Correlation between 1p deletions and aneusomy in human colorectal adenomas. Int J Cancer 1998, 75:45-50 [DOI] [PubMed] [Google Scholar]
- 47.Ried T, Knutzen R, Steinbeck R, Blegen H, Schrock E, Heselmeyer K, du Manoir S, Auer G: Comparative genomic hybridization reveals a specific pattern of chromosomal gains and losses during the genesis of colorectal tumors. Genes Chromosomes Cancer 1996, 15:1-5 [DOI] [PubMed] [Google Scholar]
- 48.Houldsworth J, Chaganti RSK: Comparative genomic hybridization: an overview. Am J Pathol 1994, 145:1253-1260 [PMC free article] [PubMed] [Google Scholar]
- 49.Bardi G, Pandis N, Fenger C, Kronborg O, Bomme L, Heim S: Deletion of 1p36 as a primary chromosomal aberration in intestinal tumorigenesis. Cancer Res 1993, 53:1895-1898 [PubMed] [Google Scholar]
- 50.Bomme L, Bardi G, Pandis N, Fenger C, Kronborg O, Heim S: Clonal karyotypic abnormalities in colorectal adenomas: clues to the early genetic events in the adenoma-carcinoma sequence. Genes Chromosomes Cancer 1994, 10:190-196 [DOI] [PubMed] [Google Scholar]
- 51.Glenny RW, Robertson HT: Fractal modeling of pulmonary blood flow heterogeneity. J Appl Physiol 1991, 70:1024-1030 [DOI] [PubMed] [Google Scholar]
- 52.Hammarberg C, Tribukait B, Rubio C, Slezak P: Application clinique de la citométrie de flux de l’ADN à la caractérisation des polypes colorectaux. Acta Endoscopica 1985, 15:265-274 [Google Scholar]
- 53.Bauer KD, Bagwell CB, Giaretti W, Melamed M, Zarbo RJ, Witzig TE, Rabinovitch PS: Consensus review of the clinical utility of DNA flow cytometry in colorectal cancer. Cytometry 1993, 14:486-491 [DOI] [PubMed] [Google Scholar]
- 54.Rabinovitch PS, Reid BJ, Haggit RC, Norwood TH, Rubin CE: Progression to cancer in Barrett’s esophagus is associated with genomic instability. Lab Invest 1988, 60:65-71 [PubMed] [Google Scholar]
- 55.Reid BJ, Sanchez CA, Blount PL, Levine DS: Barrett’s esophagus: cell cycle abnormalities in advancing stages of neoplastic progression. Gastroenterology 1993, 105:119-129 [DOI] [PubMed] [Google Scholar]
- 56.Giaretti W: Aneuploidy mechanisms in human colorectal preneoplastic lesions and Barrett’s esophagus: is there a role for K-ras and p53 mutations? Anal Cell Pathol 1997, 15:99-117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shackney SE, Smith CA, Miller BW, Burholt DR, Murtha K, Giles HR, Ketter DM, Pollice AA: Model for the genetic evolution of human solid tumors. Cancer Res 1989, 49:3344-3354 [PubMed] [Google Scholar]
- 58.Cross SM, Sanchez CA, Morgan CA, Schimke MK, Ramel S, Idzerda RL, Raskind WH, Reid BJ: A p53-dependent mouse spindle checkpoint. Science 1995, 267:1353-1356 [DOI] [PubMed] [Google Scholar]
- 59.Fukasawa K, Taesaeng C, Kuriyama R, Rulong S, Vande Woude GF: Abnormal centrosome amplification in the absence of p53. Science 1996, 271:1744-1747 [DOI] [PubMed] [Google Scholar]
- 60.Baker SJ, Preisinger AC, Jessup JM, Paraskeva C, Markowitz S, Willson JK, Hamilton S, Vogelstein B: p53 gene mutations occur in combination with 17p allelic deletions as late events in colorectal tumorigenesis. Cancer Res 1990, 50:7717-7722 [PubMed] [Google Scholar]
- 61.McCormick F: ras GTPase activating protein: signal transmitter and signal terminator. Cell 1989, 56:5-8 [DOI] [PubMed] [Google Scholar]
- 62.Krengel U, Schlichting I, Scherer A, Schumann R, Frech M, John J, Kabsch W, Pai EF, Wittinghofer A: Three-dimensional structures of H-ras p21 mutants: molecular basis for their inability to function as signal switch molecules. Cell 1990, 62:539-548 [DOI] [PubMed] [Google Scholar]
- 63.Bos JL: p21ras: an oncoprotein functioning in growth factor-induced signal transduction. Eur J Cancer 1995, 31A:1051-1054 [DOI] [PubMed] [Google Scholar]
- 64.Loeb LA: Endogenous carcinogenesis: molecular oncology into the twenty-first century (presidential address). Cancer Res 1989, 49:5489-5496 [PubMed] [Google Scholar]
- 65.Bos JL: Ras oncogenes in human cancer: a review. Cancer Res 1989, 49:4682-4689 [PubMed] [Google Scholar]
- 66.Topal MD: DNA repair, oncogenes, and carcinogenesis. Carcinogenesis 1988, 9:691-696 [DOI] [PubMed] [Google Scholar]
- 67.Capella G, Cronauer-Mitra S, Peinado MA, Perucho M: Frequency and spectrum of mutations at codons 12 and 13 of the c-K-ras gene in human tumors. Environ Health Perspect 1991, 93:125-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vousden KH, Bos JL, Marshall CJ, Phillips DH: Mutations activating human c-Ha-ras1 proto-oncogene (Hras1) induced by chemical carcinogens and depurination. Proc Natl Acad Sci USA 1986, 83:1222-1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brown K, Buchmann A, Balmain A: Carcinogen-induced mutations in the mouse c-Ha-ras gene provide evidence of multiple pathways for tumor progression. Proc Natl Acad Sci USA 1990, 87:538-542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Urosevic N, Krtolica K, Skaro-Milic A, Knezevic-Usaj S, Dujic A: Prevalence of G-to-T transversions among K-ras oncogene mutations in human colorectal tumors in Yugoslavia. Int J Cancer 1993, 54:249-254 [DOI] [PubMed] [Google Scholar]
- 71.Kinzler WK, Vogelstein B: Lessons from hereditary colorectal cancer. Cell 1996, 87:159-170 [DOI] [PubMed] [Google Scholar]
- 72.Tsao JI, Shibata D: Further evidence that one of the earliest alterations in colorectal carcinogenesis involves APC. Am J Pathol 1994, 145:531-534 [PMC free article] [PubMed] [Google Scholar]
- 73.Kouri M, Laasonen A, Mecklin JP, Järvinen H, Franssila K, Pyrhönen S: Diploid predominance in hereditary nonpolyposis colorectal carcinoma evaluated by flow cytometry. Cancer 1990, 65:1825-1829 [DOI] [PubMed] [Google Scholar]
- 74.DuBois RN: Nonsteroidal anti-inflammatory drug use and sporadic colorectal adenomas. Gastroenterology 1995, 108:1310-1314 [DOI] [PubMed] [Google Scholar]