

Abstract
β-Sheet proteins are particularly resistant to denaturation by sodium dodecyl sulfate (SDS). Here we compare unfolding of two β-sandwich proteins TNfn3 and TII27 in SDS. The two proteins show different surface electrostatic potential. Correspondingly, TII27 unfolds below the critical micelle concentration via the formation of hemimicelles on the protein surface, whereas TNfn3 only unfolds around the critical micelle concentration. Isothermal titration calorimetry confirms that unfolding of TII27 sets in at lower SDS concentrations, although the total number of bound SDS molecules is similar at the end of unfolding. In mixed micelles with the nonionic detergent dodecyl maltoside, where the concentration of monomeric SDS is insignificant, the behavior of the two proteins converges. TII27 unfolds more slowly than TNfn3 in SDS and follows a two-mode behavior. Additionally TNfn3 shows inhibition of SDS unfolding at intermediate SDS concentrations. Mutagenic analysis suggests that the overall unfolding mechanism is similar to that observed in denaturant for both proteins. Our data confirm the kinetic robustness of β-sheet proteins toward SDS. We suggest this is related to the inability of SDS to induce significant amounts of α-helix structure in these proteins as part of the denaturation process, forcing the protein to denature by global rather than local unfolding.
INTRODUCTION
Protein-detergent interactions highlight the multifarious ways in which proteins respond to changes in their environment (1). Ionic detergents can denature proteins by strong binding to charged and hydrophobic side chains at millimolar concentrations, unlike chemical denaturants such as guanidinium chloride or urea, which are only effective at molar concentrations, presumably due to weak binding to the protein backbone (2). These interactions are of great practical interest, since the majority of industrial enzyme production (both in terms of value and volume) is targeted to the detergent sector, primarily for laundering and dishwashing (3). Generally water-soluble proteins only interact with ionic detergents, since these proteins are not hydrophobic enough to compete with nonionic detergents' strong tendency to self-association. Nonionic detergents are mainly relevant for water-soluble proteins in so far as they can form mixed micelles with ionic detergents and thus attenuate the latter's denaturing properties (4). Ionic detergents typically bind to proteins through a mixture of electrostatic and hydrophobic interactions (5,6). The strong affinity of detergents for proteins means that detergent molecules can bind both as monomers and as micelles (7–9). Typically binding of monomeric detergents only leads to local changes in protein conformation, while global and cooperative unfolding occurs around the critical micelle concentration (cmc) (10). Unfolding in detergent usually leads to a denatured but not unstructured state that may contain significant amounts of α-helical structure (11).
While the thermodynamics of detergent binding to proteins has been studied intensely since the 1960s, unfolding kinetics—which can be used to deduce the mechanism(s) of unfolding—have received comparably less attention. Using S6 as a model system, we have previously shown that mixed α/β proteins unfold very rapidly (subsecond timescales) in sodium dodecyl sulfate (SDS) (12,13) and that the α-helix structures are unfolded first. All-α proteins such as myoglobin and Acyl CoA binding protein, also unfold very rapidly (K. K. Andersen and D. E. Otzen, unpublished observations). However, β-sheet proteins appear to be more robust. A thought-provoking study by Manning and Colón (14) established that oligomeric β-sheet proteins are particularly resistant to unfolding in SDS, possibly due to the higher content of nonlocal interactions; in addition, the authors suggested that there might be general selection for kinetically stable β-structures since partially unfolded β-sheet proteins are particularly susceptible to aggregation. We speculate that the increased sensitivity of all-α and α/β proteins toward SDS denaturation could be that α-helices, but not β-sheets, provide a good attack point for SDS micelles due to the ease of solvating independent folding units such as α-helices in an apolar environment. Thus the unfolding of α-helix-containing proteins but not that of β-sheet proteins should be distorted by SDS compared to the intrinsic unfolding behavior in GdmCl. To test this hypothesis, we have analyzed the response to SDS of two structurally similar β-sandwich proteins, TII27 (the 27th immunoglobulin domain from human cardiac titin) and TNfn3 (the third fibronectin type III domain from human tenascin). The secondary structure of both proteins contains exclusively β-sheet, turns and loops (Fig. 1 B). Both proteins contain a single Trp residue and no internal disulfide bridges, making them very appropriate model systems for folding studies. The equilibrium and kinetic unfolding behavior of both proteins have been extensively studied by Clarke and co-workers (15,16). Both proteins unfold under equilibrium conditions in denaturant (urea or guanidinium chloride, GdmCl) according to a two-state transition with free energies of around 7 kcal/mol (15,17). TII27 but not TNfn3 folds via a kinetic intermediate, but both proteins fold according to the nucleation-condensation mechanism (18). The two proteins differ in their electrostatic surface potential. TII27 shows several areas with positive potential where SDS' sulfate headgroup could bind, some with neighboring hydrophobic areas that could interact with the detergent alkyl chain (Fig. 1 A). TNfn3 has fewer positively charged sites, which moreover are surrounded by negatively charged residues. This suggests that TII27 but not TNfn3 should be able to attract monomeric SDS. We show that, although the two proteins behave in a broadly similar manner, there is indeed a difference in the binding interaction of SDS with the two proteins, which translates into differences in unfolding kinetics. Importantly, the two proteins only show a small increase in α-helicity upon addition of SDS; in addition, their response to mutations is similar in SDS and denaturant, consistent with our hypothesis.
FIGURE 1.

(A) Electrostatic potential of TII27 and TNfn3, illustrated using PyMOL. (B) Structures of TII27 and TNfn3, highlighting the side chains mutated in this study.
MATERIALS AND METHODS
Materials
Tris(hydroxymethyl)aminomethane (Tris) and SDS were from AppliChem (Darmstadt, Germany); n-dodecyl-β-d-maltoside (DDM) and thrombin, bovine high activity, were from Calbiochem (San Diego, CA); tris(2-carboxymethyl)phospine hydrochloride (TCEP) and pyrene were from Sigma-Aldrich (St. Louis, MO). All chemicals were molecular biology grade. Wild-type and mutant TII27 and TNfn3 were prepared as described (15,18,19). Both wild-type TII27 and all TII27 mutants have Thr-42 replaced by Ala, Ala-78 by Thr, and a C-terminal Arg-Ser extension (20). TNfn3 has a C-terminal Gly-Leu extension, which makes it more stable in comparison with the 90 amino acid long published protein sequence (17). Mutants of TII27 and TNfn3, as well as expression vectors for the wild-type proteins, were generously provided by Anette Steward and Jane Clarke, Cambridge University, Cambridge, UK.
Spectroscopic measurements
All experiments were conducted in 10 mM Tris pH 8.0 at 25°C using 11.7 μM TNfn3 or 7.7 μM TII27 unless otherwise stated. Solutions were left to equilibrate in detergent with 10 mM Tris, pH 8 at 25°C for at least 1 h. To avoid dimerization of TII27 via the single Cys-46, all experiments with TII27 were performed in the presence of 0.15 mM TCEP (millimolar TCEP concentrations led to precipitation in the presence of SDS).
Steady-state fluorescence measurements were performed on an LS-55 luminescence spectrometer (Perkin-Elmer Instruments, Wellesley, MA), using an excitation wavelength of 295 nm and measuring the emission between 315 and 375 nm. Measurements were performed in a 10-mm quartz cuvette (Hellma, Müllheim, Germany) as an average of three scans with a slit width of 10 nm and a scanning speed of 300 nm/min.
Far-ultraviolet (UV) circular dichroism (CD) spectra were recorded in a 1-mm quartz cuvette on a JASCO J-715 spectropolarimeter (Jasco Spectroscopic, Hachioji City, Japan) equipped with a Jasco PTC-423S temperature control unit. Wavelength scans were recorded in the wavelength range of 195–250 nm with a band width of 2 nm and a scanning speed of 50 nm/min. Five accumulations were averaged to yield the final spectrum. Background contributions from the buffer were subtracted.
Kinetics
For fluorescence studies, unfolding kinetics were studied either by manual mixing using the LS-55 instrument or on an SX18MV stopped-flow microanalyzer (Applied Photophysics, Leatherhead, UK) in a thermostatically controlled sample-handling unit, depending on the rapidity of the reaction. On the LS-55 instrument, samples were excited at 295 nm and the fluorescence intensities at 350 nm and 330 nm for TNfn3 and TII27, respectively, were followed until the signals reached a plateau. On the SX18MV instrument, proteins and detergent were mixed 1:10 to a final protein concentration of 1.2 μM, samples were excited at 280 nm, and the emission above 320 nm was monitored using a cut-off filter. Control experiments showed that the rate constants obtained by the two methods were identical within error.
For CD studies, samples were manually mixed and measured at 10-min intervals (average of five scans), 5-min intervals (average of three scans), and 1.5-min intervals (one scan) depending on the rapidity of the unfolding reaction.
The observed kinetics were fitted to single exponential functions with (TII27) and without (TNfn3) linear drift, leading to a first-order rate constant kobs. The linear drift was incorporated to account for photobleaching of the Trp fluorophore in TII27, which was presumably more accessible (and hence less protected from light and oxidative reactions) in TII27 than in TNfn3. The drift was insignificant in comparison to the amplitude change caused by unfolding.
Pyrene interactions
The ratio of the intensity of pyrene emission at 372.5 and 383.5nm (I3/I1) can be used to evaluate the polarity of the environment, in which pyrene is solubilized. We used pyrene emission to determine if there was binding of hemimicelles to the surface of the proteins. Pyrene was excited at 335 nm and emission between 350 and 440 nm monitored. A stock solution of 20 μM pyrene in ethanol was made and added to the samples to a final concentration of ∼0.05 μM. Each sample was recorded as the average of three emission scans.
Isothermal titration calorimetry (ITC)
The calorimetric measurements were conducted on a VP-ITC (MicroCal, Northampton, MA). The reference cell was filled with water and in a typical experiment, the sample cell was loaded with a solution of 8.7 and 69.8 μM TNfn3 or 32.3–114.8 μM TII27. The cell solution was titrated with aliquots of 2.5–4 μl of 99 mM SDS in 10 mM Tris, pH 8. All experiments were done at 22°C, where SDS demicellization is practically athermal (21). Therefore the enthalpic contribution from demicellization of SDS upon injection can be neglected in data analysis. The obtained heat signals from the ITC were integrated using the Origin software supplied by MicroCal.
Calculation of electrostatic potential
The electrostatic surface potential at pH 8 was calculated for the two Protein Data Bank files 1TEN (22) and 1TIT (23) to investigate possible interaction sites with SDS. pKa values for every titratable side chain were calculated at pH 8 by the programs PDB2PQR and Propka (24,25). Electrostatic potential values were computed by the program MEAD. MEAD uses the Poisson-Boltzman equation to determine the electric potential, and incorporate details of the atomic structure into the placement of charges and dielectric boundaries (26). Finally the electrostatic map was visualized by the program PyMOL (DeLano Scientific, San Carlos, CA).
Thermal denaturation
Thermal scans were conducted on a Cary Eclipse Fluorescence spectophotometer (Varian, Mulgrave, Australia) using a 10-mm quartz cuvette. Scans were performed from 20 to 100°C using an excitation wavelength of 295 nm and an emission wavelength of 350 nm. The scan rate was 1°C/min and slit widths of 10 nm were used. Thermal scans were fitted to a thermal transition as previously described (27).
Data analysis
The observed rate constants kobs vary in a characteristic manner with SDS concentration. From these characteristics, unfolding kinetics can be grouped into three different modes (12,13):
Mode 1
At low SDS concentrations (3–10 mM SDS), the rate constant for unfolding increases steeply and stabilizes at a plateau around 10 mM SDS. This behavior can be described by a minimal scheme (Scheme 1) involving rapid binding (complete within the dead time of stopped-flow mixing, ∼5 ms) and subsequent global unfolding:
 |
where P denotes the protein, SDS a predominantly spherical SDS micelle (below 100 mM SDS (28)), K1 the dissociation constant (K1 = [P][SDS]/[P*:SDS]), P*:SDS the protein:SDS complex, kunfmode 1 the rate constant for unfolding in mode 1, and P*:SDSmode 1 the unfolded protein:SDS complex. The magnitude of the observed rate constant kobs depends on the fraction of the protein that is complexed with SDS within the dead time.
Mode 2
At intermediate SDS concentrations (10–100 mM) before the onset of mode 3, there is a decline in the unfolding rate constants for Tnfn3. Scheme 2 models this by invoking binding of additional SDS micelles to the protein:SDS complex P*:SDS. This is analogous to uncompetitive inhibition in enzyme catalysis, whereas additional binding to unbound protein corresponds to competitive inhibition. Formation of a dead-end complex between unbound protein and SDS would not lead to a decline in unfolding rates at high SDS concentrations. In this model the dead-end complex SDS:P*:SDSmode 2 is described with a dissociation constant Kinh:
 |
For simplicity, it is assumed that there is no difference between the detergent micelles that bind to P and to P*SDS, and that only one micelle binds to P*:SDS. This means that the micellar concentration in both binding steps is described by the term ([SDS] − cuc) in units of monomers, where [SDS] is the total SDS concentration and cuc is the critical unfolding concentration. It is assumed that unfolding only proceeds above cuc. Note that Scheme 2 is merely an attempt to present a possible model, and the data do not allow for clear conclusions about the stoichiometry of the dead-end complex.
Mode 3
Above ∼100 mM SDS, the micelles gradually transform from mainly spherical to predominantly cylindrical, and the unfolding rate constants increase markedly in a manner that is linear in a double-logarithmic plot (28). This mode can also be described by rapid binding, followed by unfolding by the cylindrical micelles. Scheme 3 represents a synthesis of all three different unfolding regimes in SDS:
 |
We use two different equilibrium constants K1 and K2 as well as two different protein-detergent complexes P*:SDS and P#:SDS to emphasize the difference in micelle structure. K2 cannot be determined directly due to the lack of an obvious saturation effect in mode 3 and is merely added for formal reasons. The three different unfolding regimes lead to the following (Eq. 1):
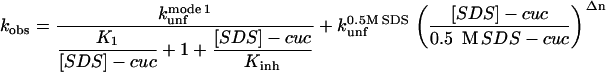 |
(1) |
where 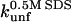 is the unfolding rate constant at 500 mM SDS, since 500 mM SDS is used as a reference concentration for unfolding in mode 3; Δn is a constant, which can be interpreted as the degree of cooperativity of mode 3. In cases where there is no inhibition (mode 2), the following (Eq. 2) comprises mode 1 + 3:
is the unfolding rate constant at 500 mM SDS, since 500 mM SDS is used as a reference concentration for unfolding in mode 3; Δn is a constant, which can be interpreted as the degree of cooperativity of mode 3. In cases where there is no inhibition (mode 2), the following (Eq. 2) comprises mode 1 + 3:
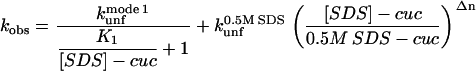 |
(2) |
The details of the models are not essential for the discussion of our results, but provide an opportunity to estimate key kinetic parameters for comparative purposes. In addition, they serve to emphasize the complexity of protein unfolding in detergent.
RESULTS
Equilibrium unfolding in SDS reveals hemimicellar unfolding of TII27 but micellar unfolding of TNfn3
To compare the unfolding of the two β-sheet proteins and provide a basis for an analysis of their unfolding mechanism in SDS, we start by describing their response to SDS under equilibrium conditions. Upon incubation with SDS, the fluorescence intensity maximum for both TII27 and TNfn3 shift from around 330 nm, typical of a buried Trp residue, to around 350 nm, indicative of increased exposure accompanying unfolding (Fig. 2 A). The fluorescence intensity of TII27 is reduced, and that of TNfn3 increased, upon unfolding. This difference probably reflects subtle conformational details relating to the immediate environment of the Trp residue in the native and denatured state.
FIGURE 2.

(A) Fluorescence and (B) Far-UV CD spectra of TII27 and TNfn3 in the absence and presence of SDS. (C) Equilibrium denaturation of TII27 and (D) TNfn3 followed by fluorescence, CD, and changes in the melting temperature as a function of SDS concentration. For both proteins, tm has been divided by 50 to be compatible with the fluorescence axis.
Far-UV CD spectra show characteristic β-sheet signatures in the absence of SDS. When SDS is added, there is a general increase in ellipticity (Fig. 2 B). Deconvolution of the spectra using the CDSSTR algorithm (29) suggest only a very slight increase in α-helicity for TNfn3 (from 7 to 9%) and a slightly larger increase for TII27 (from 5 to 17%), accompanied by ∼10% decrease in β-sheet structure and 5% increases in β-turn and unordered structure. The relatively poor accuracy of deconvolution algorithms makes it unfeasible to conclude that there are substantial differences between the conformational changes that the two proteins undergo upon unfolding in SDS.
Conformational changes associated with the stepwise addition of SDS were followed by fluorescence and far-UV CD. For TNfn3, fluorescence and CD both show a major transition around 4.8 mM SDS, which is very close to the cmc of SDS under these buffer conditions, namely 5 mM (27) (Fig. 2 D). However, for TII27, both techniques reveal that the transition occurs around 3 mM, and is complete before the cmc is reached (Fig. 2 C). Furthermore, the transition is somewhat broader for TII27, indicating a less cooperative process that involves a smaller number of SDS molecules than for TNfn3. To obtain further information about the consequences of binding of SDS at low concentrations, we also carried out thermal scans of the two proteins followed by fluorescence. For TII27, there was essentially no change in melting temperature tm until 2.5 mM SDS, after which tm dropped steeply and no thermal transition was observed above 3 mM SDS (Fig. 2 C). TNfn3 showed a constant tm until around 4 mM SDS, and no transitions were observed above 4.5 mM SDS (Fig. 2 D). These data all suggested that monomeric SDS interacted to a greater extent with TII27 than with TNfn3, with consequent earlier unfolding.
To confirm this, we followed the change in fluorescence emission of the hydrophobic probe pyrene as a function of SDS concentration in the presence of the two proteins (Fig. 3). Pyrene's fluorescence changes as it goes from a hydrophilic (aqueous) to a hydrophobic (micellar) environment, making it a good probe for the formation of micelles as well as hemimicelles formed on protein surfaces (30). In the absence of protein, the fluorescence ratio reaches a plateau around 4–5 mM, in satisfactory correspondence with the previously determined cmc of 5 mM (27). Pyrene fluorescence is not affected by the presence of TNfn3, but when TII27 is included, the fluorescence ratio rises more steeply at low SDS concentrations (0.6–3 mM) than in the absence of SDS. This suggests that SDS molecules form hydrophobic clusters on the TII27 surface in this submicellar concentration range. An alternative interpretation is that monomeric SDS induces structural changes on TII27 that allows pyrene to bind to exposed hydrophobic surfaces. This cannot be ruled out, though we consider it less likely: such surfaces are likely to be covered by SDS molecules that would have to be out-competed by pyrene despite the 1000-fold greater concentration of SDS. Nevertheless, both interpretations indicate significant interactions of submicellar SDS with TII27 but not TNfn3.
FIGURE 3.

Pyrene fluorescence as a function of SDS in the absence and presence of TII27 and TNfn3. Note the steep increase in the I3/I1 ratio of TII27 between 0.5 and 3 mM SDS compared to protein-free pyrene and pyrene in the presence of TNfn3. This suggests that hemimicellar structures form on the TII27 surface but not on TNfn3.
ITC reveals broadly similar stoichiometries of binding of SDS to the two proteins
ITC experiments on TNfn3 and TII27 with SDS reveal characteristic enthalpograms (Fig. 4). For TNfn3 they may be divided into four sections (A–D). Section A shows a very weak exothermic (negative) heat flow, which is essentially independent of the protein concentration. This suggests limited protein-surfactant interaction. At intermediate SDS concentrations (B), a strong endothermic effect sets in and peaks when [SDS] reaches 5–6 mM. This behavior has been seen for a number of other protein-SDS systems, and has been assigned the (endothermic) enthalpy change of protein denaturation (31–33) (H. L. Bagger, S. V. Hoffmann, C. C. Fuglsang, and P. Westh, unpublished data). Assuming two-state behavior for the denaturation, the maximum of the peak in section B will be close to the midpoint of the denaturation process. We will refer to this maximum as point I. Subsequent to the peak, the heat signal decreases and becomes negative (exothermic) around the boundary to section C. This type of exothermic mixing is a general observation in ITC studies on protein-SDS interactions, and it most likely reflects the association of SDS and denatured protein (31) (A. D. Nielsen, K. Borch, and P. Westh, unpublished data). The saturation of this binding is around the inflection (S-) point (point II, 8.0 mM for 69.8 μM TNfn3 in Fig. 4). This also corresponds to the apparent critical micelle concentration (cmcapp) in the presence of protein (21). SDS injected beyond this point mainly remains in micellar form and has far fewer protein molecules to interact with (31,34), leading to the sigmoidal course in section C. Finally, at high SDS concentrations (D), the heat flow levels out, indicating that the injected SDS remains in a micellar form and interacts only weakly with the existing protein-SDS complex.
FIGURE 4.

ITC enthalpograms of (left) Tnfn3 and (right) TII27 upon titration with SDS. See text for an explanations of sections A*–D.
The enthalpogram of TII27 exhibits the same four sections, preceded by a region below 1 mM SDS (denoted A*) with a strong exothermic heat flow that rapidly tapers off as [SDS] is increased. This suggests the specific binding of a few SDS molecules to TII27. Specific binding of SDS with dissociation constants in the 0.1–1 mM range and binding enthalpies of 1–2 kcal/mol has previously been found for a number of proteins (8,32). TII27 shows the same sections A–D described for Tnfn3, although the peak in section B was much smaller than for TNfn3.
To estimate the number of detergent molecules bound to the two proteins at point I in section B and point II in section C, we plot the total SDS concentrations ([SDS]tot) at points I and II against the corresponding protein concentration. Provided that the SDS concentration is low enough to neglect SDS in micellar form, SDS binding at each peak can be expressed by the mass balance (Eq. 3):
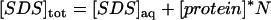 |
(3) |
where [SDS]aq denotes aqueous (nonbound) SDS and N is the average number of bound SDS per protein molecule (21). The plots give satisfactory linear relationships (Fig. 5 A) and the parameters are summarized in Table 1. Point I (interpreted as the midpoint of denaturation), shows an average binding of 14.8 ± 5.2 SDS molecules for TII27 and 16.9 ± 1.5 for TNfn3. This occurs at 4.2 ± 0.4 mM SDS (in the aqueous bulk) for TII27 and 4.9 ± 0.1 mM SDS for TNfn3, which is in reasonable correspondence with the midpoint values of ∼3 and 5 mM observed for the two proteins by fluorescence (Fig. 2). At binding saturation (point II), TII27 binds 44 ± 3.0 and TNfn3 binds 40 ± 1.8 SDS molecules. In both cases, this corresponds to ∼1.2 g SDS per gram protein, very similar to the values seen for many other proteins (35). The SDS concentration obtained by extrapolation of point II values to zero molar protein corresponds to micellization, i.e. the cmc value, which is 5.1–5.7 mM, in good accordance with the independently determined value of 5.0 mM. The binding data are summarized in Table 1.
FIGURE 5.

(A) Concentrations at which peaks I and II appear for TII27 and TNfn3 as a function of protein concentration. (B and C) Enthalpy values at peaks I and II as well as in region A* (for TII27) as a function of protein concentration. Data are summarized in Table 1.
TABLE 1.
ITC data for titration of SDS into solutions of TII27 and TNfn3
| Protein* | Region | [SDS]aq (mM)† | N‡ | Δ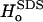 (kcal mol−1)§ (kcal mol−1)§
|
αΔH (kcal mol−1 mM−1)¶ |
|---|---|---|---|---|---|
| TNfn3 | Peak I | 4.9 ± 0.1 | 16.9 ± 1.5 | 0.117 ± 0.072 | 7.6 ± 1.8 |
| TNfn3 | Peak II | 5.7 ± 0.1 | 40.2 ± 1.8 | −0.054 ± 0.017 | −2.4 ± 0.4 |
| TII27 | A* | ‖ | ‖ | −0.102 ± 0.020 | −2.1 ± 0.2 |
| TII27 | Peak I | 4.2 ± 0.4 | 14.8 ± 5.2 | ** | ** |
| TII27 | Peak II | 5.1 ± 0.3 | 44.0 ± 3.0 | 0.010 ± 0.015 | −4.8 ± 0.1 |
All experiments in 10 mM Tris pH 8.0 at 22°C. Errors from the linear fits in Fig. 5.
Intercept from plot of the position of the peak (mM SDS) versus protein concentration.
Slope from plot of the position of the peak (mM SDS) versus protein concentration.
Intercept from plot of the height of the peak (kcal/mol) versus protein concentration.
Slope from plot of the height of the peak (kcal/mol) versus protein concentration (mM).
Not determined because no well-defined minimum available (Fig. 4 B and text).
No significant linear correlation observed (Fig. 5 C).
The amplitude of the peaks and tails also scales linearly with protein concentration although data for the comparably small peak I of TII27 is too scattered to establish an unambiguous relationship (Fig. 5 and Table 1). For peak I, it appears that the slope of these plots is much larger for TNfn3 (Fig. 5 B) than for TII27 (Fig. 5 C), and we shall return to the origin of this below. Fig. 5 C also shows that the size of the initial “tail” for TII27 is proportional to the protein concentration. This is in contrast to conventional ITC data on strongly binding pairs, and it shows that the protein (not the added “ligand”) is the limiting reactant in section A*. It implies that although clearly detectable in the enthalpograms, the specific binding is rather weak with low site occupancy in the submillimolar range.
Mixed micelles show that monomers are not required to unfold TII27
The previous sections had demonstrated that TII27 was unfolded by monomeric SDS and TNfn3 by micellar SDS. This raised the question as to whether the two proteins would respond differently from each other if SDS were predominantly presented in micellar form in the absence of significant amounts of monomeric SDS. This could be done by incorporating SDS into mixed micelles with the nonionic detergent dodecyl maltoside ((DDM) cmc 0.17 mM). We used mixed micelles with SDS mole fractions of 0.75, 0.5, and 0.25. The cmc of these mixtures has previously been determined to be ∼0.3, 0.18, and 0.15 mM, respectively (27), which means that there is not enough monomeric SDS present to effect any denaturation of TII27 (cfr. Fig. 2 C). As expected for a nonionic detergent, DDM alone does not unfold TII27 or TNfn3 (data not shown). For both proteins, titration with mixed micelles showed a clear decrease in the fluorescence ratio 330:350 nm (Fig. 6) as well as the molar ellipticity (data not shown). In addition, the aggressiveness of SDS decreases as the fraction of DM increases, with a slight deviation for TNfn3 at 0.25 mole fraction SDS. At 0.5 and 0.25 mole fraction SDS, unfolding was not completed within the probed concentration range. However, we observe slow unfolding kinetics for TII27 at all micellar mole fractions (Fig. 6 C). This confirms that the decrease in emission ratios represents bona fide unfolding and is not just an artifact due to changes in the polarity of the solvent (which would effect a change in fluorescence within a few milliseconds of mixing). Clearly TII27 is also unfolded by micellar SDS, and in fact retains an increased sensitivity to SDS compared to TNfn3; there is no significant native state baseline before unfolding at all SDS mole fractions (Fig. 6 A), whereas TNfn3 shows a cooperative unfolding in 0.75 mole fraction SDS only above ∼10 mM SDS (i.e., 13.3 mM mixed detergent) (Fig. 6 B).
FIGURE 6.

Unfolding of (A) TII27 and (B) TNfn3 in mixed micelles of SDS and DDM at SDS mole fractions 0.25–1 followed by the change in emission ratio 330:350 nm. (C) Kinetics of unfolding of TII27 in 80 mM detergent containing 0.25–1 mole fraction SDS.
Unfolding kinetics reveal a more complex unfolding behavior for TNfn3 than TII27
Up to now, we have concentrated on the conformational changes that occur before and around the cmc of SDS under equilibrium conditions. The advantage of kinetic studies is that they provide insight into the mechanism of unfolding and also allow us to access high detergent concentrations where the rates of unfolding can vary significantly with detergent concentration, although the end conformation may be spectroscopically unchanged (12). Rate constants kobs for unfolding of the two proteins as a function of SDS are shown in Fig. 7; kobs measured by CD and fluorescence are essentially similar, indicating that secondary and tertiary structure is lost in parallel. Overall, kobs is very low; in comparison, rate constants above 10 mM SDS lie around 2 and 10 s−1 for the mixed α/β proteins CI2 and S6, respectively (12), whose thermodynamic stabilities (∼7 kcal/mol in water) are comparable to the β-sheet proteins TII27 and TNfn3. Clearly the variation of kobs with [SDS] is more complex for TNfn3 than TII27. For both proteins, there is an initial increase in kobs at 3–10 mM SDS and a power-law increase above 100 mM SDS. At 10–100 mM SDS, kobs decreases slightly for TNfn3 but is essentially constant for TII27. We have observed similar behavior for S6 and CI2 (12), where we have classified the unfolding into three concentration regimes or modes (cfr. Scheme 3 in Materials and Methods). Mode 1 (here 3–10 mM SDS) displays saturation-type kinetics and corresponds to binding of spherical micelles, followed by global unfolding. Mode 2 (10–100 mM SDS), only observed for TNfn3, can be modeled as inhibition of unfolding due to additional binding of SDS micelles to TNfn3, although the model remains speculative. Finally, mode 3 is rationalized as increasingly rapid unfolding due to changes in the micellar structure from predominantly spherical to more cylindrical micelles. This has been supported by studies with the cationic detergent TTAB that does not form cylindrical micelles and does not give rise to mode 3 unfolding (12).
FIGURE 7.

Rate constants of unfolding of TII27 and TNfn3 as a function of SDS concentration. Data for TII27 are fitted to mode 1 + 3 unfolding, whereas data for TNfn3 are fitted to mode 1 + 2 + 3 unfolding (Scheme 3).
By determining kobs for a number of mutants of TII27 and TNfn3, we are able to compare the effect of mutation on unfolding in SDS with intrinsic unfolding in denaturant, i.e. GdmCl for TII27 and urea for TNfn3, compiled by Clarke and co-workers (15,43). This provides information about the nature of the transition states of unfolding. We have studied four mutants of TII27 and five mutants of TNfn3 (Fig. 8, A and B). In all cases, kobs increases compared to wild-type but the same unfolding pattern is retained. However, due to the complexity of the unfolding reaction and the existence of multiple equally valid interpretations of the unfolding mechanism, we will only focus on two parameters for each mutant, namely 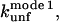 the unfolding rate constant at plateau level before inhibition sets in (typically ∼10–20 mM SDS) and
the unfolding rate constant at plateau level before inhibition sets in (typically ∼10–20 mM SDS) and 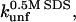 the unfolding rate constant at 500 mM SDS. These data are summarized in Table 2. For both proteins,
the unfolding rate constant at 500 mM SDS. These data are summarized in Table 2. For both proteins, 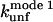 and
and 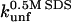 correlate significantly with each other (Fig. 8 C and Table 3), indicating that the two unfolding modes are fundamentally the same, since the transition states for unfolding by the two modes are affected in a similar manner by mutation. Similarly, both rate constants show a reasonable linear correlation with the unfolding rate kunf in water extrapolated from unfolding rate constants in GdmCl and urea (Fig. 8 D and Table 3) (15,17), suggesting an overall similar transition state of unfolding in denaturant versus detergent.
correlate significantly with each other (Fig. 8 C and Table 3), indicating that the two unfolding modes are fundamentally the same, since the transition states for unfolding by the two modes are affected in a similar manner by mutation. Similarly, both rate constants show a reasonable linear correlation with the unfolding rate kunf in water extrapolated from unfolding rate constants in GdmCl and urea (Fig. 8 D and Table 3) (15,17), suggesting an overall similar transition state of unfolding in denaturant versus detergent.
FIGURE 8.

Unfolding rate constants for mutants of (A) TII27 and (B) TNfn3 as a function of SDS. Data are summarized in Table 2. Correlation of unfolding rate constants in SDS (C) with each other and (D) with unfolding rate constants in buffer extrapolated from unfolding rate constants in denaturant. The slopes, intercepts, and correlation coefficients for the best linear fits are summarized in Table 3.
TABLE 2.
Changes in stability and unfolding kinetics for TII27, TNfn3, and their mutants in SDS and chemical denaturants
| Protein | ΔΔGD-N* |
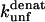 † †
|
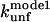 ‡ ‡
|
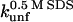 § §
|
|---|---|---|---|---|
| TNfn3 wild-type | ≡ 0 | 7.21 ± 0.26 × 10−5 | 5.81 × 10−3 | 1.3 × 10−2 |
| TN P25A | 1.73 ± 0.13 | 1.92 ± 0.46 × 10−3 | 1.02 × 10−1 | 1.04 |
| TN I48A | 2.19 ± 0.12 | 2.32 ± 0.33 × 10−4 | 9.3 × 10−3 | 8.09 × 10−2 |
| TN Y57G | 4.14 ± 0.10 | 1.29 ± 0.22 × 10−2 | 4.12 × 10−1 | 3.32 |
| TN L62A | 4.22 ± 0.11 | 7.91 ± 1.40 × 10−3 | 8.39 × 10−2 | 1.57 × 10−1 |
| TN T66A | 1.96 ± 0.12 | 8.11 ± 1.80 × 10−4 | 3.2 × 10−2 | 9.41 × 10−2 |
| TII27 wild-type | ≡ 0 | 4.9 ± 0.6 × 10−4 | 3.22 ± 2.1 × 10−4 | 3.17 ± 0.23 × 10−3 |
| TI V4A | 2.45 ± 0.10 | 1.3 ± 0.1 × 10−3 | 6.98 × 10−3 | 2.07 × 10−2 |
| TI L8A | 2.45 ± 0.10 | 1.2 ± 0.2 × 10−3 | 1.54 ± 0.54 × 10−2 | 3.72 ± 0.89 × 10−2 |
| TI L36A | 3.3 ± 0.10 | 3.9 ± 0.3 × 10−3 | 1.96 ± 0.19 × 10−2 | 1.31 ± 0.00 × 10−1 |
| TI L60A | 4.88 ± 0.08 | 3.4 ± 0.3 × 10−3 | 1.87 ± 0.22 × 10−2 | 3.26 ± 0.37 × 10−2 |
The mutation-induced change in the difference in free energy between the denatured state, D, and the native state, N, on mutation (kcal·mol−1), relative to wild-type protein. Obtained from Fowler and Clarke (15) (TII27) and Hamill et al. (43) (TNfn3).
The 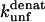 (s−1) values are obtained from Fowler and Clarke (15) (TII27, GdmCl) and Hamill et al. (43) (TNfn3, Urea). For TII27 the authors determined
(s−1) values are obtained from Fowler and Clarke (15) (TII27, GdmCl) and Hamill et al. (43) (TNfn3, Urea). For TII27 the authors determined 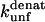 by linear extrapolation of log
by linear extrapolation of log 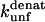 at different denaturant concentrations to zero molar denaturant;
at different denaturant concentrations to zero molar denaturant; 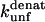 for TNfn3 mutants was calculated from Hamill et al. (43) by use of the following equation: Δ
for TNfn3 mutants was calculated from Hamill et al. (43) by use of the following equation: Δ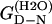 (mutant) = −RTln(ku/kf)mutant.
(mutant) = −RTln(ku/kf)mutant.
Unfolding rate constant (s−1) in SDS at the plateau level (TII27) and at the highest level before inhibition sets in (TNfn3) (cfr. Figs. 7 and 8). Errors are from the direct fits to Eq. 2 (TNfn3) or Eq. 3 (TII27). In several cases, it was not possible to obtain a stable fit, making it necessary to estimate the value by visual inspection.
Unfolding rate constant (s−1) interpolated to 500 mM SDS.
TABLE 3.
Correlations between kinetic parameters for unfolding in SDS and denaturant
| Protein | Independent variable | Dependent variable | Slope | Intercept | R |
|---|---|---|---|---|---|
| TII27 | 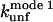 |
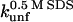 |
0.71 ± 0.17 | −0.04 ± 0.40 | 0.92 |
| TNfn3 | 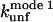 |
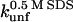 |
1.15 ± 0.23 | 0.87 ± 0.34 | 0.93 |
| TII27 |
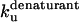 * * |
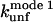 |
1.57 ± 0.52 | 2.25 ± 1.47 | 0.87 |
| TII27 |
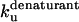 * * |
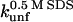 |
1.38 ± 0.42 | 2.27 ± 1.19 | 0.88 |
| TNfn3 |
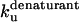 * * |
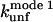 |
0.76 ± 0.12 | 0.85 ± 0.36 | 0.95 |
| TNfn3 |
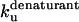 * * |
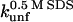 |
0.82 ± 0.28 | 1.68 ± 0.83 | 0.83 |
Unfolding rates extrapolated to zero molar denaturant. Values determined as in Table 2. Errors are from the linear fits.
DISCUSSION
Differences and similarities between two structurally similar β-sheet proteins
In this study, we have focused on the SDS-induced unfolding of two structurally similar β-sandwich proteins TII27 and TNfn3. Despite their similarity, the two proteins show significantly different electrostatic surface potential, with TII27 possessing more positively charged regions than TNfn3. This suggests that TII27 should be able to attract negatively charged detergent molecules to a greater extent than TNfn3, and this expectation is borne out by equilibrium denaturation studies, which show that TII27 denatures, and loses the ability to undergo a cooperative thermal transition, below cmc while TNfn3 only shows significant structural changes as well as a decrease in thermal stability, around the cmc. Similar effects of electrostatics have been observed for, e.g., carbonic anhydrase, whose resistance to SDS increased dramatically upon peracetylation of the lysine side chains (6).
The ability of SDS to denature TII27 at submicellar concentrations can be explained by its ability to form hemimicelles on the protein surface, as demonstrated by pyrene fluorescence experiments. Nevertheless, the presence of monomeric SDS is not essential for unfolding of TII27 but merely provides an early start, since mixed micelles with DDM (where the amount of monomeric SDS is negligible) are also able to denature TII27. Nonionic detergents only react weakly if at all with water-soluble proteins because their low cmc values indicate that detergent-detergent interactions are more thermodynamically favorable than protein-detergent interactions. The SDS-DDM micelles also have low cmc values (0.15–0.3 mM) but still interact with TII27 and TNfn3, although to a smaller extent. Clearly the concentration of negative charge in the micelle is very important for denaturing potency, since there is a fourfold increase in the amount of detergent needed to cause denaturation when the SDS mole fraction decreases 25% (from 100 to 75%).
Mixed SDS-DDM micelles are increasingly being used to quantitate membrane protein stability (36,37) and folding kinetics (4,38). The reported midpoint values of denaturation vary from 30 to 70%. However, an important difference between membrane proteins and water-soluble proteins is the former's uncompromising requirement for amphiphiles to stay in solution and consequent very high affinity for micelles. Thus, all membrane proteins will be bound in micelles even at low micelle concentrations provided there is a stoichiometric amount of detergent micelles present, and varying the absolute SDS concentration has no effect on the unfolding of diacyl glycerate kinase stability provided the mole fraction is kept constant (36). In contrast, unfolding of TII27 and TNfn3 in mixed SDS-DM micelles only occurs above a certain SDS concentration (Fig. 6), highlighting the equilibrium between free and bound protein for both TII27 and TNfn3.
Differences between the two proteins are also evident from the calorimetric titration trials. An early binding event below 1 mM SDS occurs only for TII27, and this again is in accord with the analysis of the electrostatic potentials discussed above. At higher [SDS], both proteins show an endothermic transition, which is ascribed to the surfactant-induced denaturation. In accordance with the spectroscopic measurements, this transition occurs at higher [SDS] for TNfn3. The ITC data further showed that the robustness toward SDS of these proteins did not rely on a particularly low affinity for the surfactant. In fact, the saturation binding level (∼1.2 g/g) was identical to that typically found for globular protein with no (or reduced) cysteines (35,39). The conspicuous difference in the unfolding rates (Fig.7) is not reflected in the binding stoichiometry either, since both proteins bind around 15 SDS molecules at the transition midpoint. This similarity in the binding isotherm is also relevant for the interpretation of the slopes, αΔH, in Fig. 5 (listed in Table 1). This function quantifies the change in enthalpy of the system upon a small change in the amount of both protein and surfactant. In other words it is proportional to the second derivative 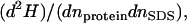 where n denotes number of moles and the subscripts defines the compound. This derivative is a measure of the enthalpy of protein-surfactant interactions in solution (40,41). The distinctive positive αΔH for peak I of TNfn3, for example, indicates a pronounced endothermic nature of the TNfn3-SDS interaction around peak I. Because the binding affinity for the two proteins are similar, we interpret this as a stronger effect of SDS on the denaturation equilibrium of TNfn3. Thus, in the transition zone, the population of denatured TNfn3 molecules increases more steeply with [SDS], than in the case of TII27. This higher degree of cooperativity for SDS denaturation of TNfn3, which is also reflected in a steeper CD and fluorescence unfolding curve than TII27 (Fig. 2), may rely on the transition zone's location close to cmc and the micelle-driven unfolding.
where n denotes number of moles and the subscripts defines the compound. This derivative is a measure of the enthalpy of protein-surfactant interactions in solution (40,41). The distinctive positive αΔH for peak I of TNfn3, for example, indicates a pronounced endothermic nature of the TNfn3-SDS interaction around peak I. Because the binding affinity for the two proteins are similar, we interpret this as a stronger effect of SDS on the denaturation equilibrium of TNfn3. Thus, in the transition zone, the population of denatured TNfn3 molecules increases more steeply with [SDS], than in the case of TII27. This higher degree of cooperativity for SDS denaturation of TNfn3, which is also reflected in a steeper CD and fluorescence unfolding curve than TII27 (Fig. 2), may rely on the transition zone's location close to cmc and the micelle-driven unfolding.
Differences in unfolding kinetics
There are two important differences between TII27 and TNfn3 as regards kinetics. Firstly, TNfn3 but not TII27 undergoes an inhibition of unfolding. Secondly, although the mutants' unfolding rate constants in SDS (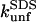 ) are linearly correlated to unfolding rate constants in denaturant (
) are linearly correlated to unfolding rate constants in denaturant (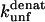 ), the slopes of the two correlations are significantly different (Fig. 8 D).
), the slopes of the two correlations are significantly different (Fig. 8 D).
The first observation clearly illustrates that inhibition of unfolding, previously reported for both S6 and CI2 (12), is not a general detergent phenomenon but is a specific reflection of protein properties. The effect can be modeled by the binding of additional SDS micelles to TNfn3—but not TII27—in the mode 2 region (10–100 mM SDS). It is difficult to come up with straightforward explanations for this difference. It is possible that the different abilities to bind monomeric SDS may affect the ability to bind more SDS molecules at higher concentrations. The ground state from which unfolding occurs could be different for the two proteins if for example TII27 is partially covered by hemimicellar structures that block access to additional (inhibiting) micelles within the dead time of mixing before unfolding occurs, in contrast to TNfn3, which does not attract submicellar structures.
The second observation relates to the nature of binding of SDS to proteins. It is safe to assume that 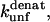 which are obtained from extrapolations from unfolding rates in denaturant (GdmCl or urea) represent the intrinsic and denaturant-independent unfolding rates. Consequently, the changes in
which are obtained from extrapolations from unfolding rates in denaturant (GdmCl or urea) represent the intrinsic and denaturant-independent unfolding rates. Consequently, the changes in 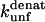 upon mutation must reflect absolute changes in the energy of the transition state. However, the degree to which changes in
upon mutation must reflect absolute changes in the energy of the transition state. However, the degree to which changes in 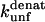 correlate to changes in
correlate to changes in 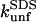 (the slopes in Fig. 8 D) will depend on how efficiently SDS can bind to and denature the protein. The fact that changes in
(the slopes in Fig. 8 D) will depend on how efficiently SDS can bind to and denature the protein. The fact that changes in 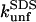 for TII27 mutants are more sensitive than those of TNfn3 to changes in
for TII27 mutants are more sensitive than those of TNfn3 to changes in 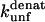 must illustrate that SDS is more efficient at destabilizing TII27 than TNfn3 (even though the total amount of SDS bound is the same, namely 1.2 g SDS per gram protein). This ties in well with our previous observations that TII27 generally is more sensitive to SDS than TNfn3, presumably facilitated by the altered electrostatic potential.
must illustrate that SDS is more efficient at destabilizing TII27 than TNfn3 (even though the total amount of SDS bound is the same, namely 1.2 g SDS per gram protein). This ties in well with our previous observations that TII27 generally is more sensitive to SDS than TNfn3, presumably facilitated by the altered electrostatic potential.
A general model for SDS unfolding of all-β versus helix-containing proteins
Generally, denaturation in SDS tends to increase the amount of α-helix structure in proteins with high native content of β-structure (11). This corresponds to the formation of a “bead-on-a-string” structure, where SDS micelles attach to individual segments of the protein and preferentially stabilize isolated structural elements with independent hydrogen-binding abilities (42). However, the SDS-unfolded states of TII27 and TNfn3 only show a relatively insignificant increase in α-helical structure. An important corollary of the linear correlations established in Fig. 8 is that the transition states for unfolding are broadly similar in SDS and denaturant. That is, we do not need to postulate a “distorted” transition state for unfolding in SDS compared to unfolding in denaturant, as opposed to, e.g., the mixed α/β protein S6 (13).
In combination with the low α-helicity of the SDS-denatured states, our data have inspired us to present the following working hypothesis for β-sheet unfolding in SDS. There are no SDS-specific attack points for the unfolding of β-sheet proteins, rather SDS has to rely on the global unfolding behavior of this class of proteins to destabilize them. In contrast, for proteins containing significant elements of α-helical structure such as S6, the α-helices may provide a first line of attack for SDS micelles. As demonstrated experimentally for S6, α-helices can quite easily be ripped out of the intact tertiary structure since they constitute independent folding units that are stable as isolated entities in micelles due to their ability to form internal hydrogen bonds, whereas β-sheet structures rely on global contacts and are therefore less easily disrupted, unless the entire protein is unfolded. This may provide an underlying structural reason for the observation by Manning and Colón on the enhanced robustness of β-sheet proteins, and particularly oligomeric structures, toward SDS (14). However, we emphasize that the present data, although consistent with this hypothesis, are insufficient to establish the validity of the model in detail. We are therefore currently pursuing experimental strategies to resolve this question.
Acknowledgments
We are very grateful to Annette Steward and Jane Clarke for generously providing mutants of TNfn3 and TII27 as well as expression vectors for the wild-type proteins.
D.E.O. is supported by the Danish National Research Foundation (inSPIN) and by the Villum Kann Rasmussen Foundation (BioNET). K.K.A. is supported by the BIOPRO Innovation Consortium. P.W. acknowledges support from Carlsberg Foundation and the Danish Research Agency (grants 26-02-0160 and 21-04-0087).
References
- 1.La Mesa, C. 2005. Polymer-surfactant and protein-surfactant interactions. J. Coll. Int. Sci. 286:148–157. [DOI] [PubMed] [Google Scholar]
- 2.Timasheff, S. 2002. Protein hydration, thermodynamic binding, and preferential hydration. Biochemistry. 41:13473–13482. [DOI] [PubMed] [Google Scholar]
- 3.Kirk, O., T. V. Borchert, and C. C. Fuglsang. 2002. Industrial enzyme applications. Curr. Opin. Biotechnol. 13:345–351. [DOI] [PubMed] [Google Scholar]
- 4.Sehgal, P., and D. E. Otzen. 2006. Thermodynamics of unfolding of an integral membrane protein in mixed micelles. Prot. Sci. 15:890–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otzen, D. E., L. Christiansen, and M. Schülein. 1999. A comparative study of the unfolding of the endoglucanase Cel45 from Humicola insolens in denaturant and surfactant. Prot. Sci. 8:1878–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gitlin, I., K. L. Gudiksen, and G. M. Whitesides. 2006. Peracetylated bovine carbonic anhydrase (BCA-Ac18) is kinetically more stable than native BCA to sodium dodecyl sulfate. J. Phys. Chem. B. 110:2372–2377. [DOI] [PubMed] [Google Scholar]
- 7.Jones, M. N., and P. Manley. 1979. Binding of n-alkyl sulphates to lysozyme in aqueous solution. J. Chem. Soc. Faraday Trans. 75:1736–1744. [Google Scholar]
- 8.Jones, M. N. 1992. Surfactant interactions with biomembranes and proteins. Chem. Soc. Rev. 21:127–136. [Google Scholar]
- 9.Reynolds, J. A., S. Herbert, H. Polet, and J. Steinhardt. 1967. The binding of divers detergent anions to bovine serum albumin. Biochemistry. 6:937–943. [DOI] [PubMed] [Google Scholar]
- 10.Tanford, C. 1980. The Hydrophobic Effect. Formation of Micelles and Biological Membranes. Wiley & Sons, New York.
- 11.Takeda, K., H. Sasaoka, K. Sasa, H. Hirai, K. Hachiya, and Y. Moriyama. 1992. Size and mobility of sodium dodecyl-sulfate bovine serum-albumin complex as studied by dynamic light-scattering and electrophoretic light-scattering. J. Coll. Int. Sci. 154:385–392. [Google Scholar]
- 12.Otzen, D. E. 2002. Protein unfolding in detergents: effect of micelle structure, ionic strength, pH, and temperature. Biophys. J. 83:2219–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otzen, D. E., and M. Oliveberg. 2002. Burst-phase expansion of native protein prior to global unfolding in SDS. J. Mol. Biol. 315:1231–1240. [DOI] [PubMed] [Google Scholar]
- 14.Manning, M., and W. Colón. 2004. Structural basis of protein kinetic stability: resistance to sodium dodecyl sulfate suggests a central role for rigidity and a bias toward beta-sheet structure. Biochemistry. 43:11248–11254. [DOI] [PubMed] [Google Scholar]
- 15.Fowler, S. B., and J. Clarke. 2001. Mapping the folding pathway of an immunoglobulin domain: structural detail from Phi value analysis and movement of the transition state. Structure. 9:355–366. [DOI] [PubMed] [Google Scholar]
- 16.Clarke, J., E. Cota, S. B. Fowler, and S. J. Hamill. 1999. Folding studies of immunoglobulin-like beta-sandwich proteins suggest that they share a common folding pathway. Structure. 7:1145–1153. [DOI] [PubMed] [Google Scholar]
- 17.Hamill, S. J., A. E. Meekhof, and J. Clarke. 1998. The effect of boundary selection on the stability and folding of the third fibronectin type III domain from human tenascin. Biochemistry. 37:8071–8079. [DOI] [PubMed] [Google Scholar]
- 18.Geierhaas, C. D., E. Paci, M. Vendruscolo, and J. Clarke. 2004. Comparison of the transition states for folding of two Ig-like proteins from different superfamilies. J. Mol. Biol. 343:1111–1123. [DOI] [PubMed] [Google Scholar]
- 19.Clarke, J., S. J. Hamill, and C. M. Johnson. 1997. Folding and stability of a fibronectin type III domain of human tenascin. J. Mol. Biol. 270:771–778. [DOI] [PubMed] [Google Scholar]
- 20.Carrion-Vazquez, M., A. F. Oberhauser, S. B. Fowler, P. E. Marszalek, S. E. Broedel, J. Clarke, and J. M. Fernandez. 1999. Mechanical and chemical unfolding of a single protein: a comparison. Proc. Natl. Acad. Sci. USA. 96:3694–3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nielsen, A. D., L. Arleth, and P. Westh. 2005. Analysis of protein-surfactant interactions—a titration calorimetric and fluorescence spectroscopic investigation of interactions between Humicola insolens cutinase and an anionic surfactant. Biochim. Biophys. Acta. 1752:124–132. [DOI] [PubMed] [Google Scholar]
- 22.Leahy, D. J., W. A. Hendrickson, I. Aukhil, and H. P. Erickson. 1992. Structure of a fibronectin type III domain from tenascin phased by MAD analysis of the selenomethionyl protein. Science. 258:987–991. [DOI] [PubMed] [Google Scholar]
- 23.Improta, S., J. K. Krueger, M. Gautel, R. A. Atkinson, J. F. Lefevre, S. Moulton, J. Trewhella, and A. Pastore. 1998. The assembly of immunoglobulin-like modules in titin: implications for muscle elasticity. J. Mol. Biol. 284:761–777. [DOI] [PubMed] [Google Scholar]
- 24.Li, H., A. D. Robertson, and J. H. Jensen. 2005. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 61:704–721. [DOI] [PubMed] [Google Scholar]
- 25.Dolinsky, T. J., J. E. Nielsen, J. A. McCammon, and N. A. Baker. 2004. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 32:W665–W667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bashford, D. 2004. Macroscopic electrostatic models for protonation states in proteins. Front. Biosci. 9:1082–1099. [DOI] [PubMed] [Google Scholar]
- 27.Sehgal, P., J. E. Mogensen, and D. E. Otzen. 2005. Using micellar mole fractions to assess membrane protein stability in mixed micelles. Biochim. Biophys. Acta. 1716:59–68. [DOI] [PubMed] [Google Scholar]
- 28.Croonen, Y., E. Geladé, M. Van der Zegel, M. Van der Auweraer, H. Vandendriessche, F. C. De Schryver, and M. Almgren. 1983. Influence of salt, detergent concentration and temperature on the fluorescence quenching of 1-methylpyrene in sodium dodecyl sulfate with m-dicyanobenzene. J. Phys. Chem. 87:1426–1431. [Google Scholar]
- 29.Lobley, A., L. Whitmore, and B. A. Wallace. 2002. DICHROWEB: an interactive website for the analysis of protein secondary structure from circular dichroism spectra. Bioinformatics Applications Note. 18:211–212. [DOI] [PubMed] [Google Scholar]
- 30.Turro, N. J., and A. Yekta. 1978. Luminescent probes for detergent solutions. A simple procedure for determination of the mean aggregation number of micelles. J. Am. Chem. Soc. 100:5951–5955. [Google Scholar]
- 31.Nielsen, A. D., L. Arleth, and P. Westh. 2005. Interactions of Humicola insolens cutinase with an anionic surfactant studied by small-angle neutron scattering and isothermal titration calorimetry. Langmuir. 21:4299–4307. [DOI] [PubMed] [Google Scholar]
- 32.Lad, M. D., V. M. Ledger, B. Briggs, R. J. Green, and R. A. Frazier. 2003. Analysis of the SDS-lysozyme binding isotherm. Langmuir. 19:5098–5103. [Google Scholar]
- 33.Kelley, D., and D. J. McClements. 2003. Interactions of bovine serum albumin with ionic surfactants in aqueous solutions. Food Hydrocolloids. 17:73–85. [Google Scholar]
- 34.Paula, S., W. Sus, J. Tuchtenhagen, and A. Blume. 1995. Thermodynamics of micelle formation as a function of temperature—a high-sensitivity titration calorimetry study. J. Phys. Chem. 99:11742–11751. [Google Scholar]
- 35.Reynolds, J. A., and C. Tanford. 1970. The gross conformation of protein-sodium dodecyl sulfate complexes. J. Biol. Chem. 245:5161–5165. [PubMed] [Google Scholar]
- 36.Lau, F. W., and J. U. Bowie. 1997. A method for assessing the stability of a membrane protein. Biochemistry. 36:5884–5892. [DOI] [PubMed] [Google Scholar]
- 37.Faham, S., D. Yang, E. Bare, S. Yohannan, J. P. Whitelegge, and J. U. Bowie. 2004. Side-chain contributions to membrane protein structure and stability. J. Mol. Biol. 335:297–305. [DOI] [PubMed] [Google Scholar]
- 38.Otzen, D. E. 2003. Folding of DsbB in mixed micelles: a kinetic analysis of the stability of a bacterial membrane protein. J. Mol. Biol. 330:641–649. [DOI] [PubMed] [Google Scholar]
- 39.Pitt-Rivers, R., and F. Impiombato. 1968. Binding of sodium dodecyl sulphate to various proteins. Biochem. J. 109:825–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westh, P., and Y. Koga. 1997. Intermolecular interactions of lysozyme and small alcohols: a calorimetric investigation. J. Phys. Chem. 101B:5755–5758. [Google Scholar]
- 41.Trandum, C., P. Westh, K. Jørgensen, and O. G. Mouritsen. 2000. A thermodynamic study of the effects of cholesterol on the interaction between liposomes and ethanol. Biophys. J. 78:2486–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ibel, K., R. P. May, K. Kirschner, H. Szadkowski, E. Mascher, and P. Lundahl. 1990. Protein-decorated micelle structure of sodium-dodecyl-sulfate-protein complexes as determined by neutron scattering. Eur. J. Biochem. 190:311–318. [DOI] [PubMed] [Google Scholar]
- 43.Hamill, S. J., A. Steward, and J. Clarke. 2000. The folding of an immunoglobulin-like Greek key protein is defined by a common-core nucleus and regions constrained by topology. J. Mol. Biol. 297:165–178. [DOI] [PubMed] [Google Scholar]