

Abstract
Advanced glycation end products (AGEs) have been implicated in the chronic complications of diabetes mellitus and have been reported to play an important role in the pathogenesis of Alzheimer’s disease. In this study, we examined the immunohistochemical localization of AGEs, amyloid β protein (Aβ), apolipoprotein E (ApoE), and tau protein in senile plaques, neurofibrillary tangles (NFTs), and cerebral amyloid angiopathy (CAA) in Alzheimer’s disease and other neurodegenerative diseases (progressive supranuclear palsy, Pick’s disease, and Guamanian amyotrophic lateral sclerosis/Parkinsonism-dementia complex). In most senile plaques (including diffuse plaques) and CAA from Alzheimer’s brains, AGE and ApoE were observed together. However, approximately 5% of plaques were AGE positive but Aβ negative, and the vessels without CAA often showed AGE immunoreactivity. In Alzheimer’s disease, AGEs were mainly present in intracellular NFTs, whereas ApoE was mainly present in extracellular NFTs. Pick’s bodies in Pick’s disease and granulovacuolar degeneration in various neurodegenerative diseases were also AGE positive. In non-Alzheimer neurodegenerative diseases, senile plaques and NFTs showed similar findings to those in Alzheimer’s disease. These results suggest that AGE may contribute to eventual neuronal dysfunction and death as an important factor in the progression of various neurodegenerative diseases, including Alzheimer’s disease.
Glucose and other reducing sugars react nonenzymatically with protein amino groups to initiate a post-translational modification process known as nonenzymatic glycosylation. 1-3 This reaction proceeds from reversible Schiff bases to stable, covalently bonded Amadori rearrangement products. 2 Once formed, the Amadori products undergo further chemical rearrangement reactions to form irreversibly bound advanced glycation end products (AGEs), 1,2 which are considered to play an important role in the pathogenesis of the chronic complications of diabetes mellitus. 1,2 AGEs are a heterogeneous group of structures with those that have already been identified, including pyrraline, pentosidine, crossline, and carboxymethyl-lysine (CML). 4
Alzheimer’s disease (AD) is the most common cause of dementia in Western countries and Japan. Pathologically, AD is characterized by the presence of neurofibrillary tangles (NFTs) and senile plaques, the major constituents of which are tau protein and amyloid β protein (Aβ), respectively. Aβ is deposited in a variety of plaques and in the cerebral vessels as cerebral amyloid angiopathy (CAA). The deposition of Aβ peptides is thought to be an early and causative event in the pathogenesis of AD and increases markedly during progression of the disease, leading in turn to the generation of NFTs and finally neuronal death. 5
It has recently been demonstrated that AGEs can be identified immunohistochemically in both senile plaques and NFTs from AD. 6 Glycation of tau, in addition to hyperphosphorylation, appears to enhance the formation of paired helical filaments, 7,8 and glycation of Aβ enhances its aggregation in vitro. 9 Plaques and NFTs are not found only in AD brains but also in the brains of patients with other neurodegenerative disorders. 5 However, little is known about whether or not AGEs are involved in the pathogenesis of other neurodegenerative diseases.
To investigate the role of AGE modification in AD and other neurodegenerative disorders, we performed immunohistochemical studies using antibodies for AGEs, Aβ, tau, ubiquitin, and apolipoprotein E (ApoE). Our data demonstrated that AGE modification was involved in pathological changes observed in both AD and other neurodegenerative disorders, implying that AGEs may be an important factor in the progression of various neurodegenerative disorders.
Materials and Methods
Subjects and Specimens
Brain tissue specimens were obtained from five pathologically verified cases of AD, three of PSP, three of Pick’s disease, three of Guamanian Parkinsonism-dementia complex (PDC), three of Guamanian amyotrophic lateral sclerosis (ALS), two of Guamanian ALS/PDC, three of diabetes mellitus (DM), and three age-matched controls. Histological sections were prepared from the cerebral cortex (temporal lobe and parietal lobe) and the hippocampus. Except for the three patients with DM, none of the subjects had diabetes. The clinical features of the subjects are summarized in Table 1 ▶ .
Table 1.
Characteristics of the Subjects
| Case | Diagnosis | Sex | Age at autopsy | Duration of disease (years) |
|---|---|---|---|---|
| 1 | AD | F | 64 | 7 |
| 2 | AD | M | 48 | 5 |
| 3 | AD | F | 68 | 8 |
| 4 | AD | M | 45 | 5 |
| 5 | AD | M | 60 | 4 |
| 6 | PSP | M | 68 | 12 |
| 7 | PSP | M | 66 | 10 |
| 8 | PSP | M | 59 | 8 |
| 9 | Pick’s disease | M | 75 | 5 |
| 10 | Pick’s disease | M | 75 | 7 |
| 11 | Pick’s disease | F | 67 | 6 |
| 12 | Guamanian PDC | F | 52 | 6 |
| 13 | Guamanian PDC | M | 56 | 2 |
| 14 | Guamanian PDC | F | 61 | 4 |
| 15 | Guamanian ALS | F | 52 | 2 |
| 16 | Guamanian ALS | M | 60 | 4 |
| 17 | Guamanian ALS | F | 51 | 3 |
| 18 | Guamanian ALS/PDC | M | 62 | 2 |
| 19 | Guamanian ALS/PDC | M | 67 | 1 |
| 20 | DM | F | 57 | |
| 21 | DM | F | 68 | |
| 22 | DM | F | 75 | |
| 23 | control | F | 73 | |
| 24 | control | M | 66 | |
| 25 | control | F | 61 |
AD, Alzheimer’s disease; PSP, progressive supranuclear palsy; PDC, Parkinsonism-dementia complex; ALS, amyotrophic lateral sclerosis; DM, diabetes mellitus.
Antibodies
The rabbit anti-AGE-modified ribonuclease antibody used was described previously. 10 The antibody detected AGE formed in vivo, such as AGE-collagen and AGE-hemoglobin, as well as glucose-derived AGE RNAse, glucose-derived AGE albumin, glucose-derived AGE low-density lipoprotein (LDL), and glucose-derived AGE collagen. 10,11 However, the antibody did not recognize unmodified RNAse, albumin, hemoglobin, LDL, acetyl LDL, or collagen as well as previously reported AGE structures such as 2-furoyl-4(5)-[2-furanyl]-1-H-imidazole (FFI), 1-alkyl-2-formyl-3,4-diglycosyl-pyrroles (AFGP), pyrraline, pentosidine, or CML. 10,11
Monoclonal antibodies against tau and Aβ were established in our laboratory and were designated as GP144 (mouse IgM) and Aβ90/12 (mouse IgG), respectively. These antibodies have been described elsewhere in detail. 12,13 Rabbit anti-ubiquitin antibody and a polyclonal goat anti-ApoE antibody were purchased from Dako (Glostrup, Denmark) and Chemicon International (Temecula, CA), respectively.
Immunohistochemical Staining
Serial paraffin sections were immunostained according to the standard streptavidin-biotin peroxidase technique using a Vectastain ABC elite kit (Vector Laboratories, Burlingame, CA). Autoclaving for 30 minutes was performed before tau and AGE immunocytochemistry to retrieve antigenicity. 14 All sections were treated with 90% formic acid for 5 minutes, and the sections used for AGE staining were also treated with 0.05% proteinase K for 60 minutes. Endogenous peroxidase was inhibited with 0.3% hydrogen peroxidase (H2O2) in methanol for 30 minutes. These sections were also incubated with 10% horse serum (for tau, Aβ, and ubiquitin), 10% rabbit serum (for ApoE), or 10% goat serum (for AGEs) to eliminate nonspecific binding. This was followed by incubation overnight at 4°C with the primary antibodies diluted 1:500 to 1:1000 in 10 mmol/L phosphate-buffered saline (PBS; pH 7.4). The sections were then sequentially incubated with the biotinylated secondary antibody for 60 minutes, with streptavidin-biotin-horseradish peroxidase for 60 minutes, and with 3,3′-diaminobenzidine/H2O2 until the reaction products were visualized (1 to 3 minutes). Then the sections were counterstained with hematoxylin. Specificity was confirmed by 1) applying PBS instead of the primary antibodies or 2) by replacing the primary antibodies with preimmune serum. As an additional control for anti-AGE immunoreactivity, positively stained tissues were subjected to absorption experiments in which anti-AGE antiserum was preincubated with AGE/bovine serum albumin (0.01 to 1 mg/ml) for 1 hour at 37°C, as described previously. 15 For double immunostaining, a combination of the peroxidase-antiperoxidase and alkaline phosphatase techniques was used. The sections were visualized with the Histofine fast blue substrate for alkaline phosphatase staining or the Histofine ACE substrate (Nichirei, Tokyo, Japan) for peroxidase-antiperoxidase staining.
Double-stained sections were prepared sequentially, first with a cell-type-specific antibody, such as Aβ90/12 and GP144, and after visualization the section was incubated with anti-AGE antibody. To confirm the specificity of anti-AGE antibody, additional double staining was performed by applying the primary antibody and secondary antibody in reverse order (first anti-AGE antibody and then the cell-type-specific antibody). These double-stained sections were not counterstained.
Semiquantification of AGE-Positive Structures
The number of senile plaques and cerebral blood vessels reacting with anti-Aβ or AGE antibodies was counted on both serial tissue sections and double-stained sections in three AD patients. In each patient, immunoreactive plaques and vessels were counted in the hippocampus and the parahippocampal gyrus using three photomicrographs (1.3 mm 2 each) at ×100 magnification. NFTs and Pick bodies (in three patients with Pick’s disease) that reacted with anti-tau or AGE antibodies were counted in the same way. Granulovacuolar degeneration granules, stained by hematoxylin and eosin (H&E) or reacted with anti-AGE antibody, were counted at ×400 magnification.
Results
Proteolytic digestion with proteinase K and autoclaving has been shown to expose cross-linked AGE moieties and enhance AGE immunoreactivity in cardiac and renal tissues. 15,16 In the present study, proteinase K pretreatment enhanced AGE immunoreactivity in both senile plaques and NFTs, but no marked improvement was observed in sections stained with other antibodies. Without autoclaving, senile plaques very weakly reacted with anti-AGE antibody, but NFTs were strongly reactive.
Figure 1 ▶ shows a representative AD brain immunostained for Aβ (Figure 1, A and D) ▶ , AGE (Figure 1, B and E) ▶ ▶ , and ApoE (Figure 1, C and F) ▶ . Double immunostaining of AD brains with anti-Aβ and AGE is also shown. In Figure 1G ▶ , the section was incubated with anti-AGE antibody before anti-Aβ antibody, whereas Figure 1H ▶ shows a section incubated with anti-Aβ antibody before anti-AGE antibody. With double staining, Aβ deposits were stained blue and AGEs were stained red, whereas co-localization of AGE and Aβ was stained purple (Figure 1, G and H) ▶ .
Figure 1.
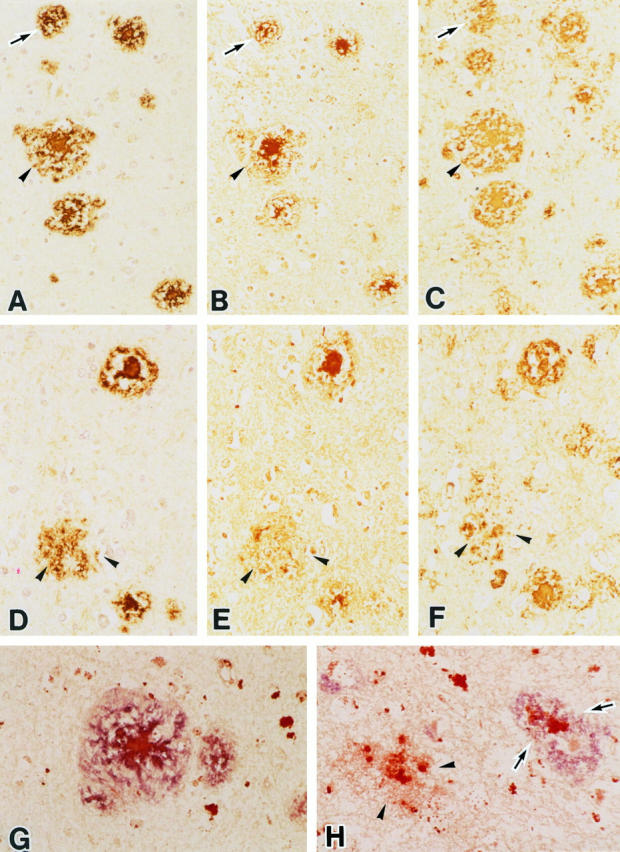
Photomicrographs of serial sections of the hippocampus from an AD brain, immunostained with anti-Aβ (A and D), anti-AGE (B and E), and anti-ApoE (C and F) antibodies. All types of plaques, diffuse plaque (arrowheads, D–F), primitive plaque (arrows, A–C), and classical plaque (arrowheads, A–C) show Aβ, AGE, and ApoE immunoreactivity. G and H: Double-staining studies with anti-Aβ (blue) and anti-AGE (red) antibodies of the hippocampus from an AD brain. G: Representative double staining in a classical plaque. Most senile plaques show both Aβ and AGE immunoreactivity (purple; arrows, H), but some plaques show only AGE immunoreactivity (red; arrowheads, H). Magnification, ×190 (A–C), ×280 (D–F), and ×380 (G and H).
All types of amyloid plaque were positive for the anti-Aβ antibody Aβ90/12, with diffuse plaques, primitive plaques, and classical plaques being all clearly demonstrated (Figure 1, A and D) ▶ .
Adjacent serial sections stained with the anti-AGE antibody also showed various types of amyloid plaques (Figure 1, B and E) ▶ . More than 90% of the Aβ-positive plaques were also stained by the anti-AGE antibody. All types of plaques (diffuse plaques, primitive plaques, and classical plaques) were recognized by anti-AGE (Figure 1, B and E) ▶ . Diffuse plaques are considered to represent an early stage of senile plaque, whereas primitive plaques and classical plaques are thought to represent mature senile plaque. 17 Thus, AGE modification was confirmed to occur in the early stage of plaque development, and the most intense AGE accumulation was observed in the cores of senile plaques (Figure 1, B and G) ▶ . The extent of AGE accumulation paralleled the development of senile plaques, indicating that a gradual and constant increase of AGE accumulation, shown as an increase of AGE immunoreactivity, occurred in AD along with the maturation of senile plaques (Figure 1, B and E) ▶ . The corona showed only weak AGE immunoreactivity (Figure 1, B and G) ▶ .
Most ApoE-positive plaques, including diffuse plaques, also showed AGE immunoreactivity (Figure 1, C and F) ▶ , but there was a subtle difference in the staining patterns of AGE and ApoE in the same plaque. ApoE stained both the corona and the core similarly, whereas AGE stained the core much more strongly than the corona (Figure 1, B and C) ▶ .
Double staining for AGE and Aβ (Figure 1G) ▶ showed that most senile plaques were stained by both antibodies. However, occasional AGE-positive plaques (approximately 5%) were recognized to be Aβ negative or very weakly Aβ positive (Figure 1H) ▶ .
Immunohistochemical studies of patients with various other neurodegenerative disorders revealed that all senile plaques, representing the aging process, observed in these disorders (three cases of Pick’s disease, one case of PSP, one case of DM, and one age-matched control) also reacted with anti-Aβ, anti-ApoE, and anti-AGE antibodies. The staining characteristics were similar to those of AD plaques.
Figure 2 ▶ shows representative staining with anti-Aβ (Figure 2A) ▶ , anti-AGE (Figure 2B) ▶ , and anti-ApoE (Figure 2C) ▶ in serial sections of CAA from an AD brain. Most vessels with CAA were positive for Aβ, AGE, and ApoE. In these vessels, the AGE-positive area corresponded with Aβ deposits, or else the Aβ deposits lay within the AGE-positive region (Figure 2B) ▶ . However, as shown in Figure 2, D–F ▶ , the vessels without amyloid deposits (approximately 10%) were stained for AGE. Among these Aβ-negative and AGE-positive vessels, approximately 30% showed ApoE immunoreactivity. Aβ-negative and AGE-/ApoE-positive vessels were also observed in other neurodegenerative diseases such as Guamanian PDC as well as in DM and control cases. In this study, we did not observe any Aβ-positive, but AGE-negative vessels and AGE immunoreactivity was present in some of the endothelial cells.
Figure 2.

Serial sections of leptomeningeal vessels (A–C) and parenchymal vessels (D–F) in the temporal lobe of an AD brain. These sections were stained with anti-Aβ (A and D), anti-AGE (B and E), and anti-ApoE (C and F) antibodies. Blood vessels with amyloid angiopathy are labeled by both anti-AGE (B) and anti-ApoE (C). Some blood vessels without amyloid deposits are labeled by both anti-AGE (E) and anti-ApoE (F). Magnification, ×125 (A–C) and ×200 (D–F).
Immunohistological studies of NFTs were performed in AD, other neurodegenerative diseases, DM, and control subjects using anti-tau, anti-ubiquitin, and anti-AGE antibodies. NFTs were detected in 14 hippocampal sections of 25 examined. Either intracellular NFTs (I-NFT) or extracellular NFTs (E-NFT) were detected in AD, PSP, Guamanian cases, DM, and controls, but not Pick’s disease. Figure 3 ▶ shows typical immunohistochemical findings with anti-tau (Figure 3, A, D, and G) ▶ , anti-AGE (Figure 3, B, E, and H) ▶ ▶ ▶ , and anti-ApoE (Figure 3, C, F, and I) ▶ ▶ ▶ in serial sections from subjects with AD (Figure 3, A–C) ▶ , Guamanian PDC (Figure 3, D–F) ▶ ▶ ▶ , and a control (Figure 3, G–I) ▶ . In these NFT-positive cases, approximately 20% of I-NFTs were stained with anti-AGE antibody, but E-NFTs were AGE negative or were only weakly stained. The AGE immunoreactivity of E-NFTs was always enhanced by autoclaving, but that of I-NFTs was obvious without autoclaving (Figure 3, B, E, and H) ▶ . In Guamanian cases, numerous I-NFTs and E-NFTs were positive for anti-tau antibody. E-NFTs showed very strong ApoE immunoreactivity, but only AGE was detected in I-NFTs (Figure 3, D–F) ▶ . There were no remarkable differences among Guamanian PDC, ALS, and ALS/PDC in the staining for AGE and ApoE. Control subjects also had NFTs, which were thought to represent the aging process, and these NFTs showed same staining pattern for AGE and ApoE (Figure 3, G–I) ▶ .
Figure 3.

Photomicrographs of serial sections of the hippocampus from an AD patient (A–C), a patient with Guamanian Parkinson-dementia complex (D–F), and a control subject (G–I). Both I-NFTs (arrows) and E-NFTs (arrowheads) are strongly labeled by anti-tau protein antibody (A, D, and G). I-NFTs show definite AGE immunoreactivity (arrows), whereas E-NFTs are virtually negative (arrowheads, B, E, and H). E-NFTs show strong ApoE immunoreactivity (arrowheads), but I-NFTs are negative (arrows, C, F, and I). Magnification, ×380.
In Pick’s disease, parts of the Pick bodies that reacted with anti-tau antibody (approximately 20%) were also labeled by anti-AGE antibody (Figure 4, A and B) ▶ ▶ . Some Pick cells were very weakly AGE positive, but the reaction varied considerably depending on the pretreatment conditions.
Figure 4.
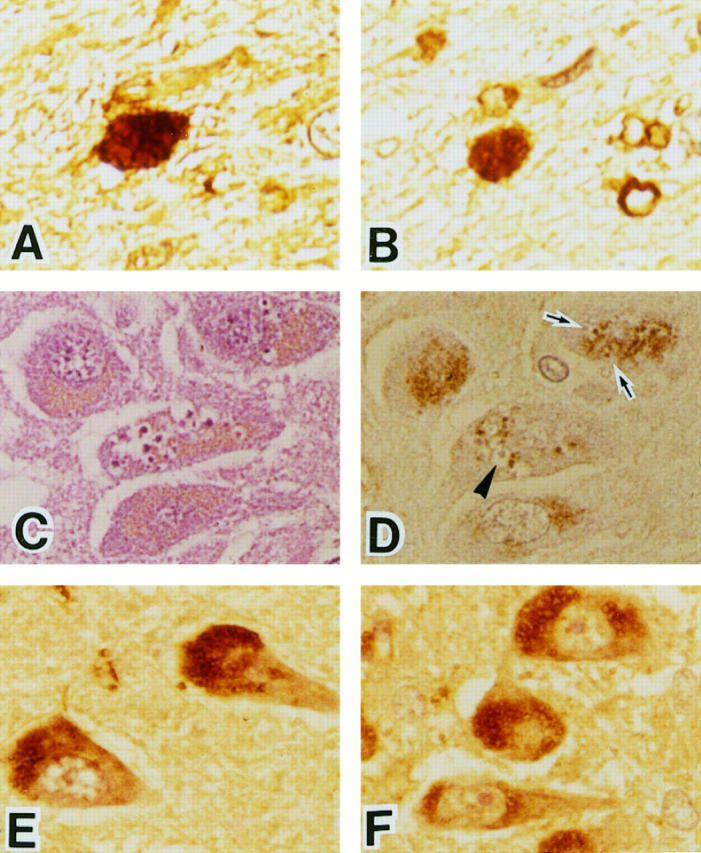
Serial sections of the temporal lobe from a Pick’s disease brain (A and B). The Pick body reacts with anti-tau protein antibody (A). The same structure is labeled by anti-AGE antibody (B). Serial sections of pyramidal neurons in the hippocampus from an AD brain (C and D). Granulovacuolar degeneration is identified by H&E staining (C). Some granules inside the vacuoles are weakly labeled by anti-AGE antibody (arrows, D), but other granules are not labeled (arrowhead, D). Sections of pyramidal neurons in the hippocampus from AD (E) and Guamanian PDC (F). Many AGE-positive granules were observed in pyramidal neurons. Magnification, ×380.
Granulovacuolar degeneration was seen in pyramidal neurons of the hippocampus in all AD and Guamanian cases, one case of PSP, and a control. The vacuoles were generally surrounded by intensely AGE-positive intracellular granules. Most of the granules inside the vacuoles were AGE negative, but some (approximately 15%) were AGE positive (Figure 4, C and D) ▶ .
Immunohistochemical comparison of AGE-positive intracellular granules was performed in hippocampal pyramidal neurons of AD (Figure 4E) ▶ , PSP, Pick’s disease, Guamanian PDC (Figure 4F) ▶ , DM, and control subjects. Numerous AGE-positive granules were present in all of these subjects and showed a characteristic granular pattern. 18 The number and intensity of AGE-immunoreactive neurons in AD and other neurodegenerative disease were similar to those in the control cases.
Discussion
AGE modification was previously reported in senile plaques, NFTs, and cerebral amyloid angiopathy from AD brains. 6 It was also reported that Aβ itself can be glycosylated in vitro, that glycosylated Aβ promotes Aβ aggregation, and that AGE-modified Aβ “seeds” promote the aggregation of soluble Aβ when compared with unmodified seed material. 9 It is an important question whether AGE modification of amyloid plaques is a primary event 19 or just a secondary consequence of Aβ deposition, 20 but this issue remains controversial. For example, Mattson et al suggested that AGE modification is a late secondary event in AD. 20 However, Smith et al suggested that AGE modification could be involved both earlier and later in the pathogenesis of AD. 19
Our immunopathological studies using serial brain sections and double staining provided the following evidence supporting a role of AGE in the pathogenesis of AD: 1) all types of plaques (diffuse, mature, and classical) showed AGE immunoreactivity, 2) AGE was present from an early stage of Aβ accumulation in diffuse plaques, and 3) approximately 5% of plaques stained by the anti-AGE antibody were very weakly stained or not stained by anti-Aβ. This was the most interesting observation from the present study and may raise two possibilities. First, AGE modification could occur secondary to Aβ deposition, but some Aβ deposits are too small to detect. The second possibility is that another protein than Aβ protein may be glycated in AGE-positive, Aβ-negative plaques. In this case, which protein undergoes glycation instead? Amyloid plaques are composed of a variety of proteins that are designated as amyloid-associated proteins. It seems possible that the protein undergoing glycation may be one of these amyloid-associated proteins or else some unidentified protein. With respect to ApoE, we have already reported the presence of ApoE-positive but Aβ-negative plaque-like formation in AD. 21 In addition, Dickson et al reported that the staining patterns of AGE and ApoE were similar, suggesting a possible relationship between AGE and ApoE in AD. 22 Tabaton et al, however, failed to detect glycated Aβ and glycated ApoE using immunoprecipitation techniques and concluded that other amyloid-associated proteins may be candidates for glycation. 23 As mentioned earlier, another possibility is that some novel or unidentified protein may be glycated. In fact, Schmidt et al very recently reported that there were novel plaque-like deposits stained by monoclonal antibodies to a 100-kd protein without Aβ deposits in AD brains. 24 Considering these reports, additional studies are needed to identify the protein undergoing glycation in amyloid plaques.
Regarding NFTs and AGEs, we found that our anti-AGE reacted mainly with I-NFTs in AD brains, whereas E-NFTs were weakly positive. In contrast, anti-ApoE reacted strongly with E-NFTs but weakly with I-NFTs. These findings were confirmed in the other neurodegenerative disorders studied and in our age-matched controls. Contrary to our observation, Dickson et al reported that AGE immunoreactivity is intense in E-NFTs but weak in I-NFTs. 22 As AGEs are a heterogeneous group of structures, 4 the difference between our findings and Dickson’s may reflect differences in the antibodies used or differences in the method of tissue preparation. AGE determinants were localized in paired helical filaments by immunoelectron microscopy, 8 and tau protein isolated from AD brains was shown to be glycated at its tubulin-binding domain. 25 Furthermore, it has been shown that AGE tau induces neuronal oxidative stress that results in increased expression of cytokine gene and amyloid precursor protein as well as release of Aβ peptide. 26
AGEs are nonenzymatic products that are considered to be relatively indigestible. It has been reported that AGEs are degraded in lysosomes after being taken up via particular cell-surface binding sites, the receptors for AGE or RAGE, and macrophage scavenger receptor, whereas proteolytic degradation releases small AGE peptides or fragments likely to contain variable portions of AGE moieties. 4 Considering these observations, our findings could also raise the possibility that weak AGE immunoreactivity in E-NFT compared with I-NFT may be related to receptor-mediated degradation. In this respect, we demonstrated that some of the granules in granulovacuolar degeneration were AGE positive in AD and other neurodegenerative diseases. Granulovacuolar degeneration is derived from autophasic mechanism involving lysosomes, 27 and our findings may provide some evidence suggesting that receptor-mediated degradation takes place in AD and other neurodegenerative disorders.
AGEs are involved in pathological structures, Pick bodies, and ballooned neurons in Pick’s disease, 28 substantia nigra neurons in Parkinson’s disease, and cortical Lewy bodies in diffuse Lewy body disease. 29 Reactive oxygen intermediates can be generated during AGE modification, 30 and the binding of AGEs to specific receptors can also generate oxidative stress 31 as well as the production of pro-inflammatory cytokines. 32 In various neurodegenerative diseases other than AD, AGEs were present in some neuropathological structures, including senile plaques, NFTs, Pick bodies, and granulovacuolar degeneration. Under these circumstances, oxidative stress may be induced by AGE generation and receptor-mediated reactions. Therefore, AGE may contribute to eventual neuronal dysfunction and death as an important factor in the progression of various neurodegenerative diseases as well as AD.
Footnotes
Address reprint requests to Dr. Nobuyuki Sasaki, Department of Neuropsychiatry, Sapporo Medical University, South 1, West 16, Chuo-ku, Sapporo, 060-8543 Japan.
Supported by grants-in-aid for scientific research 0761097 and 09470204 from the Ministry of Education, Science, and Culture of Japan.
References
- 1.Monnier VM, Cerami A: Nonenzymatic browning in vivo: possible process for aging of long-lived proteins. Science 1981, 211:491-493 [DOI] [PubMed] [Google Scholar]
- 2.Brownlee M, Vlassara H, Cerami A: Nonenzymatic glycosylation and the pathogenesis of diabetic complications. Ann Intern Med 1984, 101:527-537 [DOI] [PubMed] [Google Scholar]
- 3.Ledl F, Schleicher E: New aspects of the Maillard reaction in foods and in the human body. Angew Chem Int Ed Engl 1990, 6:565-706 [Google Scholar]
- 4.Vlassara H, Bucala R, Striker L: Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest 1994, 70:138-151 [PubMed] [Google Scholar]
- 5.Selkoe DJ: Normal and abnormal biology of the β-amyloid precursor protein. Annu Rev Neurosci 1994, 17:489-517 [DOI] [PubMed] [Google Scholar]
- 6.Smith MA, Taneda S, Richey PL, Miyata S, Yan SD, Stern D, Sayre LM, Monnier VM, Perry G: Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci USA 1994, 91:5710-5714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ledesma MD, Bonay P, Colaço C, Avila J: Analysis of microtuble-associated protein tau glycation in paired helical filaments. J Biol Chem 1994, 269:21614-21619 [PubMed] [Google Scholar]
- 8.Yan SD, Chen X, Schmidt AM, Brett J, Godman G, Zou YS, Scott CW, Caputo C, Frappier T, Smith MA, Perry G, Yen SH, Stern D: Glycated tau protein in Alzheimer disease: a mechanism for induction of oxidant stress. Proc Natl Acad Sci USA 1994, 91:7781-7791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A: Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA 1994, 91:4766-4770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Makita Z, Vlassara H, Cerami A, Bucala R: Immunochemical detection of advanced glycosylation end products in vivo. J Biol Chem 1992, 267:5133-5138 [PubMed] [Google Scholar]
- 11.Makita Z, Vlassara H, Rayfield E, Cartwright K, Friedman E, Rodby R, Cerami A, Bucala R: Hemoglobin-AGE: a circulating marker of advanced glycosylation. Science 1992, 258:651-653 [DOI] [PubMed] [Google Scholar]
- 12.Takamaru Y, Obara T, Fukatsu R, Tsuzuki K, Aizawa Y, Fujii M, Kobayashi M, Gotoda T, Yanagihara R, Garruto R, Oguma K, Takahata N: Monoclonal antibodies against paired helical filament in Alzheimer’s disease and Parkinsonism-dementia complex of Guam. Alzheimer’s and Parkinson’s Diseases, vol 1. Basic, Clinical, and Therapeutic Aspects of Alzheimer’s and Parkinson’s Diseases. New York, Plenum Press, 1990, pp 203–206
- 13.Tsuzuki K, Fukatsu R, Takamaru Y, Fujii N, Takahata N: Potentially amyloidogenic fragment of 50 kd and intracellular processing of amyloid precursor protein in cells cultured under leupeptin. Brain Res 1994, 659:213-220 [DOI] [PubMed] [Google Scholar]
- 14.Shin RW, Iwaki T, Kitamoto T, Tateishi J: Hydrated autoclave pretreatment enhances tau immunoreactivity in formalin-fixed normal and Alzheimer’s disease brain tissues. Lab Invest 1991, 64:693-702 [PubMed] [Google Scholar]
- 15.Nishino T, Horii Y, Shiiki H, Yamamoto H, Makita Z, Bucala R, Dohi K: Immunohistochemical detection of advanced glycosylation end products within the vascular lesions and glomeruli in diabetic nephropathy. Hum Pathol 1995, 26:308-313 [DOI] [PubMed] [Google Scholar]
- 16.Nakamura Y, Horii Y, Nishino T, Shiiki H, Sakaguchi Y, Kagoshima T, Dohi K, Makita Z, Vlassara H, Bucala R: Immunohistochemical localization of advanced glycosylation endproducts in coronary atheroma and cardiac tissue in diabetes mellitus. Am J Pathol 1993, 143:1649-1656 [PMC free article] [PubMed] [Google Scholar]
- 17.Ikeda S, Yanagisawa N, Allsop D, Glenner GG: Early senile plaques in Alzheimer’s disease demonstrated by histochemistry, immunocytochemistry, and electron microscopy. Hum Pathol 1990, 21:1221-1226 [DOI] [PubMed] [Google Scholar]
- 18.Li JJ, Surini M, Catsicas S, Kawashima E, Bouras C: Age-dependent accumulation of advanced glycosylation end products in human neurons. Neurobiol Aging 1995, 16:69-76 [DOI] [PubMed] [Google Scholar]
- 19.Smith MA, Sayre LM, Vitek MP, Monnier VM, Perry G: Early AGEing and Alzheimer’s. Nature 1995, 374:316. [DOI] [PubMed] [Google Scholar]
- 20.Mattson MP, Carney JW, Butterfield DA: A tombstone in Alzheimer’s? Nature 1995, 373:481. [DOI] [PubMed] [Google Scholar]
- 21.Aizawa Y, Fukatsu R, Takamaru Y, Tsuzuki K, Chiba H, Kobayashi K, Fujii N, Takahata N: Amino-terminus truncated apolipoprotein E is the major species in amyloid deposits in Alzheimer’s disease-affected brains: a possible role for apolipoprotein E in Alzheimer’s disease. Brain Res 1997, 768:208-214 [DOI] [PubMed] [Google Scholar]
- 22.Dickson DW, Sinicropi S, Yen S, Ko L, Mattiace LA, Bucala R, Vlassara H: Glycation and microglial reaction in lesions of Alzheimer’s disease. Neurobiol Aging 1996, 17:733-743 [DOI] [PubMed] [Google Scholar]
- 23.Tabaton M, Perry G, Smith M, Vitek M, Angelini G, Dapino D, Garibaldi S, Zaccheo D, Odetti P: Is amyloid β-protein glycated in Alzheimer’s disease? Neuroreport 1997, 8:907-909 [DOI] [PubMed] [Google Scholar]
- 24.Schmidt ML, Lee VM-Y, Forman M, Chiu T-S, Trajanowski JQ: Monoclonal antibodies to a 100-kd protein reveal abundant Aβ-negative plaques throughout gray matter of Alzheimer’s disease brains. Am J Pathol 1997, 151:69-80 [PMC free article] [PubMed] [Google Scholar]
- 25.Ledesma MD, Bonay P, Avila J: τ protein from Alzheimer’s disease patients is glycated at its tubulin-binding domain. J Neurochem 1995, 65:1658-1664 [DOI] [PubMed] [Google Scholar]
- 26.Yan SD, Yan SH, Chen X, Fu J, Chen M, Kuppusamy P, Smith MA, Perry G, Godman GC, Nawroth P, Zweier JL, Stern D: Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid β-peptide. Nature Med 1995, 1:693-699 [DOI] [PubMed] [Google Scholar]
- 27.Okamoto K, Hirai S, Iizuka T, Yanagisawa T, Watanabe M: Reexamination of granulovacuolar degeneration. Acta Neuropathol (Berl) 1991, 82:340-345 [DOI] [PubMed] [Google Scholar]
- 28.Kimura T, Ikeda K, Takamatsu J, Miyata T, Sobue G, Miyakawa T, Horiuchi S: Identification of advanced glycation end products of the Maillard reaction in Pick’s disease. Neurosci Lett 1996, 219:95-98 [DOI] [PubMed] [Google Scholar]
- 29.Castellani R, Smith MA, Richey PL, Perry G: Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res 1996, 737:195-200 [DOI] [PubMed] [Google Scholar]
- 30.Sakurai T, Tsuchiya S: Superoxide production from nonenzymatically glycated protein. FEBS Lett 1988, 236:406-410 [DOI] [PubMed] [Google Scholar]
- 31.Araki N, Higashi T, Mori T, Shybayama R, Kawabe Y, Kodama T, Takahashi K, Shichiri M, Horiuchi S: Macrophage scavenger receptor mediates the endocytic uptake and degradation of advanced glucation end products of the Maillard reaction. Eur J Biochem 1995, 227:408-415 [DOI] [PubMed] [Google Scholar]
- 32.Meda L, Cassatella MA, Szendrei GI, Otvos L, Baron P, Villalba M, Ferrari D, Rossi F: Activation of microglial cells by β-amyloid protein and interferon-γ. Nature 1995, 374:647-650 [DOI] [PubMed] [Google Scholar]