

Abstract
We have previously demonstrated that vascular endothelial growth factor-165 (VEGF), a tumor-secreted angiogenic factor, can acutely and chronically induce fenestrations in microvascular endothelium (Cancer Res 1997, 57:765–772). Because the morphology and function of microvascular endothelium differs from tissue to tissue, we undertook studies to examine whether the neovasculature in tumors also differed depending upon tumor location. Four tumor types implanted in the brain or subcutis in nude mice were studied: a murine rhabdomyosarcoma (M1S), a murine mammary carcinoma (EMT), and two human glioblastomas (U87 and U251). In addition, we studied Chinese hamster ovary cells stably transfected with human VEGF165. As previously reported, tumors grown in the subcutaneous space had a microvasculature that was fenestrated and had open endothelial gaps. The identical tumors when grown in the brain also had fenestrated endothelium and vessels with open endothelial gaps, but they were drastically reduced in occurrence. Open endothelial gaps were not seen in all tumors implanted in the brain (EMT and M1S), although fenestrated endothelium was always seen. VEGF and VEGF receptors were measured in tumors from both locations by immunoblotting and competitive polymerase chain reaction, respectively. VEGF amount was not significantly different between the tumor locations. Interestingly, total tumor vascular mRNA expression of both Flk-1 and Flt-1 was greater in tumor vessels derived from the brain compared with tumor vessels derived from subcutaneous tissues. These results demonstrate that the host microvascular environment determines the morphology and function of the tumor vasculature and that endothelia from different tissues vary in their ability to express the VEGF receptors given identical stimuli.
The growth of primary tumors (tumor nodules) beyond the 2-mm limit of nutrient diffusion is strictly dependent on their ability to induce vascularization by angiogenesis from the surrounding host tissue vasculature.1 In addition to tumor growth, angiogenesis is also a prerequisite for tumor spreading by metastasis.2,3 Angiogenesis in tumors, as well as in a number of physiological (vasculogenesis, wound repair, etc.) and pathological (diabetic retinopathy, macular degeneration, rheumatoid arthritis, and psoriasis) conditions, is induced by a number of factors of which the most prevalent and only endothelium-specific one is vascular endothelial growth factor (VEGF, also referred to as vascular permeability factor).4,5
In addition to its role as an angiogenic agent, VEGF also increases vascular permeability by inducing the formation of fenestrations; open gaps; and clustered, fused caveolae in endothelium.6,7 There are two identified receptors specific for VEGF that are localized on endothelium, Flk-1 and Flt-1,8,9 that bind with different affinities10 and possibly mediate different functions.4 The VEGF receptors (VEGF-Rs) are induced on proliferating neovasculature and present on fenestrated endothelium. They are either nondetectable or expressed at extremely low levels in nonproliferating, nonfenestrated endothelium.11
Because of the dependence of tumor growth on angiogenesis, the concept of targeting tumor neovasculature or preventing angiogenesis has been increasingly considered.12,13 It has been repeatedly demonstrated that by targeting the vessels directly or by inhibiting angiogenesis, tumors can be reduced and even eradicated.14-16 However, not much is known about whether or how various host tissues influence the morphology of the tumor neovascular endothelium. The microvasculature differs dramatically in morphology and function from organ to organ, eg, body wall (muscle or skin) versus central nervous system (brain). These differences provide a unique condition in which the ability of tumor-secreted factors (eg, VEGF) to modify “borrowed” host vessels can be studied. Although some relevant information exists,17,18 a more extensive comparative study could prove informative.
In principle, therefore, tumor vasculature is expected to reflect the interaction of two main (but not necessarily only) factors: 1) the type of vasculature of the host tissue and 2) the morphogenetic effects of VEGF. To assess their relative importance, we have used a set of four different tumors (M1S, EMT, U87, and U251) and Chinese hamster ovary (CHO) cells transfected with VEGF165 implanted either subcutaneously (s.c.) or in the brain (intracerebrally, i.c.), have counted on their ability to secrete VEGF, and have systematically examined the morphology of their microvasculature. In the s.c. condition, the host tissue microvasculature is provided with a continuous type of endothelium characterized by a large population of caveolae and relatively high permeability. In the i.c. condition, the microvascular endothelium has few (or no) caveolae and low permeability, thereby creating a blood-brain barrier.
In our previous work, we have studied the microvasculature of the tumors implanted s.c. and have shown that the endothelium becomes extensively fenestrated and develops numerous gaps (intercellular).19 Now we show that the same tumors implanted i.c. acquire a microvasculature that is considerably less fenestrated and has fewer (or no) gaps than in the s.c. condition. We also show that the tumors produce similar amounts of VEGF in the two conditions, but the expression of VEGF-R(s) is increased in the i.c. condition.
Materials and Methods
Tumor Growth
The tumors used were: EMT-6, experimental mammary tumor syngeneic for BALB/c mice; M1S, rhabdomyosarcoma syngeneic for DBA/2 mice; and U87 and U251, human glioblastomas. In addition, we used CHO cells transfected with cDNA encoding human VEGF165. All tumors were grown in male nude mice. Tumors were maintained by in vivo passaging and prepared as previously described.19 For tumor cell injection into the brain, 5e5 tumor cells in 5 μl of phosphate-buffered saline (PBS) were inoculated into the corpus striatum in the right hemisphere (see Ref. 20 ). s.c. tumors were removed when they reached 5 to 9 mm in diameter, and brain-implanted tumors were removed 10 to 15 days after injection (corresponding to tumors 5 to 8 mm in diameter). Animals were used in accordance with National Institutes of Health standards and an independent institutional animal care committee.
Electron Microscopy
Animals were sacrificed, the brain was exposed, and a 3% formaldehyde (freshly prepared from paraformaldehyde) solution was droppered on the surface. Brains were removed and sliced under formaldehyde fixative. Smaller pieces (1 mm3) of tumor tissue were then fixed in 1.5% glutaraldehyde and processed as previously described for electron microscopy.7 Entire preparation time from sacrifice until fixation in glutaraldehyde was less than 2 minutes for each animal. Thin (50 to 55-nm) sections were cut (Reichert-Jung Ultracut E; Vienna, Austria), picked up on copper grids, and stained with uranyl acetate and lead citrate before examination and photography (Philips CM10 electron microscope at 80 kV). Morphometric data were compiled from at least three tissue blocks from no fewer than three tumors of each type.
VEGF Immunoblotting
Tumors (s.c. or i.c.) were removed and immediately frozen on dry ice. Lysates were made by homogenizing 100 mg of frozen tumor tissue/ml lysis buffer (10 mmol/L Tris-HCl, pH 7.5, supplemented with 1% Nonidet P-40, 1 mmol/L phenylmethylsulfonylfluoride, 1 mmol/L leupeptin, and 0.2 trypsin-inhibiting units/ml aprotinin). The homogenate was passed three times through a 25-gauge needle and then centrifuged at 1000 rpm for 10 minutes. Proteins in the clarified lysate (10 to 25 μg) were resolved on a 10% polyacrylamide gel under nonreducing conditions, transferred to nitrocellulose and immunoblotted with polyclonal anti-VEGF (Santa Cruz Laboratories, Santa Cruz, CA). Blotto (5% nonfat dried milk in 0.1% Tween/PBS) was used for blocking, and 1% milk in Tween-PBS was used for all washes. Primary antibodies were incubated with the transferred electrophoretograms overnight at 4°C and positive bands were detected by enhanced chemiluminescence (Pierce, Rockford, IL) after a 1-hour incubation at room temperature with horseradish peroxidase-conjugated goat anti-rabbit antibodies (Biodesign International, Kennebunk, ME). These anti-VEGF antibodies recognize both murine and human VEGF.
Generation of Templates for VEGF-R Competitive Polymerase Chain Reaction
Flk-1 and Flt-1 templates were reverse transcription-polymerase chain reaction (RT-PCR) cloned from murine fetal total RNA isolated from murine BALB/c fetus using the Trizol reagent (Life Technologies, Inc., Gaithersburg, MD). RNA (5 μg) was reverse transcribed using Superscript II and random primers according to the manufacturer’s instruction (Life Technologies). Templates for the N-terminal region of murine Flk-1 (corresponding to amino acids 56 to 169) and murine Flt-1 (corresponding to amino acids 186 to 327) were PCR cloned using the following primers: Flk-1 (upper), 5′-GACCTGGACTGGCTTTGG-3′, and (lower), 5′-TCTCTTTTCTGGATACCT-3′; Flt-1 (upper), 5′-ACATGGGACAGTAGGAGA-3′, and (lower), 5′-ACGGAGGTGTTGAAAGAC-3′. Primers were designed using Oligo 4.0 (National Biosciences, Plymouth, MN) using sequences in GenBank. The following PCR reaction was used: 1 μl of reverse transcriptase-cDNA, 0.4 μmol/L upper and lower primers, 5 μl Pfu PCR buffer, 2.5 μl of deoxynucleotide triphosphates (2.5 mmol/L each), 38 μl of H2O, and 1 μl of Pfu polymerase (Stratagene, La Jolla, CA) using the following method (95°C for 5 minutes, 95°C for 30 seconds, 58°C for 1 minute, and 72°C for 2 minutes for 30 cycles, followed by 72°C for 10 minutes). Clones were subcloned into the pET28 expression vector (Novagen, Madison, WI) and verified by sequencing.
Competitive templates for Flk-1 and Flt-1 were generated by using AatII and AflII restriction enzymes, respectively, which cut once in the insert. Briefly, 5 μg of pET28:Flk-1 was cut with AatII and blunted with T4 DNA polymerase, whereas pET28:Flt-1 was blunted with Kleenow after restriction digestion with AflII. A 100-bp insert was cut from the pET vector (no insert) and blunted with Kleenow. The blunt-ended vectors and 100-bp insert were gel purified before overnight in-gel ligation. This generated competitive templates that were 100 bp larger but with identical primer sites to the wild-type template. For use as an internal competitive template, the plasmid containing the template was linearized with Nco1, gel purified, quantified spectrophotometrically, and serially diluted.21
VEGF-R Competitive PCR
Tumors grown either in the brain or subcutis were removed and frozen immediately on dry ice and then homogenized in a Dounce homogenizer (treated with RNase Zap (Ambion, Austin, TX)), and RNA was extracted as described above. Before reverse transcription, RNA was treated with RNase-free DNase (Promega, Madison, WI) to remove any contaminating genomic DNA.22 DNase-treated RNA was purified using the RNAeasy kit (QIAGEN, Chatsworth, CA) and quantitated spectrophotometrically, and 2 μg was reverse transcribed as described above. PCR reactions were as follows: 1 μl of tumor reverse transcriptase-cDNA, 0.4 μmol/L upper and lower primers, 5 μl of Pfu PCR buffer, 2.5 μl of deoxynucleotide triphosphate, 20 μCi [32P]dCTP, 28 μl of H2O, 1 μl of Pfu, and 10 μl of competitive template (0 to 1000 fg) using the following method (95°C for 5 minutes; 95°C for 30 seconds, 58°C for 1 minute, and 72°C for 2 minutes for 40 cycles followed by 72°C for 10 minutes). Master mixes of the PCR reaction were made and aliquoted; to these, 10 μl of each template was added. Reactions (20 μl) were separated on a 6% polyacrylamide gel made with Tris borate ethylenediamine tetraacetic acid buffer, dried and exposed to a phosphorimager screen. Sample and template bands were quantitated using ImageQuant analysis software (Molecular Dynamics, Sunnyvale, CA). Values were analyzed, and plots of template amount versus template/sample ratio were generated to calculate the amount of sample.21,23 All competitive PCR experiments were carried out in triplicate on two to five tumors. Data are presented as the average ± standard deviation.
Vascular Density Immunohistochemistry
Tumors were removed and snap frozen in ornithine carbamoyltransferase by immersion in liquid nitrogen. Sections (7 μm) were picked up on gelatin-coated slides, fixed in 1% formaldehyde; blocked with 10% fetal calf serum; and stained for endothelium using a rat anti-murine monoclonal antibody, MECA20. Sections were counterstained with hematoxylin and immunostained vessels were counted. Five fields (at ×200 magnification) were counted for each of three or four tumors.
Statistics
Statistical analysis for electron microscopic morphometry was performed as previously described using StatView (Abacus Concepts, Inc., Berkeley, CA).19
Results
Vascular Morphology
Vascular endothelium in the adult mouse brain, where the tumors were implanted, is characterized by extensive and complex tight junctions and relatively few plasmalemmal vesicles (Figure 1A)▶ . Furthermore, the majority of vessels are capillaries or very small venules and all have a well developed basement membrane. These morphological characteristics contribute to the low microvascular permeability associated with the blood brain barrier. When tumors are grown in the brain, the tumor vessels are derived from this extremely tight brain vasculature.
Figure 1.
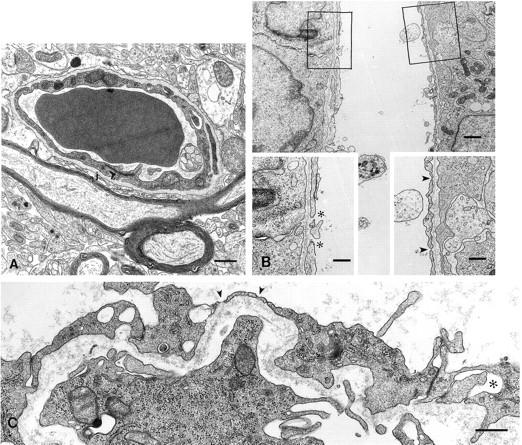
Electron micrographs of vessels in the normal brain (A), VEGF:CHO (B), and M1S (C) tumors growing in the brain. An extensive electron-dense endothelial junction is common to normal brain vessels (A, j). This junction is drastically compromised in tumor vessels, in which the intercellular gaps can be as large as 260 nm (B and C, asterisks). VEGF:CHO tumors had numerous gaps (B, asterisks), which may be remnants of fenestrations (arrowheads). B: Boxed areas are shown as insets. Bars (A, B, insets, and C), 500 nm.
The tumor microvessels underwent a remarkable transformation from one of the least permeable microvasculature to one with loose and open endothelial gaps and fenestrated endothelium (Figure 1, B and C)▶ . This transformation was most evident when VEGF-transfected CHO cells were grown in the brain. They grew into well vascularized 1-cm3 tumors (without apparent side effects to the mouse) that had the highest percentage of vessels with open gaps and fenestrated endothelium of all the tumors studied (Figure 1B▶ , insets, and Table 1▶ ). Gaps varied from 60 to 300 nm. Unlike vessels in all other tumors, vessels in the VEGF:CHO tumors had continuous and prominent basement membranes.
Table 1.
Fenestrations per Vessel Profile
| No. of vessels* | Range† | Average + SEM‡ | Median | |
|---|---|---|---|---|
| Normal skin | 124 (2.4%) | 1 to 2 | 1.33 ± 0.33 | 1 |
| Normal skeletal muscle | 251 (0%) | 0 | 0 | 0 |
| Normal brain | 144 (0%) | 0 | 0 | 0 |
| EMT-6 (murine mammary carcinoma) | ||||
| s.c. | 88 (41%) | 1 to 54 | 9.8 ± 1.8 | 6 |
| i.c. | 99 (1%) | 1 | 1 | 1 |
| M1S (murine rhabdomyosarcoma) | ||||
| s.c. | 181 (35%) | 1 to 36 | 10.8 ± 1.3 | 8 |
| i.c. | 97 (9%) | 1 to 7 | 2.4 ± 0.7 | 2 |
| U87 (human glioblastoma) | ||||
| s.c. | 62 (37%) | 1 to 44 | 10.5 ± 2.3 | 6 |
| i.c. | 110 (3%) | 1 | 1 | 1 |
| U251 (human glioblastoma) | ||||
| s.c. | 62 (37%) | 1 to 44 | 10.5 ± 2.3 | 6 |
| i.c. | 87 (4%) | 1 to 3 | 2 ± 1 | 2 |
| VEGF:CHO (VEGF165-transfected CHO cells) | ||||
| s.c. | 119 (56%) | 1 to 35 | 9.8 ± 0.9 | 8 |
| i.c. | 89 (9%) | 1 to 5 | 2.1 ± 0.5 | 2 |
*Number of vessels counted (percentage of fenestrated vessels).
†Range of fenestrations per vessel profile.
‡Average number of fenestrations per vessel profile.
Noticeably affected in most tumor vessels were loose endothelial junctions. Although junctions were not completely patent in a given plane of section, large gaps were routinely found in virtually all tumor vessels (Figure 1, B and C)▶ . There was a general lack of fused, clustered caveolae normally present in s.c. tumor vascular endothelium. However, the number of caveolae was increased in the thicker endothelial cells that had numerous mitochondria, polysomes, and rough endoplasmic reticulum, all indicative of a highly proliferative state, whereas the fenestrated endothelial cells were more attenuated (Figure 1, B and C)▶ .
Morphometric analysis was completed for all tumors to obtain a percentage of vessels with fenestrated endothelium, open gaps, and number of fenestrations per vessel profile (Table 1▶ and Figure 2▶ ). With the notable exception of the VEGF:CHO tumors, all tumors when implanted in the brain had drastically reduced percentage of vessels with fenestrated endothelium and open gaps compared with the same tumors grown s.c., but all tumors had a significantly higher percentage of vessels with fenestrated endothelium compared with the host tissue regardless of location.
Figure 2.

Tumor vascular morphometry of tumors grown in the brain (i.c.) or in the subcutis (s.c.). Bars represent the percentage of vessels with fenestrated endothelium (solid and narrow cross-hatch) and open endothelial gaps (hatch and cross-hatch). ND, not determined. All s.c. data are taken from Ref. 19 . *P < 0.001, #P < 0.005, and ∧P < 0.05 compared with host tissue vessels; see Materials and Methods for details on statistics.
No vessels were found in either the EMT or M1S i.c. tumors with open endothelial gaps, whereas open gaps were encountered in both glioblastomas and especially VEGF:CHO tumors. In fact, VEGF:CHO tumors had more vessels with open gaps when implanted in the brain than when implanted s.c. Interestingly, animals bearing large (>1 cm in diameter) VEGF:CHO tumors in their brain were less noticeably affected (eg, loss of equilibrium or loss of appetite) than animals with much smaller (<3 mm in diameter) M1S, EMT, U87, or U251 tumors, likely indicative of the relatively benign nature of the VEGF:CHO tumors. Although not taken into account in our morphometric analysis, the integrity of the endothelial intercellular junctions in vessels of all the tumors was often compromised when compared with the normal brain vessels. The junctions were not tightly joined from lumen to ablumen (compare junctions in Figure 1, A and C▶ ).
VEGF Expression
Because tumor location drastically affected tumor vascular morphology, studies were undertaken to examine whether differences in VEGF levels could account for these observed differences. Western blot analysis for VEGF on lysates from s.c. and i.c. tumors is shown in Figure 3▶ . VEGF amount in tumor lysates was not significantly different between tumor location, with the VEGF:CHO tumors having ∼10 times more VEGF compared with all other tumors. The glioblastomas, U87 and U251, had the least amount of VEGF, with U251 having the lowest of all. Although the 165-amino acid isoform appears to be most prominent, the antibody used does not recognize the other isoforms well, and the presence of other bands around 46 kd likely indicates glycosylation variants of the 165-amino acid isoform. RT-PCR was used to determine whether VEGF isoforms were differentially expressed because of tumor location or among the tumors. VEGF isoform and basic fibroblast growth factor expression were determined in the tumors using RT-PCR. The 165-amino acid isoform was the most highly expressed isoform in all tumors, but there was no significant difference among other isoforms or in basic fibroblast growth factor (bFGF) expression among the samples (data not shown).
Figure 3.

Immunoblot of tumor lysates for VEGF. Tumors were grown either s.c. or in the brain. Twenty μg of protein were resolved on a nonreducing 10% polyacrylamide gel, transferred to nitrocellulose, and blotted as described in Materials and Methods. The VEGF:CHO tumors (VEGF) always had the most VEGF, whereas the glioblastomas (U87 and U251) generated the least amount of immunodetectible VEGF. Location minimally affected the amount of VEGF in these tumors. The ∼30-kd band in the U251 i.c. lane may be a VEGF monomer, given that RT-PCR determined that VEGF 121 and 189 were not substantially produced by any tumor. This band was variably present in other samples of tumor lysates.
Flk-1 and Flt-1 Expression
In addition to tumor-secreted VEGF, we decided to investigate the amount of VEGF-Rs, Flk-1 and Flt-1, on tumor vessels using competitive PCR. Because the host microvasculature supplying the tumors is so dramatically different between s.c. tissues (primarily skeletal muscle) and brain, we hypothesized that the tumor vessels might have different levels of Flk-1 and Flt-1 in the two conditions. PCR primers and competitive templates were designed for murine receptors, because all vessels regardless of tumor type would be murine in origin. Competitive templates were designed with identical primer sites but with a 100-bp insertion. This allowed for equivalent PCR amplification efficiency and easy identification of template and sample. As template amount was decreased, the PCR product of interest (Flk-1 or Flt-1) in the tumor sample increased in intensity (Figure 4A)▶ . The data were acquired using a phosphor imager and plotted in graph form as the ratio of template signal/sample signal versus template input. When the template/sample ratio equaled 1.0, the amount of receptor mRNA in the tumor sample was calculated from the abscissa.
Figure 4.
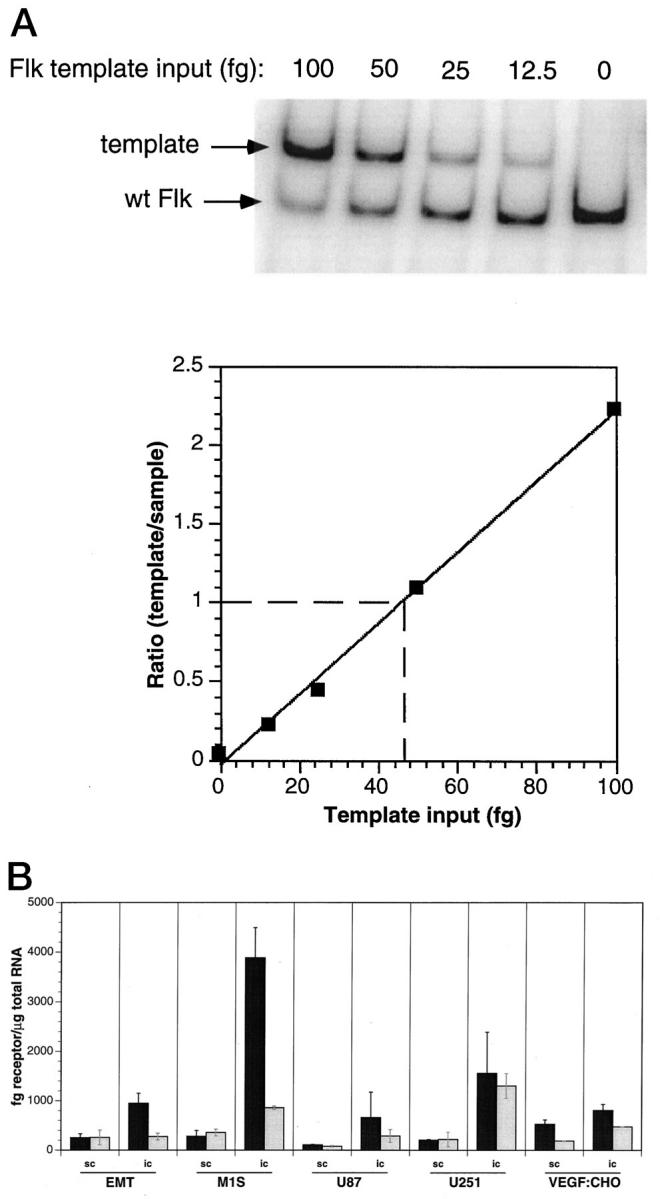
Competitive PCR analysis of VEGF receptors. Data were acquired from phosphor imager analysis and plotted to obtain fg of receptor per reaction (A). Data were then normalized to μg of total RNA and plotted (B). Flk-1 (▪) and Flt-1 (░⃞), from tumors grown in the brain (i.c.) or in the subcutis (s.c.). Flk-1 mRNA always had equal or increased expression compared with Flt-1 regardless of tumor location. Tumors grown i.c. generally had increased expression of both receptors compared with tumors grown s.c. Data are represented as average ±SD of triplicate experiments on two to five tumors.
i.c. tumors had higher levels of both Flk-1 and Flt-1 mRNA when compared with identical tumor types grown s.c. (Figure 4B)▶ . Moreover, Flk-1 mRNA amount was either the same or greater when compared with Flt-1, regardless of tumor implantation site. The increase in Flk-1 relative to Flt-1 was as great as 4.7-fold in the case of the brain-implanted M1S tumor. Generally, the s.c. tumors with the greatest amount of VEGF (Figure 3)▶ had the most receptor (either Flk-1 or Flt-1) (Figure 4B)▶ . s.c. VEGF:CHO tumors had the most immunoreactive VEGF and the most Flk-1 and Flt-1 mRNA compared with the other s.c. tumors. However, this trend was not observed when tumors were grown in the brain. U251 i.c. tumors had the least immunoreactive VEGF, but had very high levels of Flk-1 and Flt-1 mRNA expression, and VEGF:CHO i.c. tumors (which had the greatest amount of VEGF) had moderate levels expression of both VEGF-R receptors.
Vascular Density
Differences in receptor amount may be indicative of increased endothelial expression of each receptor, increased vascular density, or both. To address this question, M1S and U87 tumors, grown i.c. and s.c., were examined for vascular density by indirect immunohistochemical staining for endothelium (Table 2)▶ . Only these tumors were selected, because these tumors had the greatest and least difference in receptor expression between s.c. and i.c. tumors (Figure 4B)▶ . Although there was an increase in vascular density in both U87 and M1S tumors when grown i.c. compared with s.c. (U87, 2.1-fold increase; M1S, 1.7-fold increase, i.c. versus s.c.), this increase is not sufficient to account for the increased receptor expression observed in the competitive PCR (U87-Flt-1, 3.5-fold increase; U87-Flk-1, 5.7-fold increase; M1S-Flt-1, 2.4-fold increase; M1S-Flk-1, 14.3-fold increase, i.c. versus s.c.). Therefore, the increased receptor expression in i.c. tumors versus s.c. tumors as measured by competitive PCR is due more to the increase in endothelial expression than to vascular density.
Table 2.
Vascular Density Measured in Selected Tumors
| Vascular Density* | factor† | Flk‡ | factor† | Flt‡ | factor† | |
|---|---|---|---|---|---|---|
| M1S s.c. | 114.9 ± 16.8 | 1.72x | 0.07 | 14.3x | 0.09 | 2.44x |
| M1S i.c. | 196.1 ± 55 | 1 | 0.22 | |||
| U87 s.c. | 31.4 ± 4.5 | 2.1x | 0.03 | 5.7x | 0.02 | 3.5x |
| U87 i.c. | 67.1 ± 11.8 | 0.17 | 0.07 |
*Vascular density was measured by counting CD31-positive vessels in frozen sections (average ± SD).
†Values for i.c/s.c.
‡Values relative to M1S i.c. Flk fg receptor/μg total RNA.
Discussion
It is now well accepted that tumors derive their vasculature from surrounding tissues.24 The importance that the host microvascular bed has in regulating the morphology and physiology of the tumor neovasculature cannot be underestimated.18,25 Numerous therapies that specifically target tumor vessels or rely on tumor microvascular hyperpermeability to access the tumor cells directly are in various stages of clinical testing.12 Although some information on the interaction between host tissue environment and tumor vessel physiology is available, a comprehensive study on how this interaction affects vascular morphology and growth factor receptor expression has not been published. The microvasculature of the brain and s.c. tissues is extremely different in structure and physiology and therefore provides an excellent model system to study the influence of host microvascular environment on tumor neovascular development.
In a previous study, we demonstrated that neovascular endothelium derived from the continuous endothelium of skeletal muscle or skin became fenestrated and acquired open endothelial gaps.19 The goal of the present study was to determine whether host microvascular endothelia from two different tissues were identically modified by tumor-secreted growth factors, specifically VEGF. In other words, does the host microvasculature determine the morphology and therefore the function of the tumor vasculature, or do the tumor-secreted growth factors override all host microvascular endothelial input? To this end, a detailed morphological analysis of vessels from tumors growing in the brain was carried out. Given the recent interest in developing antiangiogenesis and vascular targeting therapies to control tumor progression, understanding the role that the host microvascular environment plays in determining the morphology and function of tumor vasculature is warranted. Moreover, a better understanding of how host microvascular endothelium determines tumor vessel morphology will influence the design and execution of therapies against primary and metastatic tumors.
Morphometric Analysis
The vasculature that supplies the majority of the central nervous system has characteristically low permeability.26 Vessels in the normal brain have such low permeability that even the smallest solutes, such as glucose and ions, must be actively transported.27 Capillaries of the normal brain consist of a continuous endothelium the cells of which cells are joined by well developed and complex tight junctions (zonulae occludentes); there are no fenestrae and very few plasmalemmal vesicles.26 Maintenance of the blood-brain barrier is generally accepted to be the result of tight endothelial junctions, lack of transcytotic vesicles, complete basement membrane, and tight junctions among astrocyte foot processes. However, the loss of the blood-brain barrier is commonly observed when tumors invade and grow into the brain.28,29 The loss of the blood-brain barrier in tumor vessels has been attributed to the generation of neovasculature with fenestrated endothelium, opened intercellular junctions, and incomplete basement membrane, all of which are seen in our tumors.30,31 Although there are anecdotal reports describing endothelial fenestrations and open gaps in vessels from tumors in the subcutis or in the brain, there are no studies that systematically characterize vascular morphology in identical tumors implanted in these two sites.32,33
The combination of drastically loose endothelial junctions, patently open gaps, and fenestrations are undoubtedly responsible for the hyperpermeability commonly associated with tumor microvasculature.34,35 Conspicuously absent in virtually every i.c. tumor vessel observed were extensive clusters of fused caveolae (vesiculo-vacuolar organelles), although fusion of two or three caveolae could be readily identified. However, it is not surprising that extensive vesicle clusters were not found, considering that brain vascular endothelium has essentially no plasmalemmal vesicles or caveolae.36 Dellian et al37 have demonstrated that neovasculature induced from brain microvasculature (pial, as well as cortical, vessels) were four to eight times more permeable to bovine serum albumin than were neovessels derived from the skin.37 However, our data demonstrate a general reduction of i.c. tumor vessels with open gaps and fenestrated endothelium compared with s.c. tumor vessels. We believe these are indications that loose endothelial junctions, which were readily observed in all our tumors but not quantitated, contribute substantially to tumor microvascular hyperpermeability. Our morphometric data describing the reduced presence of fenestrations and open endothelial gaps in i.c. tumors compared with s.c. tumors is completely consistent with functional studies looking at the pore cutoff size between tumors grown in cranial window and dorsal chamber preparations.25
It is interesting to note that, of the malignant tumors, only the glioblastomas had vessels with open gaps when implanted in the brain. In the clinical setting, glioblastoma multiforme neovasculature is characteristically hyperpermeable in comparison with other tumor types. This hyperpermeability may be an indication that glioblastomas have evolved to produce factors not made in the other tumors that allow them to modify the extremely tight blood vessels in the brain.38 This hypothesis is substantiated by experimental data demonstrating that U87 i.c. tumors had two times greater permeability to bovine serum albumin than a murine mammary carcinoma grown i.c., even though the pore cutoff size was approximately two times less, suggesting an increase in the number of pores.25
In numerous vessels, endothelial cells that were abnormally thick were found. These cells were also characterized by the increased number of mitochondria, polysomes, and rough endoplasmic reticulum in their cytoplasm, all indicative of rapidly proliferating cells. Interestingly, these cells also had caveolae, which are very rare in normal brain vessels but were generally not fenestrated. The fenestrated endothelium was always attenuated and did not appear to be highly proliferative. It is possible that the highly proliferative endothelial cells are precursors of the fenestrated endothelial cells or that they are to be found in the peripheral region of the tumors where the tumor cells are actively growing.
The intriguing finding regarding brain tumor vascular morphology was that VEGF:CHO tumors had significantly more open gaps than any other tumor, even those implanted s.c. However, these gaps may, in fact, be fenestrations without diaphragms, similar to those of the fenestrated endothelium of the kidney glomerulus, in which VEGF has been implicated in inducing and maintaining this unique endothelial phenotype.39
VEGF and VEGF Receptor Expression
VEGF expression varied greatly among the tumors. There was, however, no substantial difference in VEGF protein expression attributable to tumor location. The presence of a doublet in most tumor lysates may indicate glycosylation variants of the 165-amino acid isoform. There is a general correlation between VEGF expression and VEGF-induced morphological changes (ie, fenestrated endothelium) when the two tumor sites are viewed independently. Among the s.c. tumors, VEGF:CHO tumors secrete the most VEGF and have the highest percentage of vessels with fenestrated endothelium. Likewise, among i.c. tumors, VEGF:CHO tumors secrete the most VEGF and also have the greatest percentage of vessels with fenestrated endothelium. It would be naive to suggest that only tumor-secreted VEGF could modify the tumor vasculature, because there are other tumor-secreted factors and other microenvironmental factors (eg, pericytes, or extracellular matrix) that can affect vascular morphology.19,40 This may explain the lack of a complete positive correlation between immunoreactive VEGF in tumor lysates and endothelial modification (fenestration).
Previous reports have described a remarkable induction of Flk-1 and Flt-1 in brain tumor vessels compared with surrounding normal brain vessels.41,42 For this reason, we decided to characterize Flk-1 and Flt-1 expression in s.c. and i.c. tumors. We, therefore, established a competitive PCR assay to measure and compare Flk-1 and Flt-1 mRNA expression. Consistent with other reports, Northern blot analysis for the receptors demonstrated undetectable levels of receptor expression in normal brain and muscle, which constitute the normal host microvasculature for i.c. and s.c. tumors, respectively.11,43 We decided to develop the competitive PCR assay for the following reasons: 1) Northern blot analysis of Flk-1 and Flt-1 is extremely costly, in that it requires ∼20 μg of total RNA or 2 to 5 μg mRNA for satisfactory signals; 2) quantitative analysis of the Flk-1 and Flt-1 mRNA (or protein) expression in tumors, especially identical tumor types growing in different host tissues, is nonexistent; 3) reagents are not available to quantitate protein expression of these receptors with sufficient sensitivity; 4) except in rare circumstances, the VEGF receptors are exclusively expressed on proliferating neovasculature and are therefore excellent markers for angiogenesis and vessel density;44,45 and 5) recent evidence suggests that vessel density is an excellent prognostic indicator of patient survival and morbidity in a number of tumors.46,47 Because our competitive PCR method does not take into account varied efficiencies of reverse transcription, it can only provide semiquantitative results.48 Regardless, the competitive PCR method proves to be an excellent tool to provide relative comparisons of Flk-1 and Flt-1 expression among a number of tumor samples without necessitating exorbitant amounts of sample tissue. Interestingly, in all tumors, regardless of location, Flk-1 mRNA was expressed at equal or higher levels compared with Flt-1. Differences in function between Flk-1 and Flt-1 have been alluded to by the generation and analysis of phenotypes expressed in gene knockout animals.49,50 Furthermore, data suggest that Flk-1, not Flt-1, is primarily responsible for the mitogenic effects associated with VEGF.51,52
In general, there was a positive correlation between VEGF production and receptor expression in s.c. tumors. This was not the case with i.c. tumors. The explanation for this is uncertain, but it may be due to other factors, such as hypoxia and hypoglycemia, which differentially upregulate receptor expression.53-55 It is conceivable that identical stimuli might not induce similar responses in the vascular endothelium of muscle and brain, namely receptor mRNA upregulation. Certainly, this was the case with the tumor vascular morphology. i.c. tumors had higher expression levels of both receptors when compared with the identical tumor type grown s.c. This may represent an increased inducibility of the receptors on brain vascular endothelium (ie, higher receptor expression per endothelial cell) or increased vascular density, commonly found in brain tumors. Therefore, M1S and U87 tumors, grown i.c. and s.c., were examined for vascular density by staining for endothelium. Although there was an increase in vascular density in both U87 and M1S tumors when grown i.c. compared with s.c., this increase was not sufficient to account for the increased receptor expression. Therefore, the increased receptor expression as measured by competitive PCR seen in i.c. tumors versus s.c. tumors is due more to an increase in endothelial expression than vascular density. This suggests that different microvascular beds do not respond identically, at least in regard to the expression of the VEGF receptors, to equivalent stimuli.
Our data on the receptor expression in the brain tumors are preliminary but have generated some interesting questions: 1) How does vascular endothelium from different organs respond differently in their induction of the VEGF receptors to similar stimuli? Is brain vascular endothelium more/less susceptible to stimuli that induce Flk-1/Flt-1 expression compared with other vascular endothelium? 2) Is the higher expression of Flk-1 indicative of a more prominent role in angiogenesis? 3) Can the measurement of Flk-1 mRNA expression replace vessel counting as a prognostic indicator of tumor progression? Our results indicate that neovasculature in identical tumor types is drastically different when tumors are grown in different locations. The implications are obvious when antitumor therapies are considered, regardless of whether they are directed at the tumor cells or the tumor blood vessels. These tumor vessel morphological differences affect the tumor vascular permeability and therefore distribution of anticancer agents.5,56 Questions remain as to whether the molecular, cellular, and morphological differences in tumor vascular endothelium must be addressed individually for each tumor site (eg, primary versus metastatic) or whether universal characteristics can be found and exploited.
Footnotes
Address reprint requests to Dr. WG Roberts, Pfizer Central Research, Cancer Research, Eastern Point Rd, Groton, CT 06340.
Supported by grants from the United States Public Health Service (National Institutes of Health grant R37 HL17080-22) and the US Army Breast Cancer Research Program (DAMD 17-96-1-6203).
References
- 1.Gimbrone MA, Leapman SB, Cotran RS, Folkman J: Tumor dormancy in vivo by prevention of neovascularization. J Exp Med 1972, 136:261-276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liotta LA, Steeg PS, Stetler-Stevenson WG: Cancer metastasis and angiogenesis: an imbalance of positive and negative regulation. Cell 1991, 64:327-336 [DOI] [PubMed] [Google Scholar]
- 3.Ellis LM, Fidler IJ: Angiogenesis and metastasis. Eur J Cancer 1996, 32A:2451-2460 [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N, Davis-Smyth T: The biology of vascular endothelial growth factor. Endocr Rev 1997, 18:4-25 [DOI] [PubMed] [Google Scholar]
- 5.Dvorak HF, Brown LF, Detmar M, Dvorak AM: Vascular permeability factor/vascular endothelial growth factor, microvascular hyperpermeability, and angiogenesis. Am J Pathol 1995, 146:1029-1039 [PMC free article] [PubMed] [Google Scholar]
- 6.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF: Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219:983-985 [DOI] [PubMed] [Google Scholar]
- 7.Roberts WG, Palade GE: Increased microvascular permeability and endothelial fenestration induced by vascular endothelial growth factor. J Cell Sci 1995, 108:2369-2379 [DOI] [PubMed] [Google Scholar]
- 8.De Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N, Williams LT: The fms-like tyrosine kinase, a receptor for vascular endothelial growth factor. Science 1992, 255:989-991 [DOI] [PubMed] [Google Scholar]
- 9.Quinn TP, Peters KG, DeVries C, Ferrara N, Williams LT: Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc Natl Acad Sci USA 1993, 90:7533-7537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keyt BA, Nguyen HV, Berleau LT, Duarte CM, Park J, Chen H, Ferrara N: Identification of vascular endothelial growth factor determinants for binding KDR and Flt-1 receptors. J Biol Chem 1996, 271:5638-5646 [DOI] [PubMed] [Google Scholar]
- 11.Millauer B, Wizigmann-Voos S, Schnurch H, Martinez R, Moller NPH, Risau W, Ullrich A: High affinity VEGF binding and development expression suggest Flk-1 as a major regulator of vasculogenesis and angiogenesis. Cell 1993, 72:835-846 [DOI] [PubMed] [Google Scholar]
- 12.Folkman J: Angiogenesis and angiogenesis inhibition: an overview. Exs 1997, 79:1-8 [DOI] [PubMed] [Google Scholar]
- 13.Jain RK: Delivery of molecular medicine to solid tumors. Science 1996, 271:1079-1080 [DOI] [PubMed] [Google Scholar]
- 14.Nelson JS, Liaw L-H, Orenstein A, Roberts WG, Berns MW: Mechanism of tumor destruction following photodynamic therapy with hematoporphyrin derivative, chlorin, and phthalocyanine. J Natl Cancer Inst 1988, 80:1599-1605 [DOI] [PubMed] [Google Scholar]
- 15.Strawn LM, McMahon G, App H, Schreck R, Kuchler WR, Longhi MP, Hui TH, Tang C, Levitzki A, Gazit A, Chen I, Keri G, Orfi L, Risau W, Flamme I, Ullrich A, Hirth KP, Shawver LK: Flk-1 as a target for tumor growth inhibition. Cancer Res 1996, 56:3540-3545 [PubMed] [Google Scholar]
- 16.Moore JV, West DC: Vasculature as a target for anti-cancer therapy. Cancer Cells 1991, 3:100-102 [PubMed] [Google Scholar]
- 17.Yuan F, Salehi HA, Boucher Y, Vasthare US, Tuma RF, Jain RK: Vascular permeability and microcirculation of gliomas and mammary carcinomas transplanted in rat and mouse cranial windows. Cancer Res 1994, 54:4564-4568 [PubMed] [Google Scholar]
- 18.Fukumura D, Yuan F, Monsky WL, Chen Y, Jain RK: Effect of host microenvironment on the microcirculation of human colon adenocarcinoma. Am J Pathol 1997, 151:679-688 [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts WG, Palade GE: Neovasculature induced by vascular endothelial growth factor is fenestrated. Cancer Res 1997, 57:765-772 [PubMed] [Google Scholar]
- 20.Cheng S-Y, Huang H-JS, Nagane M, Ji X-D, Wang D, Shih CC-Y, Arap W, Hung C-M, Cavenee WK: Suppression of glioblastoma angiogenicity and tumorigenicity by inhibition of endogenous expression of vascular endothelial growth factor. Proc Natl Acad Sci USA 1996, 93:8502-8507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caputi M, Melo CA, Baralle FE: Regulation of fibronectin expression in rat regenerating liver. Nucleic Acids Res 1995, 23:238-243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rashtchian A: Amplification of RNA. PCR Methods Appl 1994, 4:S83-S91 [DOI] [PubMed] [Google Scholar]
- 23.Schneeberger C, Eder S, Speiser P, Zeillinger R: Competitive reverse transcriptase-PCR: an improved method for determination of c-erb-2 gene expression. Anticancer Res 1996, 16:849-852 [PubMed] [Google Scholar]
- 24.Folkman J, Shing Y: Angiogenesis. J Biol Chem 1992, 267:10931-10934 [PubMed] [Google Scholar]
- 25.Hobbs SK, Monsky WL, Yuan F, Roberts WG, Griffith L, Torchilin VP, Jain RK: Regulation of transport pathways in tumor vessels: role of tumor type and microenvironment. Proc Natl Acad Sci USA 1998, 95:4607-4612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Peters A, Palay SL, Webster H: Blood vessels. Peters A Palay SL Webster H eds. The Fine Structure of the Nervous System. 1991, :pp 344-355 Oxford University Press, Oxford [Google Scholar]
- 27.Virgintino D, Robertson D, Monaghan P, Errede M, Bertossi M, Ambrosi G, Roncali L: Glucose transporter GLUT1 in human brain microvessels revealed by ultrastructural immunocytochemistry. J Submicrosc Cytol Pathol 1997, 29:365-370 [PubMed] [Google Scholar]
- 28.Long DM: Capillary ultrastructure and the blood-brain barrier in human malignant brain tumors. J Neurosurg 1970, 32:127-144 [DOI] [PubMed] [Google Scholar]
- 29.McComb RD, Bigner DD: The biology of malignant gliomas: a comprehensive survey. Clin Neuropathol 1984, 3:93-106 [PubMed] [Google Scholar]
- 30.Deane BR, Lantos PL: The vasculature of experimental brain tumors. Part 1. A sequential light and electron microscopic study of angiogenesis. J Neurol Sci 1981, 49:55-66 [DOI] [PubMed] [Google Scholar]
- 31.Vick NA, Bigner DD: Microvascular abnormalities in virally-induced canine brain tumors. J Neurol Sci 1972, 17:29-39 [DOI] [PubMed] [Google Scholar]
- 32.Hirano A, Matsui T: Vascular structures in brain tumors. Hum Pathol 1975, 6:611-621 [DOI] [PubMed] [Google Scholar]
- 33.Konerding MA, Steinberg F, van Ackern C, Budach V, Streffer C: Vascular patterns of tumors: scanning and transmission electron microscopic studies on human xenografts. Strahlenther Onkol 1992, 168:444-452 [PubMed] [Google Scholar]
- 34.Kohn S, Nagy JA, Dvorak HF, Dvorak AM: Pathways of macromolecular tracer transport across venules and small veins. Lab Invest 1992, 67:596-607 [PubMed] [Google Scholar]
- 35.Yuan F, Dellian M, Fukumura D, Leunig M, Berk DA, Torchilin VP, Jain RK: Vascular permeability in a human tumor xenograft: molecular size dependence and cutoff size. Cancer Res 1995, 55:3752-3756 [PubMed] [Google Scholar]
- 36.Simionescu M, Ghinea N, Fixman A, Lasser M, Kukes L, Simionescu N, Palade GE: The cerebral microvasculature of the rat: structure and luminal surface properties during early development. J Submicrosc Cytol Pathol 1988, 20:243-261 [PubMed] [Google Scholar]
- 37.Dellian M, Witwer BP, Salehi HA, Yuan F, Jain RK: Quantitation and physiological characterization of angiogenic vessels in mice. Am J Pathol 1996, 149:59-71 [PMC free article] [PubMed] [Google Scholar]
- 38.Risau W: Induction of blood-brain barrier endothelial cell differentiation. ANYAS 1998, 633:405-419 [DOI] [PubMed] [Google Scholar]
- 39.Breier G, Risau W: The role of vascular endothelial growth factor in blood vessel formation. Trends Cell Biol 1996, 6:454-456 [DOI] [PubMed] [Google Scholar]
- 40.Hirschi KK, D’Amore PA: Pericytes in the microvasculature. Cardiovasc Res 1996, 32:687-698 [PubMed] [Google Scholar]
- 41.Plate KH, Breier G, Millauer B, Ullrich A, Risau W: Up-regulation of vascular endothelial growth factor and its cognate receptors in a rat glioma model of tumor angiogenesis. Cancer Res 1993, 53:5822-5827 [PubMed] [Google Scholar]
- 42.Plate KH, Breier G, Weich HA, Mennel HD, Risau W: Vascular endothelial growth factor and glioma angiogenesis: coordinate induction of VEGF receptors, distribution of VEGF protein and possible in vivo regulatory mechanisms. Int J Cancer 1994, 59:520-529 [DOI] [PubMed] [Google Scholar]
- 43.Jakeman LB, Winer J, Bennett GL, Altar CA, Ferrara N: Binding sites for vascular endothelial growth factor are localized on endothelial cells in adult rat tissues. J Clin Invest 1992, 89:244-253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morishita K, Johnson DE, Williams LT: A novel promoter for vascular endothelial growth factor receptor (flt-1) that confers endothelial-specific gene expression. J Biol Chem 1995, 270:27948-27953 [DOI] [PubMed] [Google Scholar]
- 45.Yamaguchi TP, Dumont DJ, Conlon RA, Breitman ML, Rossant J: flk-1, an flt-related receptor tyrosine kinase is an early marker for endothelial cell precursors. Development 1993, 118:489-498 [DOI] [PubMed] [Google Scholar]
- 46.Weidner N, Folkman J: Tumoral vascularity as a prognostic factor in cancer. DeVita VT Hellman S Rosenberg SA eds. Important Advances in Oncology 1996. 1996, :pp 167-190 Lippincott-Raven Publishers, Philadelphia [PubMed] [Google Scholar]
- 47.Craft PS, Harris AL: Clinical prognostic significance of tumour angiogenesis. Ann Oncol 1994, 5:305-311 [DOI] [PubMed] [Google Scholar]
- 48.Zimmermann K, Mannhalter JW: Technical aspects of quantitative competitive PCR. Biotechniques 1996, 21:268-279 [DOI] [PubMed] [Google Scholar]
- 49.Fong G-H, Rossant J, Gertsenstein M, Breitman ML: Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376:66-70 [DOI] [PubMed] [Google Scholar]
- 50.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu X-F, Breitman ML, Schuh AC: Failure of blood-island and vasculogenesis in Flk-1-deficient mice. Nature 1995, 376:62-66 [DOI] [PubMed] [Google Scholar]
- 51.Seetharam L, Gotoh N, Maru Y, Neufeld G, Yamaguchi S, Shibuya M: A unique signal transduction from FLT tyrosine kinase, a receptor for vascular endothelial growth factor VEGF. Oncogene 1995, 10:135-147 [PubMed] [Google Scholar]
- 52.Volm M, Koomagi R, Mattern J: Prognostic value of vascular endothelial growth factor and its receptor Flt-1 in squamous cell lung cancer. Int J Cancer 1997, 74:64-68 [DOI] [PubMed] [Google Scholar]
- 53.Brogi E, Schatteman G, Wu T, Kim EA, Varticovski L, Keyt B, Isner JM: Hypoxia-induced paracrine regulation of vascular endothelial growth factor receptor expression. J Clin Invest 1996, 2:469-476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Detmar M, Brown LF, Berse B, Jackman RW, Elicker BM, Dvorak HF, Claffey KP: Hypoxia regulates the expression of vascular permeability factor/vascular endothelial growth factor (VPF/VEGF) and its receptors in human skin. J Invest Dermatol 1997, 108:263-268 [DOI] [PubMed] [Google Scholar]
- 55.Gerber H, Condorelli F, Park J, Ferrara N: Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes: Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem 1997, 272:23659-23667 [DOI] [PubMed] [Google Scholar]
- 56.Jain RK: Barriers to drug delivery in solid tumors. Sci Am 1994, 58–65 [DOI] [PubMed]