

Abstract
It is widely accepted that PYP undergoes global structural changes during the formation of the biologically active intermediate PYPM. High-angle solution x-ray scattering experiments were performed using PYP variants that lacked the N-terminal 6-, 15-, or 23-amino-acid residues (T6, T15, and T23, respectively) to clarify these structural changes. The scattering profile of the dark state of intact PYP exhibited two broad peaks in the high-angle region (0.3 Å−1 < Q < 0.8 Å−1). The intensities and positions of the peaks were systematically changed as a result of the N-terminal truncations. These observations and the agreement between the observed scattering profiles and the calculated profiles based on the crystal structure confirm that the high-angle scattering profiles were caused by intramolecular interference and that the structure of the chromophore-binding domain was not affected by the N-terminal truncations. The profiles of the PYPM intermediates of the N-terminally truncated PYP variants were significantly different from the profiles of the dark states of these proteins, indicating that substantial conformational rearrangements occur within the chromophore-binding domain during the formation of PYPM. By use of molecular fluctuation analysis, structural models of the chromophore-binding region of PYPM were constructed to reproduce the observed profile of T23. The structure obtained by averaging 51 potential models revealed the displacement of the loop connecting β4 and β5, and the deformation of the α4 helix. High-angle x-ray scattering with molecular fluctuation simulation allows us to derive the structural properties of the transient state of a protein in solution.
INTRODUCTION
PYP is a putative photoreceptor of negative phototaxis in the purple phototropic bacterium Halorhodospira halophila (1,2). PYP is composed of four segments, namely, an N-terminal cap (residues 1–28), a PAS core (residues 29–69), a helical connector (residues 70–87), and a β scaffold (residues 88–125) (3,4). In this article, we refer to the latter three segments as the chromophore-binding region. Absorption of a photon by the chromophore of PYP, a p-coumaric acid moiety located in a hydrophobic pocket in the chromophore-binding region, triggers the isomerization of the chromophore (5) and the subsequent thermal reaction cycle (6–10). The blue-shifted reaction intermediate PYPM, which has also been referred to as I2 or pB and forms over a time scale of ∼100 μs, is assumed to be the active state. Although the downstream transducer has not been identified, structural information about PYPM is crucial for investigating the molecular mechanism of PYP-dependent photosignal transduction. According to time-resolved crystallography, the structural changes in PYPM are confined to the area near the chromophore (11,12). On the other hand, substantial conformational changes in the protein moiety of PYPM in solution have been reported in studies using spectroscopic (13,14), thermodynamic (15–17), NMR (18,19), and x-ray scattering (20) measurements. Therefore, detailed structural information about PYPM in solution is required to clarify the mechanism underlying the phototransduction.
We previously found that N-terminal truncation of the protein by chymotrypsin treatment prolongs the lifetime of PYPM without any structural perturbation in the chromophore-binding region of PYP under dark conditions (21). Prolonging the lifetime of this intermediate is useful for structural studies because the active state of the protein can be easily accumulated under continuous illumination, which allows the measurement of subtle signals from the active state. Our studies of PYPM in solution using truncated PYP variants suggested that during the formation of PYPM, the N-terminal cap moves away from the chromophore-binding region, and the chromophore-binding region is slightly swollen (22). We also observed the deformation of α-helices in the N-terminal cap and the chromophore-binding region, and structural changes in the β-sheets in the chromophore-binding region during PYPM formation (23). The recently reported NMR structure of a PYP variant that lacked the first 25 N-terminal amino-acid residues exhibits a large degree of structural disorder, especially in the chromophore-binding region, resulting in the exposure of the chromophore to the solvent (19). The NMR structure, however, does not necessarily explain the solution structural properties of the protein; the increase of the size of the protein during the formation of the active state observed in the NMR study is larger than what we observed in our previous SAXS study (19,22). Therefore, we have employed high-angle solution x-ray scattering to examine the structure of PYPM.
SAXS experiments allow the determination of the overall structural parameters of a protein, such as the radius of gyration, the maximum dimension of the particle, and the molecular shape, under various physiological conditions (24,25). Analysis using SAXS permits the construction of low-resolution structural models without any presuppositions; this method, which is called ab initio shape prediction (26,27), is widely used to characterize protein structures under physiological conditions (28,29). Recent improvements in two-dimensional x-ray detectors and the availability of third-generation synchrotron radiation sources have improved the quality of x-ray solution scattering profiles even in the higher-angle region with Q values up to 6 Å−1. High-angle profiles contain information about secondary structure packing and tertiary folds (30–32). Although the combination of MD simulation and high-angle solution scattering is expected to be useful to derive structural information (33), the systematic application of these techniques has not been reported.
In this study, to extract structural information about PYP in solution, high-angle x-ray scattering experiments were performed using N-terminally truncated PYP variants. Structural models derived with molecular fluctuation simulation were used to explain the high-angle scattering profiles. The obtained model clearly shows the loop connecting β4 and β5 and the α4 helix moving away from the chromophore-binding pocket. The obtained structural model and the scattering profile are compared with the solution structures and calculated scattering profiles reported in the NMR study (19).
Experimental
Sample preparation
Intact PYP overexpressed in Escherichia coli BL21(DE3) using the pET system (Novagen) was reconstituted by the addition of p-coumaric acid anhydride in 4 M urea buffer (34). After removal of the urea by dialysis, PYP was purified using several rounds of chromatography on DEAE Sepharose CL6B columns (Amersham Biosciences, Piscataway, NJ) until the optical purity index (absorbance at 277 nm/absorbance λmax) was less than 0.44. Truncated PYP variants lacking the N-terminal 6, 15, or 23 amino-acid residues (T6, T15, and T23, respectively) were prepared by chymotrypsin treatment and each truncated PYP variant was isolated and purified on DEAE-Sepharose columns (21). For x-ray scattering experiments, the PYP variants were suspended in MOPS buffer (10 mM MOPS and 200 mM NaCl at pH 7.0).
High-angle x-ray scattering experiments
High-angle x-ray scattering experiments were performed using the x-ray scattering spectrometer installed on the BL40B2 beamline at SPring8 (Harima, Japan) (35). The sample-to-detector distance was 100 cm, and the x-ray wavelength was set to 1 Å. The sample solution was placed into a specially designed cell with a 1-mm path length. The scattering images in the dark and under continuous illumination (wavelength > 410 nm) were collected for the same solution sample at 20°C using an on-line imaging plate detector (R-Axis IV; Rigaku). The sample solution was exchanged several times after the first minute of exposure (every 30 sec for the dark and illuminated conditions) to avoid radiation damage. The obtained two-dimensional images were circularly averaged and scaled with the incident beam monitor count. The final one-dimensional data for each sample were obtained by subtracting the profile of the buffer from that of the sample solution. To examine the protein concentration effect on the profiles, the measurements were carried out at several different protein concentrations (between 2 and 12 mg/ml). Because no significant concentration effects were observed in the high-angle region examined in this work, the profiles obtained for the highest protein concentration that showed the lowest noise level were used for the further analysis.
Calculation of the x-ray scattering profiles of atomic structural models
The x-ray scattering profiles of the crystal structures of intact PYP and the truncated PYP variants, the NMR structures, and the constructed model structures were calculated using the CRYSOL program (36); the hydration layer was not included in this analysis of the high-angle scattering. The crystal structure of the dark state of PYP (PDB ID 1NWZ) (37) was used for the calculation of the profile of intact PYP. The models of the N-terminally truncated PYP variants (T6, T15, and T23) were constructed by deleting the coordinates for the N-terminal 6-, 15-, and 23-amino-acid residues from the intact crystal structure (1NWZ), respectively. The PDB codes of the NMR structures of the dark state and the light state are 1XFN and 1XFQ, respectively (19).
CONCOORD simulation
The dependence of the fluctuation distribution on the truncation was analyzed using the CONCOORD program, which provides mutually uncorrelated structures that fulfill an empirical parameter set of upper and lower interatomic distance limits (38). Although detailed aspects of the protein dynamics were not obtained, similar results were attained for the fluctuation distributions calculated using conventional MD and CONCOORD simulations. In this study, 500 structures were generated for intact PYP, T6, T15, and T23 using the CONCOORD program with the default parameters.
RESULTS
High-angle x-ray scattering experiments with intact PYP, T6, T15, and T23 in the dark
The high-Q region (0.3 to 1 Å−1) of a scattering profile is closely related with secondary structure packing (30–33). Therefore, the N-terminal deletions of PYP were expected to affect the scattering profile. Fig. 1 a shows the experimentally observed scattering profiles of intact PYP and the various N-terminally truncated variants (T6, T15, and T23). The profile of intact PYP has two broad peaks at Q = 0.35 and 0.55 Å−1, with a valley around Q = 0.41 Å−1. The six-residue deletion (T6) increased the intensity of the peak at the lower Q value while shifting it to a higher Q value. A decrease in the intensity of the peak at the higher Q value and a shift of the valley toward a higher Q value were also observed. On the other hand, although T15 and T23 contained different deletions, they resulted in similar scattering profiles with a single maximum around Q = 0.39 Å−1. These characteristic profile changes indicate that the scattering profile in this Q region reflected intramolecular interference.
FIGURE 1.

High-angle scattering profiles of intact PYP (black line), T6 (blue line), T15 (green line), and T23 (red line) measured in solution (a) and calculated from the respective atomic structural models (b).
The experimentally observed profiles can be explained using the crystal structure of PYP (Fig. 1 b). The theoretical profile of intact PYP has two broad peaks at the same positions as those observed in the experimentally obtained curve. The theoretical profiles for T6 and T23 were also similar to the respective observed profiles. The agreement between the calculated profiles and the observed profiles indicates that the structures of T6 and T23 as well as that of intact PYP can be explained by removing the corresponding residues from the crystal structure and that the intramolecular interference scattering can be clearly separated from the small-angle scattering that results from the molecular envelope. The theoretical profile for T15 appeared to be an intermediate between those for T6 and T23 and was different from the observed profile of T15. This suggests that the degree of disorder in the N-terminal region of T15 did not allow its average structure to be defined, resulting in the disappearance of the interference between the N-terminal region and the rest of the protein.
Mean-square fluctuation of intact PYP and the truncated PYP variants
To validate the chaotic feature of the N-terminal region of T15, the mean-square fluctuations of intact PYP, T6, T15, and T23 were simulated using the CONCOORD program (38). The results are described in Fig. 2 a, whereas the crystallographic B factor of intact PYP (37) is shown in Fig. 2 b. The positions of the peaks and valleys in the mean-square fluctuation data for intact PYP are similar to those in the B-factor data; therefore, the mean-square fluctuations obtained with the CONCOORD program reflect the actual fluctuations. From Fig. 2 a, there are clear differences among T6, T15, T23, and intact PYP in the N-terminal regions and the C-terminal loops. Although T6 results in a small increase in the amplitude of the signal for the whole N-terminal region, the positions of the peaks and valleys are conserved. On the other hand, the fluctuation of the N-terminal region of T15 significantly increases, which supports the interpretation of a disordered N-terminal region in T15. It is noteworthy that the fluctuations at Phe6, Ile11, Glu12, Leu15, Asp20, and Leu23 are less pronounced than those at the other amino-acid residues in the N-terminal region. According to the crystal structure, these residues associate with the residues from the β–sheet, including Leu26, Phe28, Lys110, Ala112, Trp119, and Phe121, mainly through hydrophobic interactions (37). We believe that these residues act as nodes to stabilize the N-terminal region. In the case of T15, four of the six residues that are critical for the stabilization (Phe6, Ile11, Glu12, and Leu15) were missing, resulting in the destabilization and divergence of the N-terminal portion of T15.
FIGURE 2.

Cα fluctuation distributions of intact PYP (black line), T6 (blue line), T15 (green line), and T23 (red line) simulated using the CONCOORD program (a). For the sake of comparison, the crystallographic B-factors of the Cα positions (37) are shown in b.
X-ray scattering profiles of the PYPM intermediates of T6, T15, and T23
The x-ray scattering profiles of T6, T15, and T23 were measured under continuous illumination. Because N-terminal truncations prolong the lifetime of the M intermediate, more than 90% of the protein was expected to be in the PYPM state under continuous illumination (22). It was recently reported that the PYPM state includes various long-lived near-UV intermediates containing a protonated chromophore (39–41). Two of these substates are on the pathway of the thermal reaction cycle; the early and late substates have previously been called I2 and I2′, respectively (39,40). The equilibrium between the two substates is highly influenced by the solution condition (39–41). In our recent study, it was shown that the equilibrium under continuous illumination depends on the solution pH, and the two substates are spectroscopically and structurally distinguishable from each other (41). The substate that accumulated under acidic conditions exhibited a λmax of 367 nm, whereas the substate that amassed under alkaline conditions exhibited a λmax of 356 nm; we called these substates 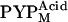 and
and 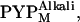 respectively. The structural change associated with
respectively. The structural change associated with 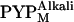 is larger than that associated with
is larger than that associated with 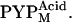 Spectral and structural similarities suggested that I2 and I2′ corresponded to
Spectral and structural similarities suggested that I2 and I2′ corresponded to  and
and 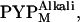 respectively. Assuming that the pKa values of the N-terminally truncated variants are identical to that of the wild-type protein, ∼80–90% of the intermediates that accumulated at this pH was assumed to be
respectively. Assuming that the pKa values of the N-terminally truncated variants are identical to that of the wild-type protein, ∼80–90% of the intermediates that accumulated at this pH was assumed to be  (41), which was supported by the fact that the λmax values of the accumulated intermediates of T6, T15, and T23 (357 nm for each) were close to that of
(41), which was supported by the fact that the λmax values of the accumulated intermediates of T6, T15, and T23 (357 nm for each) were close to that of 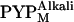 (22,41). Furthermore, even if a small amount of
(22,41). Furthermore, even if a small amount of 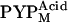 was included in the observed states, because the structural change in
was included in the observed states, because the structural change in 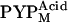 is smaller than that in
is smaller than that in 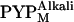 (41), the observed profile change should mainly reflect the structure of
(41), the observed profile change should mainly reflect the structure of 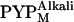 . Hereafter,
. Hereafter, 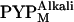 is therefore simply called PYPM. Fig. 3 shows the intensity profiles of the PYPM intermediates of the truncated PYP variants compared with those obtained for their dark states. Significant differences between the two states were observed for each truncated PYP. The profiles of the PYPM intermediates of the three truncated PYP variants are similar, with two broad peaks located at the same positions (arrowheads in the figure). The characteristic profile changes in T23, which lacks most of the N-terminal cap, indicate rearrangements of the secondary structure packing in the chromophore-binding region.
is therefore simply called PYPM. Fig. 3 shows the intensity profiles of the PYPM intermediates of the truncated PYP variants compared with those obtained for their dark states. Significant differences between the two states were observed for each truncated PYP. The profiles of the PYPM intermediates of the three truncated PYP variants are similar, with two broad peaks located at the same positions (arrowheads in the figure). The characteristic profile changes in T23, which lacks most of the N-terminal cap, indicate rearrangements of the secondary structure packing in the chromophore-binding region.
FIGURE 3.

High-angle x-ray scattering profiles of T6, T15, and T23 under illumination (circles with error bars). As a reference, the profiles of the dark states are shown (dashed lines). The characteristic bimodal profiles observed under the illumination are noted by the arrowheads.
The profiles of the PYPM intermediates of all of the truncated PYP variants are superimposed on the log-log plot in the inset of Fig. 4. The differences among the profiles appear in the valley around Q = 0.3 Å−1, where the final slope of the shape scattering and the onset of the intramolecular interference scattering overlap. To derive the contribution from the secondary structure packing, the scattering profiles that were caused by intramolecular interference were extracted by subtracting the estimated shape scattering from the original profile. In general, the final slope of the shape scattering is proportional to Q−α, where α is related to the fractal dimension (42) or the protein conformational state (43). The shape scattering profile for each sample was evaluated by extrapolation of a line fit to the final slope (0.19 Å−1 < Q < 0.21 Å−1) in the log-log plots. In the area around the valley, the slopes of the regression lines for T15 and T23 are steeper than that for T6 (see the inset in Fig. 4). The excess intensity as a result of the shape scattering was subtracted from the original intensity profile, resulting in the corrected intramolecular interference profile of the PYPM intermediate for each truncated PYP (Fig. 4). All the corrected profiles were identical within the statistical errors. This agreement suggests that the secondary structure packing of the PYPM intermediates was essentially the same, regardless of differences in the length of N-terminal region. In other words, the N-terminal regions of T6 and T15 did not influence the intramolecular interference scattering. Note that the structure of the N-terminal region of the dark state of T6 is ordered and well defined. As was the case for the dark state of T15, the N-terminal region of T6 was disordered during the formation of PYPM, which eliminated the interference.
FIGURE 4.

Corrected scattering profiles of T6 (blue line), T15 (green line), and T23 (red line), in which the excess intensity compared with the profile of T23 is subtracted from the original profiles of T6 and T15. The original profiles are shown in the log-log plot in the inset. The solid lines in the inset are the regression lines obtained by fitting the final slope.
Characterization of the solution structure of the chromophore-binding region of PYPM
The change in the profile of T23 indicates a significant rearrangement in the secondary structure packing of the chromophore-binding region during the formation of PYPM (Fig. 3 c). On the basis of the obtained profile, we attempted to construct a solution structural model of PYPM, especially for the chromophore-binding region. We attempted to generate plausible conformations from a variety of structures derived from the crystal structure of PYP using the high-angle x-ray scattering profile as a boundary condition. The structure ensemble was constructed using the CONCOORD program, which was used in the fluctuation analysis of T23. As a reference, Fig. 5 shows the calculated intensity profiles of a subset of the structure ensemble generated by randomly extracting 10 structures from the ensemble of 500 structures. Although most of the structures resulted in profiles that resembled the profile of the dark state of T23 (a single broad peak at Q = 0.39 Å−1), some structures had profiles that displayed the bimodal shape observed for the PYPM intermediate of T23 (an example of this type of profile is denoted by the thick line in Fig. 5). The structures that satisfied the following two criteria were selected as the candidate models of the PYPM structure: 1), the peak position was observed at Q < 0.39 Å−1; and 2), a clear shoulder was present around Q = 0.6 Å−1. Consequently, 51 structures from the whole structural ensemble were selected. The average of the selected structures is shown in Fig. 6 as a model of PYPM (white solid ribbon model). The crystal structure of the dark state of intact PYP without the N-terminal 23 amino-acid residues is superimposed on the model structure. The loop between β4 and β5 and the α4 helix that envelops the chromophore-binding pocket in the dark state of the protein move away from each other, opening the chromophore-binding pocket. The calculated profiles of the dark-state structure and the PYPM model structure are described in Fig. 7 b and are very close to the respective observed profiles (Fig. 3 c), indicating that the average structure satisfied the criteria required for PYPM structures. The root mean-square deviation of the model structure of PYPM from the structure of intact PYP is shown in Fig. 7 a. This result suggests that the structural changes in PYPM are localized in the N-terminal tail (residues 24–28), the α4 helix (residues 55–58), and the loop connecting β4 and β5 (residues 96–102).
FIGURE 5.

The calculated scattering profiles of 10 structures randomly extracted from the parent structure ensemble consisting of 500 structures. For the sake of clarity, each profile is shifted along the longitudinal axis. The dashed line indicates the peak position (Q = 0.39 Å−1) in the scattering profile of the dark state of T23, which was calculated from the atomic structural model. The thick line shows the features that are similar to the experimentally obtained profile of T23 under illumination, including the characteristic bimodal shape (marked with arrowheads).
FIGURE 6.

The model structures of the chromophore-binding regions of the dark state of PYP (yellow solid ribbon model) and PYPM (white solid ribbon model). The model of the dark state was built after removal of the N-terminal 23 amino-acid residues from the crystal structure (1NWZ) of intact PYP. To assess the statistical reproducibility of the observed structural differences, five average structures obtained for the subsets of ∼10 candidate structures randomly extracted from the original 51 candidate models are superposed on the solid ribbon models (white line ribbon models). This clearly demonstrates that the five average structures of the subsets are well converged on the average model of the 51 candidate structures.
FIGURE 7.

(a) The root mean-square deviation values of the Cα positions in the model structure of PYPM compared with those in the model of the dark state. (b) High-angle x-ray scattering profiles calculated from the model of the dark state (open circles) and the model of PYPM (solid circles).
We then assessed the statistical significance of the observed structural displacements between the obtained model and the dark-state structure. The 51 selected structures were randomly divided into five subsets so that each subset included ∼10 independent structures, and the average structure of each subset was calculated. These 5 average structures (white line ribbon models in Fig. 6) were superimposed on the average structure of the 51 structures. Because of the decrease in the number of structures that were averaged for each subset compared with the overall averaging, the obtained average structures of the subsets fluctuated slightly around the average structure of all 51 of the selected structures. Common displacements between each of the average structures and the dark-state structure, however, can be observed demonstrating the statistical stability of the observed displacements obtained using our selection rules.
DISCUSSION
A two-dimensional x-ray detector and third-generation synchrotron radiation enabled us to accurately measure the high-angle x-ray scattering profiles of the proteins in solution. The recent development of the CRYSOL software package (36) enabled us to calculate a reliable scattering profile from the crystal structure. It has been reported that the scattering profile in the high-angle region is sensitive to conformational changes in the protein (30–33,43–45). Therefore, the analysis of high-angle scattering should provide useful information about a protein's conformation in solution and conformational changes that occur as the protein assumes transient states.
Using PYP as a model protein, we measured significant profile changes after deletion of the N-terminal region of PYP, indicating that the high-angle scattering profile reflects intramolecular interference. The agreement between the calculated and the experimentally obtained profiles for intact PYP, T6, and T23 indicates that their structures in solution are very similar to the crystal structure. This fact further indicates that high-angle x-ray scattering can be used to examine whether or not a solution structure is the same as a crystal structure.
We observed substantial changes in the high-angle scattering profiles for the truncated PYP variants on illumination, indicating significant conformational changes in the chromophore-binding region of PYP. To characterize these conformational changes, we generated structural models of PYPM and evaluated the validity of these atomic structural models with high-angle scattering.
A recent study revealed that equilibrium fluctuation in the ligand-free state (inactive state) is closely related with structural changes that occur on ligand binding (active state formation) (46). Furthermore, computational and experimental studies have proposed that the structure of an active state can be found in the structural ensemble of the inactive state (47–49). Therefore, it can be assumed that the structural ensemble that includes the structure of the active state of PYPM can be substituted with the structural ensemble obtained by MD simulation of the inactive state of PYP. Because the solution structure of PYPM was predicted to undergo large conformational changes, the ensemble should include large-amplitude fluctuations. MD simulation of large conformational changes is limited by computational resources. In this study, we used the CONCOORD program to build the ensemble instead of MD simulation. This program produces a structural ensemble that is generated using the empirical restriction parameters of the interatomic distances. The restriction using the empirical parameters is not as stringent as that used in MD simulations, and the obtained structure ensemble includes a wide range of structures. It was reported that although the fluctuation distribution obtained using CONCOORD is similar to that obtained with MD simulations, the amplitudes of the fluctuations tend to be larger (38). In this analysis, 51 candidates for the structure of PYPM were selected from the structural ensemble based on the calculated scattering profiles. The scattering profile for the average structure of the 51 selected structures retains two required characteristics of the PYPM structure (Fig. 7 b). We can conclude that the selected structures possess the common principal components of the structural displacements from the dark-state structure, which reflect the structural change that occurs during the formation of PYPM.
This structural analysis based on the intramolecular interference scattering observed in a high-angle region has some limitations. Highly disordered regions in an object cannot clearly exhibit intramolecular interference. Therefore, it is difficult to determine the structure of such regions, as was seen in the cases of the N-terminal regions of the dark state of T15 and of the PYPM intermediates of T6 and T15. Furthermore, mixed states that include several structurally distinguishable states make it cumbersome to determine the intrinsic structures of each state. To handle cases involving equilibria among several thermodynamically stable states, the observed profile should be decomposed into the individual profiles of each component. To overcome this problem, one simple solution is to find solution conditions that drastically shift the equilibrium toward the desired state. If such a condition cannot be found, a series of scattering profiles should be measured under various pHs, temperatures, and salt concentrations to slightly shift the equilibrium. Singular value decomposition analysis can then be applied to the data set to reconstruct a scattering profile of each component.
For PYPM of T23, the high-angle scattering profile exhibited substantial changes during the formation of the intermediate. Whereas a single broad peak at Q = 0.39 Å−1 was observed for the dark state of T23, the light state of T23 showed the characteristic bimodal shape. Consequently, we were able to select the recommended structural models using very qualitative selection rules. We are interested in the structural characterization of large structural changes in proteins, which can be assumed to be a crucial event for protein function. Because large structural changes produce significant alterations in scattering profiles, qualitative selection procedures are widely used to characterize these structural changes. Improving the structural selection criteria, however, may produce additional structural information. In this article, it was shown that the calculated profile was quite similar to the observed profiles in regard to the peak position and the overall profile shape. Carefully comparing the profiles, however, reveals slight differences between the calculated and observed profiles; the peak widths of the observed profiles are slightly broader than that of the calculated profile (e.g., comparing Fig. 3 and Fig. 7 b). This broadening is mainly caused by fluctuation around the average structure. It was reported that the scattering profile calculated from a crystal structure can be improved to match to the observed profile by employing the Debye-Waller expression in the calculation to account for fluctuations in the protein (45). Quantitative selection of the structures using this modified calculation method should also provide dynamic information about the protein.
The solution structures of PYP lacking the N-terminal 25 residues under the dark and illuminated conditions were clarified in NMR studies (19). The NMR structure demonstrates the partially unfolded nature of PYPM. In particular, the three regions at residues 42–58, 63–78, and 96–103 (the amino-acid positions in intact PYP) are highly disordered and adopt various orientations that expose the hydrophobic chromophore to the solvent. Although the structural changes in the α4 helix (residues 55–58) and the loop connecting β4 and β5 (residues 96–102) revealed in this study are conserved in the NMR structure, there are significant differences in the amplitudes of the structural displacements. In our model, the disordered region in the NMR structure does not appear to be as elongated, and the chromophore is buried inside the molecule. The 20 NMR structures of PYPM and the dark state of the protein are listed in the 1ODV and 1XFQ PDB files, respectively. The scattering profiles of the NMR structures were calculated using CRYSOL. The profiles of the 20 NMR structures of the dark state of PYP are similar to the profile of the dark state of T23 (not shown). On the other hand, the calculated profiles of the NMR structures of PYPM are quite different from each other and demonstrate ambiguous intramolecular interference scattering (Fig. 8). Although some structures resulted in two peaks with relatively low intensities, the positions of the two peaks are widely separated from each other and are not consistent with the peak positions obtained for T23. The calculated profiles also suggest that the NMR structures are different from each other. The spatial average of these profiles does not show the characteristic properties of the experimental scattering curve. The increases in the calculated radii of gyration of the NMR structures (>2 Å) are also larger than the observed value (∼0.7 Å) (22), suggesting that the NMR structures are not as compact as the native solution structure.
FIGURE 8.

The calculated scattering profiles of the NMR structures of PYPM (1ODV). Each profile is shifted along the longitudinal axis, and the profiles for model 1 to model 20 are shown from the bottom to the top of the figure. The dashed line indicates the peak positions (Q = 0.36 Å−1 and 0.55 Å−1) in the scattering profile of the PYPM intermediate of T23.
It is possible that the elongation of the α-helix-rich region (residues 42–78) and the degree of disorder are overestimated in the NMR structures of PYPM. Although the reason that NMR produced such highly disordered structures is unclear, the poor distance restraints in these regions may not yield good convergent structures, resulting in the divergent features of the obtained models. Our circular dichroism study on N-terminally truncated variants of PYP, however, suggested that a portion of the helices involved in chromophore binding region unfolds during the formation of PYPM (23). Thus, it is possible that the structure of PYPM actually fluctuates, particularly in the α-helix-rich region, causing the poor definition of the distance restrictions observed in the NMR study. The scattering profile of the PYPM intermediate of T23, however, clearly shows intramolecular interference, indicating that the fluctuation is not large and is not stochastic; i.e., the average structure can be defined. Therefore, the NMR structure may simply exaggerate each displacement.
X-ray scattering and NMR experiments provide different but complementary structural information about a protein in solution. NMR measurements reveal subsets of high-resolution interatomic distances in well-structured regions of an object. The number of distance restrictions, especially long-distance correlations, however, is rapidly reduced by slight fluctuations, resulting in an ensemble of divergent structures. On the other hand, high-angle x-ray scattering measurements provide global structural information through the superposition of distance correlations among all interatomic distances as long as an average structure of the object can be determined. However, it is difficult to extract each interatomic distance from the observed profile. Because the two measurements were performed using comparable solution conditions, combination of these two methods compensates for the drawbacks associated with each method. In this work, the structural ensemble was built from the dark-state structure of PYP using the CONCOORD software package with the default empirical restriction parameters for interatomic distances. If the subset of distance restrictions determined by NMR measurements is used in addition to the empirical parameters in the construction of a structural ensemble, the obtained structures that make up the ensemble necessarily satisfy the experimentally determined restrictions. Consequently, the structures selected from the ensemble using high-angle x-ray scattering criteria satisfy the restrictions determined by both methods, resulting in improvements of the accuracy and the reliability of the obtained model structure.
The structure of the N-terminal region of T6 is similar to that of the dark state of the wild-type protein. It, however, undergoes large structural changes during the formation of PYPM that abrogate the intramolecular interference between the N-terminal and chromophore-binding regions. The lack of interference suggests that the N-terminal region primarily moves stochastically and assumes a variety of conformations in PYPM. A schematic structural model for wild-type PYPM was built by combining the structural model of the chromophore-binding region of the PYPM intermediate of T23 with the structural fluctuation of the N-terminal region predicted by the results for T6 (Fig. 9). Note the curvature change of the β-sheet that accompanies the movement of the β4-β5 loop. Our previous study using Fourier transform infrared spectroscopy revealed that the prominent amide I peak assigned to the antiparallel β-sheet structure changed as the protein became the PYPM intermediate, although the precise mechanism underlying this change was unclear (23). The observed curvature change would cause the change in the amide I peak. Although the movement of the β4-β5 loop cannot be seen in a crystal (11,12), a similar structural change was predicted in an MD study, in which this loop was found to move ∼3 Å away from the other loop (50). The movements of the β4-β5 loop and the α4 helix are likely to be involved in regulating the accessibility of the chromophore-binding domain to solvent water molecules, resulting in a fully protonated chromophore during the formation of PYPM.
FIGURE 9.

A schematic model of the PYPM intermediate of intact PYP (solid ribbon model). The crystal structure of the dark state of intact PYP (1NWZ; line ribbon model) is superimposed on the model. The regions colored with red undergo large structural displacements during the formation of PYPM. The curvature change can be observed in the β5-β6 region (purple). The N-terminal region of PYPM (blue) is structurally disordered, which is predicted from T6, and does not represent a definable structure. Several residues in the N-terminal cap that are close to each other in the dark state are represented by ball-and-stick models on the crystal structure of intact PYP. The movement of Leu26 and Phe28 on the linker between the α2 helix and the β1 strand (red color) would perturb the adjacent Phe6, Leu15, and Leu23 residues. Because these residues are thought to participate in the stabilization of the N-terminal region, movement of the linker would cause the destabilization of PYPM.
The fluctuation analysis indicated that several residues, including Phe6, Ile11, Glu12, Leu15, Asp20, and Leu23, stabilize the N-terminal region of the protein (Fig. 2). According to the crystal structure of the dark state of PYP, these residues associate with the Leu26, Phe28, Lys110, Ala112, Trp119, and Phe121 from the β-sheets in the chromophore-binding region (37). The solution structural model of the chromophore-binding region of PYPM shows a large structural change in the N-terminal tail (residues 24–28) of T23, which is the linker between the α2 helix and the β1 strand in intact PYP. Assuming that this structural change in the N-terminal tail also occurs in intact PYP, Leu26 and Phe28 on the linker would be perturbed. Because these residues are close to the several residues involved in the N-terminal cap (Phe6, Leu15, and Leu23; Fig. 9), it is plausible that the structural change of the linker region involving Leu26 and Phe28 influences the stability of the N-terminal cap of intact PYP, leading to the formation of the chaotic N-terminal structure in PYPM.
High-angle x-ray scattering allows us to measure structural properties under various solution conditions. Moreover, it permits us to derive structural changes in the transient states of proteins as well as their resting structures in solution. Fluctuation analysis combined with high-angle x-ray scattering is a powerful method for deducing structural changes in a protein's transient states.
Acknowledgments
This work was performed with the approval of the JASRI Program Advisory Committee (Proposal No. 2001B0361-NLS-np, 2002A0387-NL2-np, and 2002B0689-NL2-np). It was supported in part by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology of Japan.
Abbreviations used: PYP, photoactive yellow protein; PYPM, the M intermediate of PYP; Tn, a PYP variant in which the N-terminal n residues were removed; Q, momentum transfer (= 4πsinθ/λ where θ is the scattering angle and λ the wavelength of the incident x-ray); SAXS, small-angle x-ray scattering; MD, molecular dynamics.
References
- 1.Meyer, T. E. 1985. Isolation and characterization of soluble cytochromes, ferredoxins and other chromophoric proteins from the halophilic phototrophic bacterium Ectothiorhodospira halophila. Biochim. Biophys. Acta. 806:175–183. [DOI] [PubMed] [Google Scholar]
- 2.Sprenger, W. W., W. D. Hoff, J. P. Armitage, and K. J. Hellingwerf. 1993. The eubacterium Ectothiorhodospira halophila is negatively phototactic, with a wavelength dependence that fits the absorption spectrum of the photoactive yellow protein. J. Bacteriol. 175:3096–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Borgstahl, G. E., D. R. Williams, and E. D. Getzoff. 1995. 1.4 A structure of photoactive yellow protein, a cytosolic photoreceptor: unusual fold, active site, and chromophore. Biochemistry. 34:6278–6287. [DOI] [PubMed] [Google Scholar]
- 4.Pellequer, J. L., K. A. Wager-Smith, S. A. Kay, and E. D. Getzoff. 1998. Photoactive yellow protein: a structural prototype for the three-dimensional fold of the PAS domain superfamily. Proc. Natl. Acad. Sci. USA. 95:5884–5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imamoto, Y., Y. Shirahige, F. Tokunaga, T. Kinoshita, K. Yoshihara, and M. Kataoka. 2001. Low-temperature Fourier transform infrared spectroscopy of photoactive yellow protein. Biochemistry. 40:8997–9004. [DOI] [PubMed] [Google Scholar]
- 6.Meyer, T. E., E. Yakali, M. A. Cusanovich, and G. Tollin. 1987. Properties of a water-soluble, yellow protein isolated from a halophilic phototrophic bacterium that has photochemical activity analogous to sensory rhodopsin. Biochemistry. 26:418–423. [DOI] [PubMed] [Google Scholar]
- 7.Hoff, W. D., I. H. van Stokkum, H. J. van Ramesdonk, M. E. van Brederode, A. M. Brouwer, J. C. Fitch, T. E. Meyer, R. van Grondelle, and K. J. Hellingwerf. 1994. Measurement and global analysis of the absorbance changes in the photocycle of the photoactive yellow protein from Ectothiorhodospira halophila. Biophys. J. 67:1691–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imamoto, Y., M. Kataoka, and F. Tokunaga. 1996. Photoreaction cycle of photoactive yellow protein from Ectothiorhodospira halophila studied by low-temperature spectroscopy. Biochemistry. 35:14047–14053. [DOI] [PubMed] [Google Scholar]
- 9.Ujj, L., S. Devanathan, T. E. Meyer, M. A. Cusanovich, G. Tollin, and G. H. Atkinson. 1998. New photocycle intermediates in the photoactive yellow protein from Ectothiorhodospira halophila: picosecond transient absorption spectroscopy. Biophys. J. 75:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imamoto, Y., M. Kataoka, F. Tokunaga, T. Asahi, and H. Masuhara. 2001. Primary photoreaction of photoactive yellow protein studied by subpicosecond-nanosecond spectroscopy. Biochemistry. 40:6047–6052. [DOI] [PubMed] [Google Scholar]
- 11.Genick, U. K., G. E. Borgstahl, K. Ng, Z. Ren, C. Pradervand, P. M. Burke, V. Srajer, T. Y. Teng, W. Schildkamp, D. E. McRee, K. Moffat, and E. D. Getzoff. 1997. Structure of a protein photocycle intermediate by millisecond time-resolved crystallography. Science. 275:1471–1475. [DOI] [PubMed] [Google Scholar]
- 12.Ihee, H., S. Rajagopal, V. Srajer, R. Pahl, S. Anderson, M. Schmidt, F. Schotte, P. A. Anfinrud, M. Wulff, and K. Moffat. 2005. Visualizing reaction pathways in photoactive yellow protein from nanoseconds to seconds. Proc. Natl. Acad. Sci. USA. 102:7145–7150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brudler, R., R. Rammelsberg, T. T. Woo, E. D. Getzoff, and K. Gerwert. 2001. Structure of the I1 early intermediate of photoactive yellow protein by FTIR spectroscopy. Nat. Struct. Biol. 8:265–270. [DOI] [PubMed] [Google Scholar]
- 14.Xie, A., L. Kelemen, J. Hendriks, B. J. White, K. J. Hellingwerf, and W. D. Hoff. 2001. Formation of a new buried charge drives a large-amplitude protein quake in photoreceptor activation. Biochemistry. 40:1510–1517. [DOI] [PubMed] [Google Scholar]
- 15.Van Brederode, M. E., W. D. Hoff, I. H. van Stokkum, M. L. Groot, and K. J. Hellingwerf. 1996. Protein folding thermodynamics applied to the photocycle of the photoactive yellow protein. Biophys. J. 71:365–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohishi, S., N. Shimizu, K. Mihara, Y. Imamoto, and M. Kataoka. 2001. Light induces destabilization of photoactive yellow protein. Biochemistry. 40:2854–2859. [DOI] [PubMed] [Google Scholar]
- 17.Khan, J. S., Y. Imamoto, M. Harigai, M. Kataoka, and M. Terazima. 2006. Conformational changes of PYP monitored by diffusion coefficient: effect of N-terminal alpha-helices. Biophys. J. 90:3686–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubinstenn, G., G. W. Vuister, F. A. Mulder, P. E. Dux, R. Boelens, K. J. Hellingwerf, and R. Kaptein. 1998. Structural and dynamic changes of photoactive yellow protein during its photocycle in solution. Nat. Struct. Biol. 5:568–570. [DOI] [PubMed] [Google Scholar]
- 19.Bernard, C., K. Houben, N. M. Derix, D. Marks, M. A. van der Horst, K. J. Hellingwerf, R. Boelens, R. Kaptein, and N. A. van Nuland. 2005. The solution structure of a transient photoreceptor intermediate: Delta25 photoactive yellow protein. Structure. 13:953–962. [DOI] [PubMed] [Google Scholar]
- 20.Shimizu, N., H. Kamikubo, K. Mihara, Y. Imamoto, and M. Kataoka. 2002. Effect of organic anions on the photoreaction of photoactive yellow protein. J. Biochem. (Tokyo). 132:257–263. [DOI] [PubMed] [Google Scholar]
- 21.Harigai, M., S. Yasuda, Y. Imamoto, K. Yoshihara, F. Tokunaga, and M. Kataoka. 2001. Amino acids in the N-terminal region regulate the photocycle of photoactive yellow protein. J. Biochem. (Tokyo). 130:51–56. [DOI] [PubMed] [Google Scholar]
- 22.Imamoto, Y., H. Kamikubo, M. Harigai, N. Shimizu, and M. Kataoka. 2002. Light-induced global conformational change of photoactive yellow protein in solution. Biochemistry. 41:13595–13601. [DOI] [PubMed] [Google Scholar]
- 23.Harigai, M., Y. Imamoto, H. Kamikubo, Y. Yamazaki, and M. Kataoka. 2003. Role of an N-terminal loop in the secondary structural change of photoactive yellow protein. Biochemistry. 42:13893–13900. [DOI] [PubMed] [Google Scholar]
- 24.Glatter, O., and O. Kratky. 1982. Small Angle X-ray Scattering. Academic Press, New York.
- 25.Feigin, L. A., and D. I. Svergun. 1987. Structure Analysis by Small-Angle X-Ray and Neutron Scattering. Plenum Press, New York.
- 26.Svergun, D. I. 1999. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76:2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Svergun, D. I., M. V. Petoukhov, and M. H. J. Koch. 2001. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 80:2946–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Funari, S. S., G. Rapp, M. Perbandt, K. Dierks, M. Vallazza, C. Betzel, V. A. Erdmann, and D. I. Svergun. 2000. Structure of free Thermus flavus 5 S rRNA at 1.3 nm resolution from synchrotron x-ray solution scattering. J. Biol. Chem. 275:31283–31288. [DOI] [PubMed] [Google Scholar]
- 29.Kato, R., M. Kataoka, H. Kamikubo, and S. Kuramitsu. 2001. Direct observation of three conformations of MutS protein regulated by adenine nucleotides. J. Mol. Biol. 309:227–238. [DOI] [PubMed] [Google Scholar]
- 30.Fedorov, B. A. 1975. A “block” approach to the study of highly helical proteins by x-ray scattering in solution. J. Mol. Biol. 98:341–353. [DOI] [PubMed] [Google Scholar]
- 31.Ueki, T., Y. Inoko, M. Kataoka, Y. Amemiya, and Y. Hiragi. 1986. X-ray scattering study on hemoglobin solution with synchrotron radiation: a simple analysis of scattering profile at moderate angles in terms of arrangement of subunits. J. Biochem. (Tokyo). 99:1127–1136. [DOI] [PubMed] [Google Scholar]
- 32.Kataoka, M., I. Nishii, T. Fujisawa, T. Ueki, F. Tokunaga, and Y. Goto. 1995. Structural characterization of the molten globule and native states of apomyoglobin by solution x-ray scattering. J. Mol. Biol. 249:215–228. [DOI] [PubMed] [Google Scholar]
- 33.Pickover, C. A., and D. M. Engelman. 1982. On the interpretation and prediction of x-ray scattering profiles from biomolecules in solution. Biopolymers. 21:817–831. [Google Scholar]
- 34.Mihara, K., O. Hisatomi, Y. Imamoto, M. Kataoka, and F. Tokunaga. 1997. Functional expression and site-directed mutagenesis of photoactive yellow protein. J. Biochem. (Tokyo). 121:876–880. [DOI] [PubMed] [Google Scholar]
- 35.Miura, K., M. Kawamoto, K. Inoue, M. Yamamoto, T. Kumasaka, M. Sugiura, A. Yamano, and H. Moriyama. 2000. SPring-8 User Experiment Report. 4:168.
- 36.Svergun, D. I., C. Baberato, and M. H. J. Koch. 1995. CRYSOL—a program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinate. J. Appl. Crystallogr. 28:768–773. [Google Scholar]
- 37.Getzoff, E. D., K. N. Gutwin, and U. K. Genick. 2003. Anticipatory active-site motions and chromophore distortion prime photoreceptor PYP for light activation. Nat. Struct. Biol. 10:663–668. [DOI] [PubMed] [Google Scholar]
- 38.de Groot, B. L., D. M. van Aalten, R. M. Scheek, A. Amadei, G. Vriend, and H. J. Berendsen. 1997. Prediction of protein conformational freedom from distance constraints. Proteins. 29:240–251. [DOI] [PubMed] [Google Scholar]
- 39.Borucki, B., J. A. Kyndt, C. P. Joshi, H. Otto, T. E. Meyer, M. A. Cusanovich, and M. P. Heyn. 2005. Effect of salt and pH on the activation of photoactive yellow protein and gateway mutants Y98Q and Y98F. Biochemistry. 44:13650–13663. [DOI] [PubMed] [Google Scholar]
- 40.Borucki, B., C. P. Joshi, H. Otto, M. A. Cusanovich, and M. P. Heyn. 2006. The transient accumulation of the signaling state of photoactive yellow protein is controlled by the external pH. Biophys. J. 91:2991–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shimizu, N., Y. Imamoto, M. Harigai, H. Kamikubo, Y. Yamazaki, and M. Kataoka. 2006. pH-dependent equilibrium between long lived near-UV intermediates of photoactive yellow protein. J. Biol. Chem. 281:4318–4325. [DOI] [PubMed] [Google Scholar]
- 42.Schmidt, P. W. 1991. Small-angle scattering studies of disordered, porous and fractal systems. J. Appl. Crystallogr. 24:414–435. [Google Scholar]
- 43.Kataoka, M., and Y. Goto. 1996. X-ray solution scattering studies of protein folding. Fold. Des. 1:107–114. [DOI] [PubMed] [Google Scholar]
- 44.Zhang, R., P. Thiyagarajan, and D. M. Tiede. 2000. Probing protein fine structures by wide angle solution x-ray scattering. J. Appl. Crystallogr. 33:565–568. [Google Scholar]
- 45.Tiede, D. M., R. Zhang, and S. Seifert. 2002. Protein conformations explored by difference high-angle solution x-ray scattering: oxidation state and temperature dependent changes in cytochrome c. Biochemistry. 41:6605–6614. [DOI] [PubMed] [Google Scholar]
- 46.Goh, C. S., D. Milburn, and M. Gerstein. 2004. Conformational changes associated with protein-protein interactions. Curr. Opin. Struct. Biol. 14:104–109. [DOI] [PubMed] [Google Scholar]
- 47.Kumar, S., B. Ma, C. J. Tsai, N. Sinha, and R. Nussinov. 2000. Folding and binding cascades: dynamic landscapes and population shifts. Protein Sci. 9:10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bosshard, H. R. 2001. Molecular recognition by induced fit: how fit is the concept? News Physiol. Sci. 16:171–173. [DOI] [PubMed] [Google Scholar]
- 49.Ikeguchi, M., J. Ueno, M. Sato, and A. Kidera. 2005. Protein structural change upon ligand binding. Linear response theory. Phys. Rev. Lett. 94: article 078102. [DOI] [PubMed]
- 50.Shiozawa, M., M. Yoda, N. Kamiya, N. Asakawa, J. Higo, Y. Inoue, and M. Sakurai. 2001. Evidence for large structural fluctuations of the photobleached intermediate of photoactive yellow protein in solution. J. Am. Chem. Soc. 123:7445–7446. [DOI] [PubMed] [Google Scholar]