

Abstract
Intravenous immunoglobulin (IVIg) is increasingly used in the treatment of autoimmune and inflammatory diseases, including vasculitides and Kawasaki disease. However, the outcome of IVIg interaction with endothelial cells of the vascular bed is not clear as yet. We have investigated the effect of IVIg on the in vitro activation of human endothelial cells, as assessed by cell proliferation and reverse transcription-polymerase chain reaction-detected expression of mRNA coding for adhesion molecules (intercellular adhesion molecule-1 and vascular cellular adhesion molecule-1), chemokines (monocyte chemoattractant protein-1, macrophage colony-stimulating factor, and granulocyte-macrophage colony-stimulating factor), and proinflammatory cytokines (tumor necrosis factor-α, interleukin-1β, and interleukin-6). IVIg inhibited proliferation of endothelial cells in a time-dependent manner. This effect was dependent on both Fc and F(ab′)2 fragments of the immunoglobulin molecule and was fully reversible. Tumor necrosis factor-α and interleukin-1β also inhibited thymidine incorporation, but to a lesser degree. IVIg had no effect on basal levels of mRNA coding for the adhesion molecules, chemokines, and proinflammatory cytokines. IVIg fully down-regulated the expression induced by tumor necrosis factor-α or interleukin-1β of mRNA coding for these molecules. Thus, blockade of cellular proliferation and of cytokine-induced expression of adhesion molecules, chemokines, and cytokines may explain the therapeutic effect of IVIg in vascular and inflammatory disorders.
Endothelium is not merely a barrier between the bloodstream and tissue, but, by virtue of their location, endothelial cells (ECs) are continuously facing humoral factors and, in some circumstances, actively participate in inflammatory and immunomodulatory responses (for review, see Refs. 1 and 2 ). During inflammation, activated ECs rapidly synthesize and release chemokines and cytokines and express, in a few minutes or in a few hours, new adhesion molecules involved in the adhesion, rolling, and diapedesis of leukocytes. 1-4 Among these newly synthesized molecules, monocyte chemoattractant protein-1 (MCP-1), macrophage colony-stimulating factor (M-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF), intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1), interleukins 1 and 6 (IL-1 and IL-6), and tumor necrosis factor-α and -β (TNF-α and TNF-β) play a key role and can be induced in vitro by numerous agents such as proinflammatory cytokines IL-1β and TNF-α, 5-7 modified low-density lipoproteins, 8 and bacterial lipopolysaccharide. 9
Intravenous immunoglobulin (IVIg) is therapeutic immunoglobulin (Ig) prepared from pools of plasma of several thousand healthy blood donors. In addition to its use as substitutive therapy for primary and secondary antibody deficiencies, IVIg exhibits immunomodulatory effects in diseases mediated by autoantibodies and in diseases believed to be primarily mediated by autoaggressive T cells in humans and in experimental animals. 10,11 IVIg has been used effectively in the treatment of autoimmune cytopenias, 12-17 the acute Guillain-Barré syndrome, 18,19 myasthenia gravis, 20 and anti-factor VIII autoimmune disease. 21 Patients suffering from systemic inflammatory conditions such as dermatomyositis, 22 and, particularly, Kawasaki syndrome greatly benefit from IVIg treatment (for review, see Refs. 23 and 24 ). IVIg has also been used in the treatment of anti-neutrophil cytoplasmic antigen-associated systemic vasculitis. 25,26 The mechanisms of action of IVIg are, as yet, poorly understood, although several mutually nonexclusive hypotheses have been proposed. 11 These include the blockade of Fcγ receptors on phagocytic cells, 27 interference with activated complement, 28,29 modulation of production and release of cytokines and their inhibitors, 30,31 modulation of T- and B-lymphocyte functions, 32-34 suppression of autoantibody production, and selection of immune repertoires. 35,36 However, little is known on the direct interaction between IVIg and ECs of the vascular bed. The present study was undertaken to address the potential role of IVIg on EC function by following both EC proliferation and EC expression of key adhesion molecules, chemokines, and cytokines. We have shown that IVIg inhibited EC proliferation in a dose- and time-dependent manner and down-regulated the expression of adhesion molecule mRNA (ICAM-1 and VCAM-1), chemokine mRNA (MCP-1, M-CSF, and GM-CSF), and proinflammatory cytokine mRNA (TNF-α, IL-1β, and IL-6) induced by TNF-α or IL-1β. These results may explain, at least in part, the therapeutic effect of IVIg in vascular and inflammatory disorders.
Methods
Antibodies
IVIg (Sandoglobulin; Sandoz, Basel, Switzerland), was a kind gift from the Central Laboratory of the Swiss Red Cross Blood Transfusion Service (Bern, Switzerland). Two other preparations of IVIg were Gammagard (N. V. Baxter S. A., Lessines, Belgium) and Endobulin (Immuno AG, Vienna, Austria). F(ab′)2 fragments were prepared from IgG by digestion with 2% (w/w) pepsin (Sigma Chemical Co., St Louis, MO) in acetate buffer, pH 4.1, for 18 hours at 37°C, followed by chromatography on protein A-Sepharose. F(ab′)2 fragments were free of IgG and Fc fragments as assessed by sodium dodecyl sulfate-polyacrylamid gel electrophoresis and enzyme-linked immunosorbent assay. Concentrations of purified IgG and F(ab′)2 fragments were determined spectrophotometrically at 280 nm. Fc fragments were a gift from Dr. M. C. Bonnet (Pasteur Merieux, Lyon, France). Human albumin was obtained from the Laboratoire Français de Fractionnement et de Biotechnologies, L. F .B. (Les Ulis, France). No endotoxin contamination was detected in IVIg preparations using the limulus amebocyte assay. 31
Cell Culture
Umbilical cords were collected from healthy newborns after normal pregnancy and delivery (Notre-Dame de Bon Secours Hospital, Paris, France). Human umbilical vein ECs (HUVECs) were obtained after a 4-minute treatment of the umbilical vein with 0.15% collagenase I (300 U/mg, Sigma) in phosphate-buffered saline (PBS; 13.8 mmol/L NaCl, 0.5 mmol/L Na2HPO4, 4.1 mmol/L KCl, and 0.2 mmol/L KH2PO4) supplemented with 11.1 mmol/L glucose. The cells were cultured in 0.2% gelatin-coated (Sigma) 75-cm 2 tissue culture flasks (Costar, Cambridge, MA) in medium M199 (Life Technologies, Inc., Grand Island, NY) containing Earle’s salts, l-glutamine, and 25 mmol/L HEPES and supplemented with 20% fetal calf serum (FCS; Dutscher, Brumath, France), 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.25 μg/ml fungizone (Life Technologies). At confluency, primary cultured cells were harvested after 6 minutes of trypsinization using 33% trypsin (Biological Industries, Kibbutz Beit Haemek, Israel) in PBS and plated again in three 75-cm 2 tissue culture flasks from one flask. The ECs were characterized by their typical cobblestone morphology and by the presence of factor VIII antigen. For experiments, cells were used at passages two and three.
Growth-Inhibition Studies
After trypsinization, cells were seeded in 96-well plates (Costar) coated with 0.2% gelatin, at a concentration of 1.5 × 10 4 cells/well and grown for 3 to 4 days in M199 containing 20% FCS. To synchronize cells in a G0/G1 stage, FCS concentration was reduced to 1% for 24 hours. At the end of the depletion period, cells were replaced in M199/20% FCS and stimulated for various periods of time in the presence of 0.5 μCi [3H]thymidine/well (Amersham Life Science, Little Chalfont, United Kingdom), with the following agents: 1) IVIg [10, 20, 30, and 40 mg/ml (0.10, 0.13, 0.20, and 0.26 mmol/L, respectively)] for 6, 18, 24, 30, or 48 hours; 2) human albumin (40 mg/ml) for 48 hours; 3) F(ab′)2 (0.20 and 0.26 mmol/L) and Fc (0.20 and 0.26 mmol/L) fragments of IVIg for 24 hours; and 4) purified human recombinant TNF-α (0.5 or 50 ng/ml; Alexis Corp., San Diego, CA), purified human recombinant IL-1β (0.5 or 50 ng/ml; Sigma), TNF-α (50 ng/ml) plus IVIg (40 mg/ml), and IL-1β (50 ng/ml) plus IVIg (40 mg/ml) for 24 hours. At the end of incubation times, radioactivity was measured by liquid scintillation counter (1450 MicroBeta Plus; Wallac, Turku, Finland). The choice of IVIg concentrations in these in vitro studies was based on the average concentrations of therapeutic IgG found in the plasma of patients treated with IVIg.
Cell Viability
For determination of cell number, cells were seeded at a concentration of 5 × 10 3 cells/well and processed as above. After a 24- or 48-hour incubation period with 0, 20, 30, or 40 mg/ml IVIg, cells were harvested by a 6-minute trypsinization. Both live and dead cells were counted in a Malassez chamber using trypan blue. Cell viability was also assessed by FACScan after incubation in presence of IVIg (20, 30, or 40 mg/ml) or human albumin (40 mg/ml) for 24 or 48 hours. At the end of the incubation time, nonadherent cells were harvested, and adherent cells were trypsinized, washed twice with PBS, and suspended with the nonadherent cells in PBS at a final concentration of 1.5 × 10 5 cells/ml. Propidium iodide (10 μl; 500 mg/ml) (Sigma) was added to the 0.5 ml cell suspension, and the uptake of the dye by dead cells was immediately followed by fluorescence analysis using a FACScan (Becton Dickinson, San Jose, CA).
Reversibility Assay
Cells were seeded and starved as described. For reversibility assays, they were incubated in M199/20% FCS with or without IVIg as follows: 1) IVIg (20, 30, or 40 mg/ml) for 24 or 48 hours, and 2) IVIg (20, 30, or 40 mg/ml) for 24 or 48 hours followed by withdrawal of IVIg for the next 24 or 48 hours. In all cases, 0.5 μCi [3H]thymidine/well was added to the cultures for the last 24 hours of incubation period, and the radioactivity was measured.
Reverse Transcription and Polymerase Chain Reaction (PCR) Analysis
ECs were plated in 25-cm 2 flasks in M199/20% FCS for 3 to 4 days to confluency. The medium was then replaced with control medium (M199/20% FCS) and the agonists as follows: IVIg (1, 10, or 40 mg/ml), IL-1β (0.5 or 50 ng/ml), TNF-α (0.5 or 50 ng/ml), IL-1β (50 ng/ml) plus IVIg (40 mg/ml), and TNF-α (50 ng/ml) plus IVIg (40 mg/ml). After 4 hours, mRNAs were extracted with 1 ml of TRIzol (Life Technologies) for 5 to 10 × 10 6 cells, according to the technique provided by the manufacturer. They were reverse transcribed into cDNA with oligo(dT) and Moloney murine leukemia virus reverse transcriptase (Life Technologies). For each experiment, the volume of sample of cDNA was adjusted in such a way as to yield identical levels of glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The reverse transcription products were amplified with primers listed in Table 1 ▶ . All sequences were found in the GenBank database (National Center for Biotechnology Information, National Institutes of Health, Bethesda, MD). All PCRs were carried out in 25 μl of a mixture containing 10 mmol/L deoxynucleotide triphosphate, 1× PCR buffer (10 mmol/L Tris-HCl, pH 8.3, 50 mmol/L KCl, 40% dimethyl sulfoxide, 0.001% gelatin, MgCl2 (see concentrations in Table 1 ▶ ), and 2.5 U AmpliTaq polymerase (Perkin-Elmer Corp., Norwalk, CT)). Each sample was incubated in a DNA thermal cycler (Perkin-Elmer Corp.) at 52°C to 58°C for 30 to 40 cycles, depending on the primer (Table 1) ▶ . The PCR fragments were analyzed by electrophoresis on 2% agarose gels and visualized by ethidium bromide staining (Eurobio, Les Ulis, France). Polaroid photographs of ethidium bromide-stained gels were digitized into 512 × 512-pixel gray-scale images. The amount of nucleic acid, determined by densitometric analysis of the dots, was proportional to the logarithm of the optic density. Analysis was performed using the public domain NIH Image 1.51 program. The intensities of the cDNA bands for each protein were normalized to the GAPDH band intensities. All experiments represent at least three umbilical cords in culture. For each cell extract, PCR was run five times.
Table 1.
Primer List and Characteristics of Polymerization
| Upper primer | Lower primer | Fragment size (bp) | Annealing temperature (°C) | MgCl2 (mmol/L) | |
|---|---|---|---|---|---|
| GAPDH | 5′-GTG AAG GTC GGA GTC AAC G-3′ | 5′-GGT GAA GAC GCC AGT GGA CTC-3′ | 299 | 55 | 1 |
| MCP-1 | 5′-AGT CTC TGC CGC CCT TCT GTG-3′ | 5′-TGC TGC TGG TGA TTC TTC TAT-3′ | 167 | 54 | 1 |
| M-CSF | 5′-GCT CCC TGC TGT TGT TGG TCT-3′ | 5′-TTG GCG GTG TTA TCT CTG AAG-3′ | 265 | 56 | 0.75 |
| GM-CSF | 5′-CAG CCA CTA CAA GCA GCA CT-3′ | 5′-AGG GGA TGA CAA GCA GAA AG-3′ | 113 | 52 | 1 |
| ICAM-1 | 5′-CGA CTG GAC GAG AGG GAT TGT-3′ | 5′-ATT ATG ACT GCG GCT GCT ACC-3′ | 290 | 58 | 0.75 |
| VCAM-1 | 5′-GAC CTT CAT CCC TAC CAT TG-3′ | 5′-ACT TCC TTT CTG CTT CC-3′ | 372 | 53 | 1 |
| IL-1β | 5′-TAC AGT GGC AAT GAG GAT GAC-3′ | 5′-AAG ATG AAG GGA AAG AAG GTG-3′ | 251 | 56 | 1 |
| IL-6 | 5′-ATG TGT GAA AGC AGC AAA GAG-3′ | 5′-TCC AAA AGA CCA GTG ATG ATT-3′ | 134 | 51 | 1 |
| TNF-α | 5′-CCC TGA GTC CTG CCA TCT AAC-3′ | 5′-AGC CAA GCC TGT AAA GTC TGT-3′ | 259 | 55 | 1 |
Statistical Analysis
Proliferative response experiments were performed on at least three umbilical cords, with each value being measured in triplicate. Results are expressed as mean ± standard error of the mean. Statistical analysis was carried out using two-way analysis of variance with time of incubation or concentration of agents and treatment as factors. Statistical significance was achieved if P was < 0.05. In cases of interaction between the factors, one-factor analysis of variance was used at one level of the other factor. Data were analyzed using the Statview 4.0 software (Abacus Concepts, Inc., Berkeley, CA).
Results
As shown in Figure 1 ▶ , IVIg (Sandoglobulin) significantly inhibited EC proliferation, as assessed by [3H]thymidine incorporation. The inhibition was time dependent and highly effective during the first 6 hours of treatment. Between 6 and 30 hours, 10 mg/ml IVIg had no effect any longer, whereas other concentrations induced, at a lower rate, an inhibition of thymidine incorporation. After 30 hours of incubation, despite the presence of IVIg, inhibition partly disappeared, except at the highest IVIg concentration. A similar pattern of inhibition of the proliferative response of HUVECs was also observed with two other sources of IVIg, namely, Gammagard and Endobulin (Figure 2) ▶ . The inhibitory effect of all of the Ig preparations studied was dose dependent (Figures 1 and 2) ▶ ▶ . Gammagard and Endobulin were more potent inhibitors than Sandoglobulin (Figure 2) ▶ .
Figure 1.

Kinetics of the inhibitory effect of IVIg on EC proliferation. IVIg added to the culture medium for various periods of time inhibited [3H]thymidine incorporation in a dose- and time-dependent manner. Maximal inhibition was attained at 6 hours when low concentration of IVIg was used, or after 30-hour incubation for higher concentrations, and partly disappeared afterwards, except for 40 mg/ml IVIg. Time effect, P = 0.001; group effect, P = 0.001 (n = 6 cords).
Figure 2.

Comparative effect of different sources of IVIg on EC proliferation. Sandoglobulin, Gammagard, and Endobulin added to the culture medium for 24 hours inhibited [3H]thymidine incorporation in a dose-dependent manner. Gammagard and Endobulin were more potent inhibitors than Sandoglobulin. Group effect: P = 0.034 (n = 4 cords).
The number of live cells was significantly reduced by IVIg in a dose-dependent manner, as assessed by trypan blue dye exclusion (Figure 3) ▶ . When cells were incubated in the presence of 20 or 30 mg/ml IVIg, but not with 40 mg/ml IVIg, the live cell number was always higher after 48 hours than after 24 hours of incubation, a feature that reflects the partial removal of inhibition of thymidine incorporation seen after 30 hours. The reduction of cell number is also partly due to the death of the cells that increased with IVIg concentration, which, however, did not exceed 8% at the end of the treatment of cells with IVIg (duration of treatment effect, P = 0.004; concentration effect, P = 0.001; n = 3). When cell viability was assessed by FACScan analysis (Figure 4) ▶ , the number of dead cells as determined by propidium iodide uptake did not exceed 6%, whatever the time of incubation and/or the concentration of agonists (duration of treatment effect, P = 0.45; group effect, P = 0.38; n = 3). Therefore, the significant decrease in the number of live cells and the small number of dead cells during the entire period of treatment with IVIg indicated that the inhibition of proliferation of cells is associated with a blockade of cell division rather than only being due to cell death. To check whether this inhibitory effect of IVIg was not simply due to the presence of high amounts of nonspecific protein, 40 mg/ml of human albumin instead of IVIg was added to the culture medium for 48 hours. Thymidine incorporation remained similar to control under such conditions (data not shown).
Figure 3.

Inhibitory effect of IVIg on EC proliferation. As assessed by trypan blue exclusion, the number of live cells was significantly reduced by IVIg in a dose-dependent manner. Nevertheless, 20 or 30 mg/ml of IVIg had less effect when added for 48 hours than for 24 hours. The number of dead cells slightly increased with IVIg concentration but did not exceed 8% at the end of the incubation period. Live cells: time effect, P = 0.001; concentration effect, P = 0.001; dead cells: time effect, P = 0.004; concentration effect, P = 0.001 (n = 3 cords).
Figure 4.

FACScan analysis of cell viability after IVIg treatment. The number of dead cells as determined by propidium iodide uptake did not exceed 6%, whatever the time of incubation and/or the concentration of agonists. Each curve represents a typical experiment. For each condition, the ratio of dead cells to live cells (mean ± standard error of the mean) is given above the graph. Duration of treatment effect, P = 0.4527; group effect, P = 0.3841 (n = 3 cords).
Figure 5 ▶ illustrates the effect of removal of IVIg from culture medium on EC proliferation. After a 24- or a 48-hour incubation period of cells with IVIg, [3H]thymidine incorporation was monitored at 0, 24, or 48 hours after IVIg withdrawal. When cells were incubated for 24 hours with 20 and 30 mg/ml IVIg (Figure 5A) ▶ , the inhibition of cell proliferation disappeared and the uptake of thymidine was fully restored 24 hours after IVIg had been withdrawn. However, when IVIg was used at a 40 mg/ml concentration, a 24-hour washout was unable to overcome the inhibition, which was lifted if recovery was pursued for another 24-hour period. When IVIg was used for 48 hours at the concentration of 20 or 30 but not 40 mg/ml (Figure 5B) ▶ , thymidine incorporation was partially restored at the time of IVIg withdrawal (t = 0), as already seen in Figure 1 ▶ . The inhibition level was then 1.5- to 2-fold less pronounced than just after a 24-hour stimulation. Twenty four hours after IVIg removal, the uptake of thymidine was fully recovered, whatever the concentrations of IVIg.
Figure 5.

Effect of IVIg washout on EC proliferation inhibition. After a 24-hour (A) or 48-hour (B) incubation period of cells with IVIg, [3H]thymidine incorporation was monitored at 0, 24, or 48 hours after IVIg was withdrawn. When cells were incubated for 24 hours (A) with 20 and 30 mg/ml IVIg, but not with 40 mg/ml, the uptake of thymidine was fully restored 24 hours after IVIg had been withdrawn. When IVIg was used for 48 hours (B), the uptake of thymidine was fully recovered 24 hours after IVIg removal. After a 24- or 48-hour IVIg incubation: time effect, P = 0.001; group effect, P = 0.001 (n = 3 cords).
To determine whether the inhibitory effect of IVIg was dependent on their F(ab′)2 or Fc fragments, ECs were incubated for 24 hours with purified F(ab′)2 or Fc fragments of IVIg. Both F(ab′)2 and Fc significantly inhibited the [3H]thymidine uptake in a similar manner (Table 2) ▶ . Figure 6 ▶ shows the effect of TNF-α and IL-1β on cell proliferation. After 24-hour incubation, TNF-α significantly reduced [3H]thymidine incorporation in a dose-dependent manner, whereas IL-1β was fully effective at a dose as low as 0.5 ng/ml. However, for both cytokines, the maximum inhibitory effect was twofold less pronounced than the effect induced by 40 mg/ml IVIg (44.7 ± 1.8, 43.2 ± 3.9, and 80.6 ± 2% inhibition induced by TNF-α, IL-1β, and IVIg, respectively; n = 3). HUVECs were then either coincubated with a mixture of TNF-α or IL-1β and IVIg, prepared just before addition onto the cells (Figure 6 ▶ , lane A), or were pretreated for 15 minutes with TNF-α or IL-1β alone, before the addition of IVIg into the culture medium (Figure 6 ▶ , lane B). Under both conditions, IVIg plus cytokines led to a more efficient inhibition of cell proliferation than IVIg alone (group effect, P = 0.0012; n = 3), but no difference was seen depending on the way in which IVIg was added to the cytokines.
Table 2.
Effect of F(ab)′2 and Fc Fragments of IVIg on [3H]Thymidine Incorporation by HUVEC Cells in Culture (n = 6 Cords)
| IVIg | F(ab′)2 | Fc | ||||
|---|---|---|---|---|---|---|
| 0.20 mmol/L (30 mg/ml) | 0.26 mmol/L (40 mg/ml) | 0.20 mmol/L | 0.26 mmol/L | 0.20 mmol/L | 0.26 mmol/L | |
| CPM (% of control) | 31.59 ± 1.50 | 18.50 ± 0.47 | 46.22 ± 2.87* | 41.80 ± 2.88* | 41.84 ± 3.57* | 41.37 ± 0.68* |
*No significant difference among effects. Treatment effect: P = 0.0001. CPM: count per minute.
Figure 6.

TNF-α- and IL-1β-induced inhibition of EC proliferation. Both cytokines inhibited [3H]thymidine incorporation in a dose-dependent manner. The maximum inhibitory effect was twofold less pronounced than the IVIg effect. When HUVECs were coincubated with a mixture of TNF-α or IL-1β and IVIg prepared just before addition onto the cells (A columns) or were pretreated for 15 minutes with TNF-α or IL-1β alone before the addition of IVIg into the culture medium (B columns), IVIg plus cytokines led to a more efficient inhibition of cell proliferation than IVIg alone (group effect, P = 0.0012; n = 3), but no difference was seen depending on the way in which IVIg was added to the cytokines (P = 0.082; n = 3).
The expression of mRNA for GAPDH, ICAM-1, and VCAM-1 is shown in Figure 7 ▶ . In concentrations ranging from 1 to 40 mg/ml, IVIg had no significant effect on the synthesis of mRNA encoding ICAM-1 and VCAM-1. Both IL-1β (0.5 and 50 ng/ml) and TNF-α (0.5 and 50 ng/ml) induced a synthesis of ICAM-1 and VCAM-1 mRNA in a dose-dependent manner. Again, IL-1β was efficient at doses as low as 0.5 ng/ml. As compared with control, IL-1β (50 ng/ml) and TNF-α (50 ng/ml) increased twofold the expression of VCAM-1 and ICAM-1 mRNA. Although IVIg had no substantial effect on the basal level of expression of ICAM-1 and VCAM-1, IVIg (40 mg/ml) significantly down-regulated to control values the expression of VCAM-1 and ICAM-1 mRNA induced by IL-1β or TNF-α.
Figure 7.

Expression of mRNA encoding VCAM-1 and ICAM-1 adhesion molecules. IL-1β and TNF-α induced an overexpression of mRNA coding for VCAM-1 (B) and ICAM-1 (C) in a dose-dependent manner. Although IVIg alone had no significant effect on this synthesis, it down-regulated the mRNA expression induced by IL-1β or TNF-α. The intensities of the cDNA bands for each protein were normalized to the GAPDH band intensities (A). Experiments were run five times for each cell extract, with cell lines coming from three to five different umbilical cords.
We then examined the effect of IVIg on levels in ECs of mRNA encoding the chemokines (MCP-1, M-CSF, and GM-CSF) and proinflammatory cytokines (TNF-α, IL-6, and IL-1β). At rest, the expression of mRNA coding for the chemokines (Figure 8) ▶ and cytokines (Figure 9) ▶ of ECs was minimal. IVIg had little or no effect on the basal level of chemokines and proinflammatory cytokines. As shown in Figures 7 and 8 ▶ ▶ , IL-1β and TNF-α significantly increased the levels of all chemokines and cytokines studied. IVIg at 40 mg/ml down-regulated the expression of the chemokines MCP-1, M-CSF, and GM-CSF and of the cytokines TNF-α, IL-6, and IL-1β, induced by IL-1β and TNF-α.
Figure 8.

Expression of mRNA encoding MCP-1, M-CSF, and GM-CSF chemokines. IL-1β and TNF-α induced an overexpression of mRNA coding for MCP-1 (A), M-CSF (B), and GM-CSF (C) in a dose-dependent manner. Although IVIg alone had no significant effect on this synthesis, it down-regulated the mRNA expression induced by IL-1β or TNF-α. The intensities of the cDNA bands for each protein were normalized to the GAPDH band intensities depicted in Figure 7A ▶ .
Figure 9.
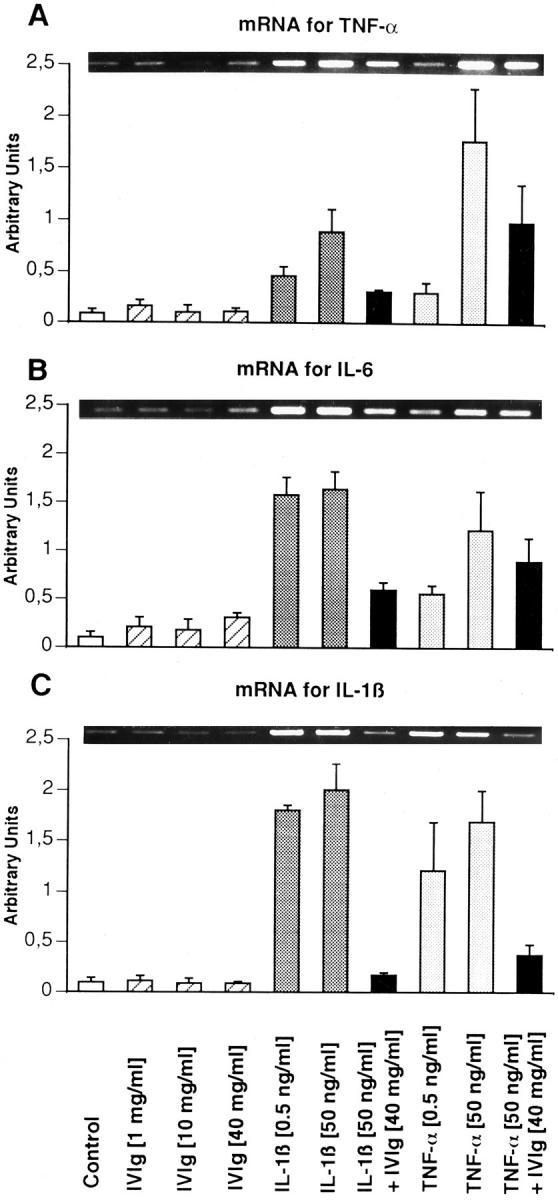
Expression of mRNA encoding proinflammatory cytokines TNF-α, IL-6, and IL-1β. IL-1β and TNF-α induced an overexpression of mRNA coding for TNF-α (A), IL-6 (B), and IL-1β (C) in a dose-dependent manner. Although IVIg alone had no significant effect on this synthesis, it down-regulated the mRNA expression induced by IL-1β or TNF-α. The intensities of the cDNA bands for each protein were normalized to the GAPDH band intensities depicted in Figure 7A ▶ .
Discussion
Our results provide an insight into the critical events underlying the immunoregulatory function of IVIg in diseases in which severe inflammation of vascular tissue is a hallmark of the pathology. Using HUVECs as target cells, we show in the present study that IVIg from different commercial sources modulates the function of ECs. IVIg inhibited EC proliferation in a dose- and time-dependent manner. It also down-regulated the TNF-α- or IL-1β-induced expression of mRNA encoding major adhesion molecules, chemokines, and proinflammatory cytokines, which are significantly implicated in the leukocyte recruitment observed in several inflammatory diseases. 4
The observed inhibitory effect of IVIg on EC proliferation, as assessed by [3H]thymidine incorporation and cell enumeration, was not merely due to a high concentration of protein, because human albumin at 40 mg/ml had no effect on the thymidine uptake by the cells. The inhibitory effect of IVIg was also reversible, in a dose- and time-dependent manner. At 48 hours of culture of ECs in the presence of IVIg however, there was an abolition of the inhibitory effect of IVIg, and the cell number progressively increased. Although the mechanism underlying this escape of cells from the antiproliferative action of IVIg after 48 hours is not clear, the phenomenon reflects the clinical picture after therapy with IVIg: a transient decrease of leukocyte count followed by a recovery in the count has been observed in volunteers infused with IVIg. 37 The drop in the number of live cells and the small increase in the number of dead cells clearly indicated that the inhibition of cell proliferation induced by IVIg is associated with an arrest of the cell cycle at the G0/G1 phase, rather than only being due to a mortality of the cells. Therefore, the changes in osmolarity provoked by the presence of sugar as a stabilizing agent and the acidic pH used to prevent the precipitation of commercial IVIg preparations had little effect on HUVEC viability. The mechanisms involved in the cell cycle arrest and mortality of cells remain unknown. We dissected the role of the variable region of Ig (F(ab′)2 fragments) and the constant Fc portion of Ig in the antiproliferative effect of IVIg. We found that both F(ab′)2 and Fc were able to inhibit significantly, in a similar level, the proliferation of HUVECs. Although the underlying mechanisms involved in the analogous effect observed with both F(ab′)2 and Fc portions of Ig are not clear as yet, a receptor-mediated mechanism cannot be ruled out. In vivo, it is most likely that both F(ab′)2 and Fc portions of Igs are involved in the immunomodulatory functions. 38
The endothelium plays a central role in the immunopathology of several vascular disorders in many inflammatory conditions such as Kawasaki disease, Wegener’s granulomatosis, or vasculitides in which use of IVIg has been shown to be beneficial (for reviews, see Refs. 23, 39, and 40). It may act either as a target for injury or by encouraging the development of lesions because of its anatomical position and physiological function. Bound anti-EC antibodies in Kawasaki disease have the potential to mediate EC injury and lysis via either complement or by more subtle changes in EC functions. It is well established that the onset of inflammation provokes the expression of adhesion molecules on ECs. 4,7,41,42 As the basal level of expression of adhesion molecules, chemokines, and cytokines by HUVECs under the experimental conditions used in this study is low (Figures 7 through 9) ▶ ▶ ▶ , we have induced the expression of the adhesion molecules ICAM-1 and VCAM-1; the chemokines MCP-1, M-CSF, and GM-CSF; and the proinflammatory cytokines TNF-α, IL-1β, and IL-6 by two proinflammatory cytokines, IL-1β and TNF-α, to evaluate the effect of IVIg on the expression of these molecules. The expression of TNF-α by HUVECs has been a matter of debate. 43-48 Our observation suggests that TNF-α induces an autocrine up-regulation of the mRNA expression of TNF-α. The role of such an autocrine production of TNF-α on EC function is, however, not known. IVIg significantly down-regulated the cytokine-induced expression of all these molecules involved in an inflammatory process, although IVIg alone, in concentrations ranging from 1 to 40 mg/ml, had no effect. One of the reasons for this blocking effect may be the anti-cytokine nature of IVIg, as IVIg contains antibodies directed against cytokines. 49,50 However, we did not observe any difference in the proliferative responses of HUVECs, either when IVIg was mixed with TNF-α or IL-1β before addition to the cells or when it was added after a preincubation of cells with the cytokines for 15 minutes. We therefore believe that the inhibitory effect of IVIg on the cytokine-induced activation of HUVECs may not be exclusively due to neutralization of TNF-α or IL-1β by antibodies directed against these cytokines. Another possibility is that IVIg contains soluble receptors that “soak up” the TNF-α or IL-1β stimulators that abrogate the stimulatory effects of these cytokines. Further, it is also possible that in IVIg, anti-idiotypic antibodies bearing internal images of molecules that mimic such receptors may exist. The molecular mechanisms involved in the modulation of the function of ECs warrant further investigation.
Over the last decade, IVIg has been used in the treatment of several posttransplantation complications including cytomegalovirus (CMV)-associated diseases, including transplant arteriosclerosis, obliterative bronchiolitis, CMV-induced myocarditis, CMV-associated graft versus host disease, CMV-retinitis, and CMV-hepatitis (acute and chronic). 51-55 An immune-mediated vascular disease is associated with acute and chronic allograft rejection, in which T-lymphocyte activation and the release of interferon-γ by activated T cells in turn induce the expression of class II molecules on vascular ECs. Underlying mechanisms point out a relationship between CMV infection and inducible expression of human leukocyte antigens on allograft vascular ECs and on specific lymphocyte subpopulations that infiltrate the graft. 56 Such activation of ECs results in an enhanced cellular adhesion and in lymphocyte-mediated tissue damage. Our results suggest that some of the anti-inflammatory effects observed in patients treated with IVIg are related to a decreased ability of ECs to proliferate and to a down-regulation of the expression of molecules involved in the onset and progression of inflammation. Although the relevance of these findings in in vivo situation needs further investigation, a possible beneficial effect of IVIg lies in the control of EC activation in the inflammatory conditions, because generation of microvessels is a salient feature of neoplasia and inflammation. 57,58
Acknowledgments
We acknowledge the technical assistance of Emmanuelle Bonnin and the skillful help of Michel Paing in photography.
Footnotes
Address reprint requests to Dr. Jacques Chevalier, Immunopathologie Rénale et Vasculaire, INSERM U 430, Hôpital Broussais, 96 Rue Didot, 75674 Paris Cedex 14, France. E-mail: chevalier@hbroussais.fr.
Supported in part by grant 97043 from Rhône-Poulenc Rorer (Paris, France) and the Central Laboratory of the Swiss Red Cross (Bern, Switzerland). CX was supported by 24-month fellowship no. 3Q4/044 from the French Foreign Ministry and the French Embassy in Beijing, China.
This work was presented in abstract form at the European Renal Association, European Dialysis and Transplant Association Annual Congress, Geneva, Switzerland, September 21–24, 1997, and at the American Society of Nephrology Meeting in San Antonio, TX, November 2–5, 1997.
CX’s present address is Department of Histology and Embryology, Shanghai Second Medical University, Shanghai, China. NL’s present address is Laboratory of Experimental Surgery, Department of Clinical Surgery, Federal University of Santa Catarina, Florianopolis, Brazil.
References
- 1.Carlos TM, Harlan JM: Leukocyte-endothelial adhesion molecules. Blood 1994, 84:2068-2101 [PubMed] [Google Scholar]
- 2.Demuth K, Myara I, Moatti N: Biologie de la cellule endothéliale et athérogenèse. Ann Biol Clin 1995, 53:171-189 [PubMed] [Google Scholar]
- 3.Matsushima KV, Oppenheim JJ: Interleukin-8 and MCAF: novel inflammatory cytokines induced by TNF and Il-1. Cytokines 1989, 1:2-10 [DOI] [PubMed] [Google Scholar]
- 4.Brady HR: Leukocyte adhesion molecules and kidney diseases. Kidney Int 1994, 45:1285-1300 [DOI] [PubMed] [Google Scholar]
- 5.Munro JM, Pober JS, Cotran RS: Tumor necrosis factor and interferon-γ induce distinct patterns of endothelial activation and associated leukocyte accumulation in skin of Papio anubis. Am J Pathol 1989, 135:121-133 [PMC free article] [PubMed] [Google Scholar]
- 6.Osborn LR, Hession R, Tizard R, Vassala C, Luhowskyj S, Chi-Rosso G, Lobb R: Direct expression cloning of vascular cell adhesion molecule-1 and cytokine induced endothelial protein that binds lymphocytes. Cell 1989, 248:415-423 [DOI] [PubMed] [Google Scholar]
- 7.Scholz D, Devaux B, Hirche A, Potzsch B, Kropp B, Schaper W, Schaper J: Expression of adhesion molecules is specific and time-dependent in cytokine-stimulated endothelial cells in culture. Cell Tissue Res 1996, 284:415-423 [DOI] [PubMed] [Google Scholar]
- 8.Rajavashisth TB, Analibi A, Territo MC, Berliner JA, Navab M, Fogelman AM, Lusis AJ: Induction of endothelial cell expression of granulocyte and macrophage colony stimulating factors by modified low density lipoproteins. Nature 1990, 344:254-257 [DOI] [PubMed] [Google Scholar]
- 9.Jirik FR, Podor TJ, Hirano T, Kishimoto T, Loskutoff DJ, Carson DA, Lotz M: Bacterial lipopolysaccharide and inflammatory mediators augment IL-6 secretion by human endothelial cells. J Immunol 1989, 142:144-147 [PubMed] [Google Scholar]
- 10.Dwyer JM: Manipulating the immune system with immune globulin. N Engl J Med 1992, 326:107-116 [DOI] [PubMed] [Google Scholar]
- 11.Kazatchkine MD, Dietrich G, Hurez V, Ronda N, Bellon B, Rossi F, Kaveri SV: V region-mediated selection of autoreactive repertoires by intravenous immunoglobulin (IVIg). Immunol Rev 1994, 139:79-107 [DOI] [PubMed] [Google Scholar]
- 12.Imbach P, Barandun S, d’Apuzzo V, Baumgartner C, Hirt A, Morell A, Rossi E, Schoni M, Vest M, Wagner HP: High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet 1981, i:1228-1230 [DOI] [PubMed] [Google Scholar]
- 13.McIntyre EA, Linch DC, Macey MG, Newland AC: Successful response to intravenous immunoglobulin in autoimmune hemolytic anemia. Br J Haematol 1985, 60:387-388 [DOI] [PubMed] [Google Scholar]
- 14.Oda H, Honda A, Sugita K: High dose intact IgG infusion in refractory autoimmune hemolytic anemia (Evans syndrome). J Pediatr 1985, 107:744-746 [DOI] [PubMed] [Google Scholar]
- 15.Lalezari P, Korshidi M, Petrosova M: Autoimmune neutropenia of infancy. J Pediatr 1986, 109:764-769 [DOI] [PubMed] [Google Scholar]
- 16.McGuire WA, Yang HH, Bruno E, Brandt J, Briddell R, Coates TD, Hoffman R: Treatment of antibody-mediated pure red-cell aplasia with high-dose intravenous gammaglobulin. N Engl J Med 1987, 317:1004-1008 [DOI] [PubMed] [Google Scholar]
- 17.Blanchette VS, Imbach P, Andrew M: A prospective randomized trial of intravenous immunoglobulin G, oral prednisolone and intravenous anti-D in childhood acute idiopathic thrombocytopenic purpura. Lancet 1994, 344:703-707 [DOI] [PubMed] [Google Scholar]
- 18.Van der Meché FGA, Smith PIM, : Dutch Guillain-Barré Study Group: A randomized trial comparing intravenous immune globulin and plasma exchange in Guillain-Barré syndrome. N Engl J Med 1992, 326:1123-1129 [DOI] [PubMed] [Google Scholar]
- 19.Hughes R: Plasma exchange versus intravenous immunoglobulin for Guillain-Barré syndrome. Jpn J Apheresis 1996, 15:S12. [DOI] [PubMed] [Google Scholar]
- 20.Gajdos P, Outin H, Elkharrat D, Brunel D, de Rohan-Chabot P, Raphael JC, Goulon M, Goulon-Goeau C, Morel E: High-dose intravenous gammaglobulin for myasthenia gravis. Lancet 1984, i:406-407 [DOI] [PubMed] [Google Scholar]
- 21.Sultan Y, Kazatchkine MD, Maisonneuve P, Nydegger UE: Anti-idiotypic suppression of autoantibodies to Factor VIII (antihaemophilic factor) by high-dose intravenous gammaglobulin. Lancet 1984, ii:765-768 [DOI] [PubMed] [Google Scholar]
- 22.Dalakas MC, Illa I, Dambrosia JM, Soueidan SA, Stein DP, Otero C, Dinsmore ST, McCrosky S: A controlled trial of high-dose intravenous immune globulin infusions as treatment for dermatomyositis. N Engl J Med 1993, 329:1993-2000 [DOI] [PubMed] [Google Scholar]
- 23.Möller G: Immunoglobulin treatment: mechanisms of action. Immunol Rev. 1994, 139:1-188 [Google Scholar]
- 24.Kazatchkine MD, Kaveri SV: Therapeutic immunomodulation with normal polyspecific immunoglobulin G (intraveinous immunoglobulin, IVIg). Capra JD Zanetti M eds. The Antibodies. 1997, :pp 141-173 Harwood Academic Publishers, Amsterdam [Google Scholar]
- 25.Jayne DR, Lockwood CM: Pooled intravenous immunoglobulin in the management of systemic vasculitis. Adv Exp Med Biol 1993, 336:469-472 [DOI] [PubMed] [Google Scholar]
- 26.Lockwood CM: New treatment strategies for systemic vasculitis: the role of intravenous immune globulin therapy. Clin Exp Immunol 1996, 1:77-82 [PubMed] [Google Scholar]
- 27.Fehr J, Hofmann V, Kappeler U: Transient reversal of thrombocytopenia in idiopathic thrompocytopenic purpura by high-dose intravenous γ globulin. N Engl J Med 1982, 306:1254-1258 [DOI] [PubMed] [Google Scholar]
- 28.Basta M, Fries LF, Frank MM: High doses of intravenous Ig inhibit in vitro uptake of C4 fragments onto sensitized erythrocytes. Blood 1991, 77:376-380 [PubMed] [Google Scholar]
- 29.Basta M, Dalakas MC: High-dose intravenous immunoglobulin exerts its beneficial effect in patients with dermatomyositis by blocking endomysial deposition of activated complement fragments. J Clin Invest 1994, 94:1729-1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andersson UG, Björk L, Skansen-Saphir U, Andersson JP: Down-regulation of cytokine production and interleukin-2 receptor expression by pooled human IgG. Immunology 1993, 79:211-216 [PMC free article] [PubMed] [Google Scholar]
- 31.Ruiz de Souza V, Carreno MP, Kaveri SV, Ledur A, Sadeghi H, Cavaillon JM, Kazatchkine MD, Haeffner-Cavaillon N: Selective induction of interleukin-1 receptor antagonist and interleukin-8 in human monocytes by normal polyspecific IgG (intravenous immunoglobulins). Eur J Immunol 1995, 25:1267-1273 [DOI] [PubMed] [Google Scholar]
- 32.Stohl W: Cellular mechanisms in the in vitro inhibition of pokeweed mitogen-induced B cell differentiation by immunoglobulin for intravenous use. J Immunol 1986, 136:4407-4413 [PubMed] [Google Scholar]
- 33.Abe J, Kotzin BL, Meissner C, Melish ME, Takahashi M, Fulton D, Romagne F, Malissen B, Leung DYM: Characterization of T cell repertoire changes in acute Kawasaki disease. J Exp Med 1993, 177:791-796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kaveri SV, Vassilev T, Hurez V, Lengagne R, Lefranc C, Cot S, Pouletty P, Glotz D, Kazatchkine MD: Antibodies to a conserved region of HLA Class I molecules, capable of modulating CD8 T cell-mediated function, are present in pooled normal immunoglobulin for therapeutic use. J Clin Invest 1996, 97:865-869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dietrich G, Varela FJ, Hurez V, Bouanani M, Kazatchkine MD: Selection of the expressed B cell repertoire by infusion of normal immunoglobulin G in a patient with autoimmune thyroiditis. Eur J Immunol 1993, 23:2945-2950 [DOI] [PubMed] [Google Scholar]
- 36.Kaveri SV, Dietrich G, Ronda N, Hurez V, Ruiz de Souza V, Rowen D, Vassilev T, Kazatchkine MD: Suppression of autoimmunity through manipulation of immune network with normal immunoglobulin G. Gergely J eds. Progress in Immunology. 1993, :pp 643-649 Springer Verlag, Berlin [Google Scholar]
- 37.Schnorf J, Arnet B, Burek-Kozlowska A, Gennari K, Rohner R, Spath PJ, Spycher MO: Laboratory parameters measured during infusion of immunoglobulin preparations for intravenous use and related tolerability. Kazatchkine MD Morell A eds. Intravenous Immunogobulin: Research and Therapy. 1996, :pp 312-313 Parthenon, New York [Google Scholar]
- 38.Fridman WH, Teillaud JL, Sautès C: Role of Fc receptors in immunomodulation by intravenous immunoglobulin. Kazatchkine MD Morell A eds. Intravenous Immunogobulin: Research and Therapy. 1996, :pp 73-80 Parthenon, New York [Google Scholar]
- 39.Savage COS, Cooke SP: The role of endothelium in systemic vasculitis. J Autoimmun 1993, 6:237-249 [DOI] [PubMed] [Google Scholar]
- 40.Leung DYM: Kawasaki disease. Curr Opin Rheumatol 1993, 5:41-50 [DOI] [PubMed] [Google Scholar]
- 41.Wellicome SM, Thornhill MH, Pitzalis C, Thomas DS, Lanchbury JSS, Panayi GS, Haskard DO: A monoclonal antibody that detects a novel antigen on endothelial cells that is induced by tumor necrosis factor, IL-1, or lipopolysaccharide. J Immunol 1990, 144:2558-2565 [PubMed] [Google Scholar]
- 42.Gasic AC, McGuire G, Krater S, Farhood AI, Goldstein MA, Smith CW, Entman ML, Taylor AA: Hydrogen peroxide pretreatment of perfused canine vessels induces ICAM-1 and CD18-dependent neutrophil adherence. Circulation 1991, 84:2154-2166 [DOI] [PubMed] [Google Scholar]
- 43.Pober JS, Cotran RS: Immunologic interactions of T lymphocytes with vascular endothelium. Adv Immunol 1991, 50:261-301 [DOI] [PubMed] [Google Scholar]
- 44.Toborek M, Hennig B: Is endothelial cell autocrine production of tumor necrosis factor a mediator of lipid-induced endothelial dysfunction? Med Hypotheses 1996, 47:377-382 [DOI] [PubMed] [Google Scholar]
- 45.Cendan JC, Moldawer LL, Souba WW, Copeland EM, Lind S: Endotoxin-induced nitric oxide production in pulmonary artery endothelial cells is regulated by cytokines. Arch Surg 1994, 129:1296-1300 [DOI] [PubMed] [Google Scholar]
- 46.Kahaleh MB, Zhou S: Induction of tumor necrosis factor (TNF) synthesis by endothelial cells upon exposure to r-TNF. Arthritis Rheum 1989, 32:S124 [Google Scholar]
- 47.Nagura H, Ohtani H: Expression of major histocompatibility class-II antigens by vascular endothelial cells leads to amplified immunoinflammatory processes. Acta Histochem Cytochem 1992, 25:653-660 [Google Scholar]
- 48.Ueta E, Yoneda K, Yamamoto T, Osaki T: Influence of SNN-6010, an elemental diet, on the generation of cytokines, NO, and chemiluminescence from leukocytes and umbilical vein endothelial cells in man. Br J Clin Pharmacol 1997, 44:385-391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abe Y, Horiuchi A, Miyake M, Kimura S: Anti-cytokine nature of human immunoglobulin: one possible mechanism of the clinical effect of intravenous therapy. Immunol Rev 1994, 139:5-19 [DOI] [PubMed] [Google Scholar]
- 50.Hansen MB, Svenson M, Abbell K, Yasukawa K, Diamant M, Bendt-zen K: Influence of interleukin-6 autoantibodies on IL-6 binding to cellular receptors. Eur J Immunol 1995, 25:348-354 [DOI] [PubMed] [Google Scholar]
- 51.Sullivan KM, Kopecky KG, Jocom J, Fischer L, Buckner CD, Meyers JD, Counts JW, Bowden RA, Petersen FB, Witherspoon RP, Budinger MD, Schwartz RS, Applebaum FR, Clift RA, Hansen JA, Sanders JE, Thomas ED, Storb R: Immunomodulatory and anti-microbial efficacy of intravenous immunoglobulin in bone marrow transplantation. N Engl J Med 1990, 323:705-709 [DOI] [PubMed] [Google Scholar]
- 52.Bass EB, Powe NR, Goodman SN, Graziano SL, Griffiths RI, Kickcler TS, Wingard JR: Efficacy of immune globulin in preventing complications of bone marrow transplantation: a meta-analysis. Bone Marrow Transplant 1993, 12:273-282 [PubMed] [Google Scholar]
- 53.Glowacki LS, Smaill FM: Use of immune globulin to prevent symptomatic cytomegalovirus disease in transplant recipients: a meta-analysis. Clin Transplant 1994, 8:10-18 [PubMed] [Google Scholar]
- 54.Messori A, Rampazzo R, Scroccaro G, Martini N: Efficacy of hyperimmune anti-cytomegalovirus immunoglobulins for the prevention of cytomegalovirus infection in recipients of allogeneic bone marrow transplantation: a meta-analysis. Bone Marrow Transplant 1994, 13:163-167 [PubMed] [Google Scholar]
- 55.Wittes JT, Kelly A, Plante KM: Meta-analysis of CMVIG studies for the prevention and treatment of CMV infection in transplant patients. Transplant Proc 1996, 6:17-24 [PubMed] [Google Scholar]
- 56.Knight DA, Waldman WJ: Cytokine-mediated induction of endothelial adhesion molecules and histocompatibility leukocyte antigen expression by cytomegalovirus-activated T cells. Am J Pathol 1994, 148:105-119 [PMC free article] [PubMed] [Google Scholar]
- 57.Folkman J: Tumor angiogenesis. Adv Cancer Res 1985, 43:175-203 [DOI] [PubMed] [Google Scholar]
- 58.Furcht LT: Critical factors controlling angiogenesis: cell products, cell matrix, and growth factors. Lab Invest 1986, 55:505-509 [PubMed] [Google Scholar]