

Abstract
In cardiac ventricular myocytes, events crucial to excitation-contraction coupling take place in spatially restricted microdomains known as dyads. The movement and dynamics of calcium (Ca2+) ions in the dyad have often been described by assigning continuously valued Ca2+ concentrations to one or more dyadic compartments. However, even at its peak, the estimated number of free Ca2+ ions present in a single dyad is small (∼10–100 ions). This in turn suggests that modeling dyadic calcium dynamics using laws of mass action may be inappropriate. In this study, we develop a model of stochastic molecular signaling between L-type Ca2+ channels (LCCs) and ryanodine receptors (RyR2s) that describes: a), known features of dyad geometry, including the space-filling properties of key dyadic proteins; and b), movement of individual Ca2+ ions within the dyad, as driven by electrodiffusion. The model enables investigation of how local Ca2+ signaling is influenced by dyad structure, including the configuration of key proteins within the dyad, the location of Ca2+ binding sites, and membrane surface charges. Using this model, we demonstrate that LCC-RyR2 signaling is influenced by both the stochastic dynamics of Ca2+ ions in the dyad as well as the shape and relative positioning of dyad proteins. Results suggest the hypothesis that the relative placement and shape of the RyR2 proteins helps to “funnel” Ca2+ ions to RyR2 binding sites, thus increasing excitation-contraction coupling gain.
INTRODUCTION
Contraction of the cardiac myocyte results from a process known as excitation-contraction coupling (ECC). ECC is initiated when individual L-type calcium (Ca2+) channels (LCCs) open in response to membrane depolarization, producing Ca2+ flux into a microdomain known as the dyad. The resulting increase in dyadic Ca2+ leads to opening of Ca2+-sensitive Ca2+-release channels known as ryanodine receptors (RyR2s) located in the closely apposed junctional sarcoplasmic reticulum (JSR) membrane, producing additional flux of Ca2+ from the JSR into the dyad (Fig. 1 A). These two sources of Ca2+ flux generate the intracellular Ca2+ transient that triggers cardiac muscle contraction. Understanding the molecular basis of this Ca2+-induced Ca2+-release (CICR) process is therefore of fundamental importance to understanding cardiac muscle function in both health and disease.
FIGURE 1.

(A) A schematic representation of calcium-induced calcium release in the dyad. Ca2+ ions pass through L-type Ca2+ channels (LCCs) and enter the dyad where they bind to ryanodine receptors (RyRs), triggering their opening leading to Ca2+ release from the junctional sarcoplasmic reticulum (JSR). Ca2+ ions released from the SR diffuse out of the dyad and into the bulk myoplasm, where they can bind to myofilaments, and initiate cell shortening. (B) Layout of LCCs and RyR2s within the dyad. The quasicrystal array of RyR2s (gray shaded boxes with crosshair) lie in the JSR membrane with a smaller number of LCCs (green circles) randomly distributed in the opposing T-tubule membrane.
Recent measurements indicate that there are ∼20–100 RyR2s per dyad and that dyad diameter and height (i.e., sarcolemmal-JSR membrane spacing) are ∼100–200 nm and ∼15 nm, respectively (1,2). A range of computational models predict that during an action potential (AP), peak Ca2+ concentration in the dyad may range from 100 to 1000 μM (3,4). A simple calculation shows that this corresponds to 10–100 free Ca2+ ions in the dyad (2). Thus, it is clear that both feed forward and feed back signaling between RyR2s and LCCs in the dyad is likely mediated by relatively few Ca2+ ions. Additionally, these qualitative estimates of the number of Ca2+ ions involved in LCC-RyR2 signaling suggest that conclusions based on simulations that determine gradients in dyad Ca2+ concentration using models based on laws of mass action may be problematic (see Bhalla (5) for further discussion). Rather, it is likely that the stochastic motions of Ca2+ ions within the dyad impart a degree of “signaling noise” between LCCs and RyR2s and that this signaling noise may in turn affect macroscopic properties of CICR at the cell and tissue level (e.g., see Tanskanen et al. (6)).
The small dimensions, in particular the limited height, of the dyad imply that the structure of proteins located within the dyad may also serve to restrict motion of Ca2+ ions. The largest protein within the dyad is RyR2. The structure of RyR2 has been measured at a resolution of 3.0 nm (7), whereas that of RyR1 (the skeletal muscle isoform, sharing ∼70% sequence identity to RyR2 (8)) has been measured at 1.0 nm resolution (9). RyR2 is a large protein composed of four 565-kDa subunits. The cytoplasmic portion of the protein has dimensions ∼27 × 27 × 12 nm (9,10), where the 12-nm height spans nearly the full distance between JSR and T-tubule membranes (1,2,11). The clustering of RyR2s opposite each LCC therefore presents a considerable Ca2+ diffusion barrier. The crystal structure of the cardiac LCC has also been measured at ∼0.4-nm resolution (12). The LCC is ∼19 nm in height and ∼14.5 nm in width, protruding from the T-tubule membrane ∼2 nm into the dyad (12). In addition, the structure of the Ca2+-binding protein calmodulin (CaM), one molecule of which is tethered to the inner pore of the LCC (13), has been measured at 0.1-nm resolution (14). CaM is a dual-lobed protein of approximate dimensions 4.5 × 4.5 × 6.5 nm. Given the small dimensions of the dyad, it is likely that the physical location and dimensions of these important dyad proteins have a considerable influence on motion of Ca2+ ions and thus properties of CICR.
In this study, we present a computational, molecularly and structurally detailed model of the cardiac dyad and use this model to address the following questions: 1), What is the number of Ca2+ ions that are present in the dyad during CICR? 2), How does the physical arrangement of large proteins within the dyad influence the process of CICR? 3), How does “signaling noise” due to the small number of Ca2+ ions within the dyad affect the nature of CICR. The results demonstrate that: 1), at the level of the single dyad, CICR may be mediated by as few as 20–50 Ca2+ ions; 2), even though the number of free Ca2+ ions in a single dyad during CICR is small and highly variable, ECC gain is consistently high (measured over the population of dyads in a single cell) and is a robust feature of local Ca2+ signaling; and 3), the structure of proteins that reside in the dyad help to funnel Ca2+ toward RyR2 binding sites, and in doing so, enhance ECC gain.
METHODS
The motion of individual Ca2+ ions in the dyad is influenced by the following factors: a), interactions between Ca2+ ions and other mobile ions and molecules within the dyad; b), the physical boundary imposed by membranes bounding the dyad and the location and shape of proteins within the dyad; c), the presence of an electric field near the T-tubule membrane; d), stochastic gating of LCCs and RyR2s, producing a stochastic boundary flux; e), location of Ca2+ binding sites (such as those on LCCs, RyR2s, and CaM); and f), the nature of the interface between the dyad and the surrounding myoplasm. To properly simulate Ca2+ dynamics, these influences must be included in a detailed model of the dyad.
Structure of the dyad
The volume of the dyad is represented using a 200 × 200 × 15 nm lattice (Fig. 1 B; gray boxes represent RyR2; green dots represent LCCs) with 1-nm spacing between lattice points. The lower boundary of the dyad is limited by the T-tubular membrane and the upper boundary is limited by the JSR membrane (Fig. 1 A). Ca2+ can diffuse across the lateral boundary of the dyad between the dyadic volume and the myoplasm (Fig. 1 A). The geometry of the dyadic volume that is accessible to diffusing Ca2+ ions is determined by the presence, shape, and location of proteins, which due to their large size relative to the dyad, occupy a significant fraction of the dyadic volume. Chief among these are LCCs, RyR2s, and CaM. RyR2s are located in the JSR membrane in quasicrystalline arrays of tens of RyR2s (Fig. 1 B) (1). In this study, we employ a structural model of the cytosolic (i.e., dyadic) assembly of the RyR2 based on experimental cryo-electron microscopy (cryo-EM) measurements (Fig. 2 A) (10,15). The overall dimensions of the cytosolic assembly of the RyR2 are ∼27 × 27 × 12 nm and the model exhibits fourfold symmetry with four “feet” structures. Substantial evidence indicates the pore of the RyR2 complex is located in the center of the tetrameric structure (Fig. 2 A, green dot) (9). Each model dyad is assumed to contain 20 RyR2s arranged in an asymmetric 4 × 5 quasicrystal (Fig. 1 B), where neighboring RyR2s come into contact with each other near their corners, with an overlap of 12 nm along their edges (16). A 5:1 RyR2/LCC ratio is assumed consistent with previous measures of the relative density of RyR2 and LCC proteins (2,17). Thus, this model dyad shares some fundamental features with the calcium release unit model studied previously by Greenstein and Winslow (18).
FIGURE 2.

(A) Model RyR2 structure as seen from its cytosolic face based on experimentally measured structure (10,15,28). Red spheres indicate the positions of the baseline model Ca2+-binding activation sites, blue spheres indicate the positions of alternate hypothetical activation sites, yellow spheres indicate the positions of model inactivation sites, and the green sphere depicts the assumed position of the RyR2 pore. (B) Side view of a portion of the dyad showing a single RyR2 and opposing LCC with tethered CaM. The assumed position of the Ca2+-binding sites for CDI on CaM are indicated by yellow spheres.
The precise locations of the Ca2+-binding sites mediating RyR2 activation and inactivation are not yet known. Using computational methods, Takeshima et al. (19) identified a region of RyR1 (residues 4364–4529) containing three predicted high-affinity Ca2+ binding sites located near segment M1 (Zorzato nomenclature (20)). Gel overlay assays were used to show that these sites bound 45Ca2+, and an antibody to residues 4478–4512 (the third predicted binding site) increased RyR1 open probability (21). In addition, RyR1 residues within the region 4254–4631 influence Ca2+ binding affinity (22). Both of these loci are within divergent region 1 (DR1) of RyR1 and RyR2 proteins. This region has been mapped to physical domain 3 (the “handle” domain) (7,23). However, a more recent study investigating the functional consequences of mutations of the predicted Takeshima EF-hand binding sites demonstrated little effect on Ca2+ binding, calling into question the validity of these locations as Ca2+ activation binding sites (24). In subsequent studies, Li and Chen demonstrated that a single point mutation of the highly conserved glutamate 3987 in segment M2 of mouse RyR2 dramatically reduces Ca2+ sensitivity (25). Segment M2 is adjacent to DR1 and is likely within physical domain 3 (26). Considered together, these results suggest that physical domain 3 may play an important role in the Ca2+ activation process and that the RyR2 glutamate at position 3987 lies within a region that remains a candidate as a Ca2+ activation binding site. We therefore assume that each of the four RyR2 subunits contains a single activation site in a position corresponding to domain 3 (Fig. 2 A, red dots). To better understand how the location of the RyR2 Ca2+ activation binding sites influence CICR (see Fig. 6), we also test a hypothesized alternative location for these sites on the surface of domain 1, within ∼3 nm of the RyR2 pore (Fig. 2 A, blue dots).
FIGURE 6.

Histograms of Ca2+ release latency (defined as the time between opening of the first LCC and opening of the first RyR2) from a population of 1500 dyads simulated at 0 mV under the following conditions: (A) geometric protein structures included, electric field present (i.e., membrane surface charges present); (B) geometric protein structures excluded, electric field present; (C) geometric protein structures included, electric field absent; and (D) geometric protein structures excluded, electric field absent.
The existence of a mechanism for Ca2+-mediated RyR2 inactivation under physiological conditions remains uncertain. Ca2+ binding motifs have been identified between amino acids 3726 and 5037 in the C-terminus region of a RyR2 monomer and have been suggested as possible inactivation sites (24,27). However, these amino acid residues are located predominantly in the transmembrane region of the RyR2 protein and are obscured by the much larger cytoplasmic assembly (28). Recently, Thomas et al. (29) suggested that binding sites may instead be localized to N-terminal and central domains. Functional characterization of three types of RyR2 mutations linked to arrhythmogenic right ventricular dysplasia type 2 (ARVD2) (30) demonstrate profound alterations to Ca2+ sensitivity and Ca2+-dependent inactivation when compared with wild-type (WT) channels. In particular, WT RyR2 exhibited biphasic Ca2+-dependent Ca2+ release with high affinity Ca2+ activation and low affinity Ca2+ inactivation. Mutant channels expressing L433P (domain 10, N-terminus) revealed significant reduction in Ca2+-dependent inactivation. The N2386I mutation (domain 9, central domain) displayed heightened Ca2+ sensitivity. The combined mutations at domains 10 and 9 (R176Q/T2504M) demonstrated total ablation of Ca2+-dependent inactivation. Cryo-EM reveals that domains 9 and 10 lie in close proximity to each other and partially comprise the portion of the RyR2 tetramer that extends into the cytosol known as the clamp (28). Therefore, in this study, we localized the low affinity Ca2+-binding inactivation sites to the cleft regions between domains 10 and 9 on the clamp portions of the RyR2 tetramer (Fig. 2 A, yellow dots), supporting the observation that mutations to either domain may inhibit Ca2+-dependent inactivation.
The locations of LCCs within the dyad are shown in Fig. 1 B (green dots). The cytosolic region of each LCC structure occupies an area of 10 × 13 nm and extends 2 nm from the inner surface of the T-tubule membrane. A constitutively tethered CaM acts as the Ca2+ sensor for Ca2+-dependent inactivation of LCCs (31,32). It has recently been shown that a single CaM molecule is both necessary and sufficient to produce Ca2+-dependent inactivation of its associated channel (13). The model therefore includes a single CaM molecule associated with each LCC (Fig. 2 B). Because the precise location and orientation of this CaM with respect to the LCC is not known, we assume, for simplicity, that the CaM is located adjacent to the LCC along the sarcolemma. A geometric model of CaM is created by approximating the crystal structure of its Ca2+-unbound form (identified as 1CFD in Protein Data Bank), as measured experimentally by Kuboniwa et al. (33), at 1-nm resolution (Fig. 2 B). A single CaM molecule contains four Ca2+ binding sites, of which the two sites located on the carboxyl tail have been shown to be responsible for Ca2+-dependent inactivation (CDI) of the associated LCC (31,32). In this model, CDI can proceed when both of the carboxyl-tail binding sites are occupied (Fig. 2 B, yellow dots). It has been assumed that the carboxyl tail of CaM and the associated CDI binding sites are oriented toward the LCC. LCC facilitation is thought to be regulated by the two Ca2+ binding sites located on the amino-terminal lobe of CaM (34) (see Anderson (35) for a review), however the process of LCC facilitation was not included in this model, and the amino-terminal binding sites were therefore not implemented in the model CaM molecule.
RyR2 and LCC kinetic models
A comprehensive description of Ca2+ dynamics in the dyad requires kinetic models of LCC and RyR2 gating to quantitatively describe the source fluxes of Ca2+ into the dyad. LCCs and RyR2s are modeled using continuous-time, discrete-state Markov processes. In many previous models (e.g. (18,36)), the incorporation of Ca2+-dependent ion channel gating kinetics was accomplished by allowing model state transition rates to be defined by expressions that depend upon the relevant Ca2+ concentration. This approach is an approximation in that it combines the Ca2+-binding step and the conformational change of the ion channel (e.g., inactivation) into a single state transition where the conformational change of the channel protein has been assumed to be rate-limiting (e.g., see Peterson et al. (32)). In the model presented here, Ca2+ binding to the channel protein must be considered separately from the subsequent conformational change of the channel protein. In order for a Ca2+-dependent state transition to occur, the required Ca2+-binding sites must first be occupied by Ca2+ ions (e.g., Ca2+-dependent inactivation of an LCC requires that the two carboxyl-terminal Ca2+ binding sites of the associated CaM are occupied). When a freely diffusing Ca2+ ion enters a lattice position adjacent to an available Ca2+-binding site, that Ca2+ ion has the opportunity to bind to the site. The relative magnitude of the binding rate compared to the rate of diffusion determines the probability that the Ca2+ ion either binds to the site or diffuses away. Ca2+-binding transitions are incorporated into the overall model of Ca2+ movement in the dyad (see below). Ion channel transition rates are therefore defined as functions that depend upon the occupancy of the Ca2+ binding sites (rather than Ca2+ concentration). Rates for binding and unbinding of Ca2+ to sites on both the LCCs and RyR2s are given in the Appendix.
The kinetic model of the LCC is described using an 11-state continuous time Markov chain model (Fig. 3 A) developed previously (18,36,37). Briefly, the upper row of states describes the LCC normal gating mode (Mode Normal) and the lower row of states describes gating when the LCC undergoes Ca2+-dependent inactivation (Mode CDI). The “downward” transition rates from Mode Normal to Mode CDI become nonzero only when both Ca2+-binding sites of the associated CaM molecule are occupied. The rate for CDI was adjusted based on that used in a previous implementation of this channel model in the presence of 100 μM [Ca2+] (18). Transition rates from Mode CDI to Mode Normal (i.e., recovery from Ca2+-mediated inactivation) do not depend on whether or not Ca2+ ions are bound to the associated CaM. Voltage-dependent inactivation of the LCC is incorporated as a separate gating variable (not shown in Fig. 3) with rates identical to those described previously (18).
FIGURE 3.

(A) Kinetic model of LCC gating based on the model of Greenstein and Winslow (18). (B) Kinetic model of RyR2 gating based on that of Stern et al. (38). The processes of Ca2+ binding/unbinding to activation and inactivation sites and the process of voltage-dependent inactivation are not depicted in this figure (see text for details). Transition rate constants are provided in the Appendix.
The kinetics of RyR2 gating are described by a four-state Markov model (Fig. 3 B) originally developed by Stern et al. (38,39). In this model, state 1 is the closed resting state; state 2 is the open state; and states 3 and 4 represent Ca2+ inactivated states. Based on the fourfold symmetry of the RyR2 protein, the original model of Stern et al. (38,39) has been modified to include the assumption that channel opening requires Ca2+ binding to all four activation sites (one on each subunit). This is implemented by allowing the opening rate from state 1 to state 2 to become nonzero only when all four activation sites are Ca2+ bound. In addition to being consistent with RyR2 tetrameric structure, the assumption of four Ca2+ activation sites agrees with experimental studies where the application of fast Ca2+ spikes (generated via flash photolysis) demonstrated a steep Ca2+-dependence of activation, indicating that multiple Ca2+ ions (n ∼ 4) must bind the channel to enable opening (40). It is assumed that RyR2 inactivation can proceed when Ca2+ is bound to at least one of the four inactivation sites. In accordance with previous studies (39), it is assumed that the rate of RyR2 inactivation is dependent upon the activation state of the channel (i.e., whether it is open or closed). This was implemented by allowing the rate of Ca2+ binding/unbinding to the RyR2 inactivation sites to depend upon its conformational state. All RyR2 transition rates, and Ca2+-binding/unbinding rates are given in the Appendix.
The unitary Ca2+ current of a single RyR2 is assumed to be 1.24 pA under physiological conditions (38,39). This corresponds to an influx rate of 3870 Ca2+ ions per millisecond. LCC permeability is set such that the unitary Ca2+ current through a single LCC is 0.12 pA at 0 mV (41), which corresponds to an influx rate of 375 Ca2+ ions per millisecond. LCC open-channel permeability, and hence Ca2+ influx rate, varies with membrane potential as described previously (see Fig. 2 G of Greenstein and Winslow (18)). The entry of Ca2+ ions into the dyad via an open RyR2 or LCC is simulated by a Poisson process with a rate set equal to the rate of Ca2+ ion entry for each LCC or RyR2 as described above. It is assumed that ion entry via an LCC or RyR2 is blocked if a Ca2+ ion occupies the lattice point that is adjacent to the channel mouth.
Membrane Ca2+ binding
Ca2+ buffering plays an important role in cardiac myocyte Ca2+ dynamics (2). In the dyad, membrane phospholipid headgroups act as fixed Ca2+ buffers. The density of slow SR and sarcolemmal buffering sites is large, typically assumed to be ∼0.1–0.2 site/nm2 (3,4). Binding site density and Ca2+ binding/unbinding rates in this model are based on the work of Langer and Peskoff (3) and Soeller and Cannell (4) and are given in the Appendix. It has been assumed that both low-affinity and high-affinity Ca2+ binding sites are present on both the SR membrane and sarcolemma. Ca2+ binding to buffer sites is implemented in the same manner as described above for Ca2+ binding to ion channel proteins. In addition to fixed Ca2+ buffers, mobile Ca2+ buffers are also known to be present in the dyad. Freely diffusing calmodulin is the main mobile Ca2+ buffer in the dyad, however, the typically assumed CaM concentration of ∼15 μM (42) translates into a single molecule in a dyad of radius 50 nm. Recently, however, it has been suggested that there may be local enrichment of CaM molecules in the vicinity of the LCC, resulting in as many as 25 free CaM molecules at the site of each LCC (13). It remains unclear how these CaM molecules would be targeted to the LCC, and whether they can freely diffuse in this restricted space. Because the local dynamics of free CaM are not yet understood, and because CaM is a large, rather slowly diffusing molecule, freely diffusing CaM molecules were not included in this dyad model. Similarly, the role of ATP as a mobile Ca2+ buffer was not included in this model.
Ca2+ dynamics in the dyad
The motion of a mobile Ca2+ ion in the dyad is influenced by the Brownian random force from the surrounding solvent and the electrostatic potential stemming from proteins, membranes, and other ions, including other Ca2+ ions. Indeed, in the confined space of the dyad, collisions of Ca2+ ions with other free ions are likely to be frequent and therefore correlations between the ions should be considered. Consequently, we model the joint positions (r1,…,rN) of N Ca2+ ions present in the dyad as a 3N-dimensional Brownian motion in a potential field, which describes both interactions of Ca2+ ions with other Ca2+ ions as well as electrostatic potentials. The time evolution of the joint probability density of these Ca2+ ions to be present at positions (r1,…,rN) in the dyad at time t, P(r1,…,rN,t), is described by the Fokker-Planck equation (FPE; see, e.g., Risken (43))
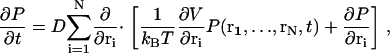 |
(1) |
where ri = (ri,1,ri,2,ri,3) is a vector indicating the position of the i-th ion, D is the diffusion constant, kB is Boltzmann's constant, T is temperature, and the notation ∂/∂ri is defined as
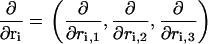 |
The total potential energy of the system, V, is given as a function of the ion positions,
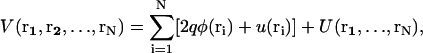 |
(2) |
where q is the elementary charge. The total potential energy V has several contributions: a), φ(ri) is the electrostatic potential of the i-th ion due to charges on the surrounding lipids and proteins (for example, φ contains the potential due to surface charge density, ρ, from the negatively charged phospholipid headgroups); b), u(ri) is known as a hard-core potential that becomes nonzero at the location of impenetrable structures such as proteins within the dyad. This potential simply defines the space accessible for the mobile ions and is ultimately determined by the structural model of the dyad; and c), U(r1,…,rN) is the mutual interaction potential between the Ca2+ ions. This potential is determined by the physical size (hard-core repulsion) of the ions, and the dielectric/buffering conditions in the dyad. For two ions, U depends on the ionic separation and the range of U is determined by the Debye length, κ, within the dyad. For Ca2+ ions in the dyad, κ is ∼1 nm (4). In effect, the ions feel a strong repulsion if they are within distance κ and no interaction otherwise. Therefore κ serves as a natural correlation length between the ions.
The electrostatic potential, φ, is dominated by membrane surface charges in the dyad. We use the Debye-Hückel model of charge-charge interaction, in which φ is given by
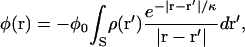 |
(3) |
where the integral is taken over the surface S of the membrane, ρ is the membrane surface charge density, and κ is again the Debye length. The constant φ0 depends on the dielectric constant and ionic conditions in the dyad. We follow Soeller and Cannel (4) and approximate φ as a monoexponential function arising from sarcolemmal surface charges
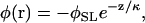 |
(4) |
where h is the height of the dyad and z is the vertical distance from the sarcolemma at point r (see Fig. 1 of Soeller and Cannell (4)).
The volume within the dyad that is accessible to a particular Ca2+ ion is assumed to include any space not occupied by an LCC, RyR2, CaM, or other Ca2+ ions. It is assumed that diffusing Ca2+ ions cannot penetrate the surface of any protein (including the RyR2 foot processes), the JSR membrane, or the sarcolemma. These surfaces/membranes are therefore considered as reflecting (i.e., no-flux boundary conditions). The lateral boundary at the interface between the dyadic volume and myoplasm is treated as an absorbing boundary for ions flowing from the dyad to the myoplasm (see below). In addition, a baseline rate of Ca2+ entry into the dyad from the myoplasm has been implemented based on the assumption of a constant myoplasmic Ca2+ concentration of 100 nM. These boundary conditions ensure that the dyads are statistically independent.
Discrete approximation
The multidimensional FPE (Eq. 1) describes the evolution of the probability density function for the positions of Brownian particles subject to a potential. Rather than solving the time-dependent joint probability density from the FPE, we generate paths of Ca2+ ion movement in the dyad using an algorithm described by Wang et al. (44). This method produces a finite differencing of the FPE that can be interpreted as a spatially discrete Markov process, which can then be simulated using Monte Carlo methods (45). The dyadic space is discretized into a lattice consisting of 1 nm3 boxes. Because the timescale of Ca2+ diffusion is sufficiently more rapid than other kinetic events such as channel gating, the system can be considered to be in a local steady state within any single box. Furthermore, if the potential changes linearly within the box, then an analytic local solution is possible within the box (44,46). Using the local solution, and continuity requirements, Brownian motion in continuous space can be discretized into a discrete-state Markov process. Consequently, the FPE is converted into a master equation
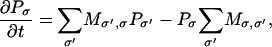 |
(5) |
where σ = (r1, r2,…, rN) labels the composite state of the system where each ri describes the position of the i-th ion. Pσ is the probability that the system is in state σ, and 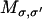 is the transition rate from state σ to state σ′. The transition rates for Ca2+ movement can be directly obtained from the local equilibrium solution of the FPE (44). Transitions can only occur between connected states of the system (i.e., states that differ in the position of a single ion by a distance equivalent to a single lattice point). Therefore if σ = (r1, r2,…,ri,…, rN) and σ′ = (r1, r2,…,ri′,…, rN), then
is the transition rate from state σ to state σ′. The transition rates for Ca2+ movement can be directly obtained from the local equilibrium solution of the FPE (44). Transitions can only occur between connected states of the system (i.e., states that differ in the position of a single ion by a distance equivalent to a single lattice point). Therefore if σ = (r1, r2,…,ri,…, rN) and σ′ = (r1, r2,…,ri′,…, rN), then
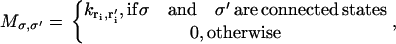 |
(6) |
where 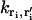 is the transition rate from the initial location ri of the ith ion to the final location ri′ of the ith ion. Transition rate formulas for a variety of boundary conditions have been derived previously (44) as:
is the transition rate from the initial location ri of the ith ion to the final location ri′ of the ith ion. Transition rate formulas for a variety of boundary conditions have been derived previously (44) as:
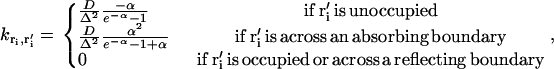 |
(7) |
where D is the diffusion constant of Ca2+, Δ is the lattice spacing (= 1 nm), and α is the change in the local potential given by
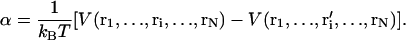 |
(8) |
Note that if there is no potential difference between two adjacent boxes, 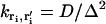 , which is equal to the inverse of the expected time for the ion to diffuse to the adjacent box in the absence of an electric field. Under conditions where α would be large and negative (e.g., when an ion occupies the adjacent box or there is an adjacent reflecting boundary),
, which is equal to the inverse of the expected time for the ion to diffuse to the adjacent box in the absence of an electric field. Under conditions where α would be large and negative (e.g., when an ion occupies the adjacent box or there is an adjacent reflecting boundary), 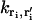 is automatically set to zero. Thus, no more than one Ca2+ ion can occupy a single lattice point at a time and Ca2+ ions cannot transition into inaccessible region of the dyad (e.g., space filled by protein structures). On the lateral boundaries of the simulation grid, absorbing boundary conditions are applied (Eq. 7), allowing ions to escape the dyadic region and be absorbed into the bulk cytosolic compartment. These definitions completely specify the transformation from the continuous-space FPE description to the discrete-space Markov description.
is automatically set to zero. Thus, no more than one Ca2+ ion can occupy a single lattice point at a time and Ca2+ ions cannot transition into inaccessible region of the dyad (e.g., space filled by protein structures). On the lateral boundaries of the simulation grid, absorbing boundary conditions are applied (Eq. 7), allowing ions to escape the dyadic region and be absorbed into the bulk cytosolic compartment. These definitions completely specify the transformation from the continuous-space FPE description to the discrete-space Markov description.
In discretized form, the description of Ca2+ dynamics in the dyad can be coupled with the processes of Ca2+ binding and gating dynamics of RyR2s and LCCs. As described above, gating dynamics for each channel are described by a Markov process where the binding and unbinding of Ca2+ ions may affect ion channel transition rates. The Markov processes for Ca2+ diffusion, Ca2+ binding, and channel gating can be combined. Thus, the state space described by σ is expanded to include both the positions of Ca2+ ions and the states of the ion channels. The process of Ca2+ binding to the channels is incorporated by allowing ions in lattice boxes adjacent to a binding site to bind with rates kon and koff (see Appendix for rates governing each type of binding site). The incorporation of these processes allows the entire dyad to be simulated by a single all-inclusive Markov process, in which the dynamics of the RyR2s and LCCs are coupled to the dyadic Ca2+ dynamics of the model.
To compute the time-dependent solution, we perform a kinetic Monte Carlo sampling algorithm to generate paths of mobile ions. The general algorithm of sampling continuous time Markov processes was first proposed by Bortz et al. (45). This method has been used extensively in the study of biochemical networks and molecular motors (44,46,47). Briefly, for each given state σ, all possible destination states, σ ′, and their associated transition rates, 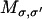 are tabulated. The net exit rate for the current state is obtained by summing over σ ′,
are tabulated. The net exit rate for the current state is obtained by summing over σ ′,
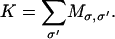 |
(9) |
A random number Δt is then picked from the waiting time distribution 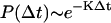 and is assigned as the time the system will remain in its current state. To determine the destination state for the following transition, another uniform random number λ in the interval (0,K) is generated. The subinterval in which λ resides, where the size of each subinterval corresponds to the magnitude of each individual exit rate from the current state, determines the destination state. In this fashion, trajectories/paths that include ion diffusion, opening and closing of channels, and binding of Ca2+ to channels can be computed. Time-dependent distributions of Ca2+ ions can be obtained by analyzing sample paths generated using the Monte Carlo procedure. Average quantities such as ion current, Ca2+ flux, and ECC gain are computed by summing/averaging over a large number of dyad simulations. In addition to calculating average quantities, the Monte Carlo approach used here allows for analysis of the degree of variance in relevant measures such as ECC gain.
and is assigned as the time the system will remain in its current state. To determine the destination state for the following transition, another uniform random number λ in the interval (0,K) is generated. The subinterval in which λ resides, where the size of each subinterval corresponds to the magnitude of each individual exit rate from the current state, determines the destination state. In this fashion, trajectories/paths that include ion diffusion, opening and closing of channels, and binding of Ca2+ to channels can be computed. Time-dependent distributions of Ca2+ ions can be obtained by analyzing sample paths generated using the Monte Carlo procedure. Average quantities such as ion current, Ca2+ flux, and ECC gain are computed by summing/averaging over a large number of dyad simulations. In addition to calculating average quantities, the Monte Carlo approach used here allows for analysis of the degree of variance in relevant measures such as ECC gain.
A typical simulation involves an ensemble of 400 dyads, each of which is clamped to test potentials in the range of −40 mV to +50 mV in 10-mV increments where each voltage clamp step is held for 100 ms. This simulation requires ∼40 h of runtime on an IBM BladeCenter with 62 nodes, each with two Intel Xeon 2.8 Ghz processors. Although the computational task of simulating this model is substantial compared to most conventional myocyte models, the runtime is several orders of magnitude faster than a typical molecular dynamics simulation. This model allows for simulation of a large number of dyads with timescales measured in seconds. A 64-bit version of the Mersenne Twister algorithm (48,49) was employed as the random number generator.
RESULTS
Excitation-contraction coupling gain
Macroscopic SR Ca2+ release is often quantified by measuring ECC gain. The characteristic voltage-dependent shape of the ECC gain function arises as a result of local control of Ca2+ release at the level of the dyad (18). ECC gain measures the relative magnitude of JSR Ca2+ release via RyR2s to Ca2+ influx via LCCs. Fig. 4 A shows the voltage dependence of peak LCC Ca2+ flux (JLCC, solid circles) and peak RyR2 Ca2+ flux (JRyR, open circles) obtained in response to 100-ms duration depolarizing voltage-clamp steps from −40 to 50 mV from a holding potential of −100 mV. In Fig. 4 B, the peak fluxes of Fig. 4 A have been normalized based on their respective maxima. Although both peak Ca2+ fluxes are essentially bell shaped, JRyR reaches its peak value at a potential that is ∼10 mV more negative than that where JLCC reaches its peak (i.e., the curves are shifted with respect to each other), as was also observed in experiments (50,51). The consequence of this shift is that peak ECC gain, defined as the ratio of peak JRyR to peak JLCC, is a monotonically decreasing function of membrane potential, as shown in Fig. 4 C. In Fig. 4 C, the simulated peak ECC gain is compared to the experimental results of Song et al. (50) and Wier et al. (51). The simulated peak ECC gain curve exhibits gain values that are within the range of the experimentally measured values and demonstrates a similar shape. Only at potentials more negative than −20 mV do the simulation results differ from the experimental measurements. However, this is not unexpected because both experimental and model (see below) measures of ECC gain exhibit higher variability in this range of potentials compared to positive potentials. In Fig. 4 D, integrated ECC gain (defined here as the ratio of the total number of Ca2+ ions that enter the dyad via RyR2s to the total number that enter via LCCs over the duration of a 100-ms voltage clamp) is shown for comparison to peak ECC gain. Both measures of gain display the characteristic monotonic decay with increasing membrane potential. Each data point in the panels of Fig. 4 was calculated as the average of three simulations, each consisting of a population of 400 dyads.
FIGURE 4.

Voltage dependence of LCC Ca2+ influx (JLCC), JSR Ca2+ release flux (JRyR), and ECC gain. (A) Peak values of JLCC (solid circles) and JRyR (open circles) as a function of clamp membrane potential. (B) Peak Ca2+ fluxes (data of panel A) normalized by their respective maxima. (C) ECC gain based on peak Ca2+ fluxes (data of panel A) for the model (solid line) are compared to experimental data of Song et al. (dark gray dashed line (50)) and Wier et al. (light gray dashed line (51)). (D) Comparison of peak ECC gain (solid line) and integrated ECC gain (dashed line).
The model was used to further explore the functional influence of protein structures in the dyad on ECC gain. This was done by comparison of control model simulations to simulations in which the geometric models of protein structure were not included in the dyad (i.e., properties of LCC and RyR2 gating that determine Ca2+ fluxes entering the dyad remain intact, but the space-filling models of LCCs, RyR2s, and CaM that act as Ca2+ diffusion barriers are absent, effectively increasing the volume of the dyad accessible to Ca2+ ions). Peak ECC gain obtained for the baseline model (solid line) is compared to that for the model in which LCC, RyR2, and CaM molecule structures were omitted (dashed line) in Fig. 5. Ca2+ binding site locations for the no-structure simulations remain at the same positions in space as when the protein structures are included. The rates for all channel gating and Ca2+-binding processes are identical for both cases; only the structural obstacles to Ca2+ diffusion within the dyad have been altered. Over a wide range of potentials, the peak ECC gain is decreased upon removal of the protein structures from the dyad. To demonstrate that the difference in gain is significant, and not simply a consequence of variability in the output of the stochastic simulation, three independent simulations of gain both including (circles) and excluding (triangles) protein structure models are shown for potentials −20 mV, 0 mV, and +20 mV. At each of these three potentials, all values of simulated gain are higher when protein structures are intact (average gain of 20.1, 14.7, and 12.9, for −20 mV, 0 mV, and +20 mV, respectively) compared to when protein structures are excluded (average gain of 16.5, 8.7, and 6.9, respectively). The protein structures occupy ∼15% of the dyad volume. To show that the difference in ECC gain in the presence versus absence of protein structures is not simply attributable to the difference in Ca2+-accessible dyad volume, peak ECC gain was obtained in a model in which protein structures were absent and dyad volume was reduced by ∼15% by decreasing the height of the dyad from 15 to 13 nm (Fig. 5, dotted line). Throughout the full range of clamp potentials, ECC gain values for this model were similar to those of the baseline model in the absence of protein structures (gain of 15.0, 9.9, and 5.5, for −20 mV, 0 mV, and +20 mV, respectively).
FIGURE 5.

The role of dyadic protein structures on ECC gain. Peak ECC gain as a function of membrane potential for the baseline model, which includes space-filling geometric models of protein structure in the dyad (solid line), for the model with protein structures excluded (dashed line), and for a modified model with dyad height reduced from 15 to 13 nm and protein structures excluded (dotted line). Three independent simulations of gain including (circles) and excluding (triangles) geometric protein structures are shown at −20, 0, and +20 mV.
The data of Fig. 5 indicate that the physical shape and configuration of dyad proteins influences Ca2+ diffusion during CICR in such a way as to enhance ECC gain. The effect of protein structures on latency of Ca2+ release (defined as the time between opening of the first LCC and opening of the first RyR2) in response to a voltage-clamp step to 0 mV is shown in Fig. 6 A (protein structures included) and B (protein structures excluded). Data were collected from a population of 1500 dyads in each case. The simulated latencies for Ca2+ release displayed a similar distribution both with (mean = 7.5 ± 8.0 ms) and without (mean = 7.2 ± 8.0 ms) protein structures. However, latency values were determined only from dyads in which a release event was triggered (defined as an opening of duration >1.0 ms of at least one RyR2). The probability of triggering a release event was 0.152 (228 out of 1500 dyads) with protein structures intact and 0.105 (158 out of 1500 dyads) with protein structures excluded, a 44% increase. These simulations were repeated in the absence of the electric field that is normally associated with sarcolemmal surface charges in Fig. 6, C (protein structures included) and D (protein structures excluded). Under these conditions Ca2+ release latencies were considerably shorter than in the presence of the electric field, however, they still displayed a similar distribution both with (mean = 0.89 ± 0.51 ms) and without (mean = 0.90 ± 0.44 ms) protein structures. In the absence of the electric field, the probability of triggering a release event was 0.25 (127 out of 500 dyads) with protein structures excluded and 0.36 (181 out of 500 dyads) with protein structures intact. Although these values are ∼2.5-fold greater than in the presence of the electric field, the probability of triggering a release event remained 44% greater with protein structures excluded than with protein structures intact. The data of Figs. 5 and 6 indicate that even though the time necessary for an LCC opening to trigger RyR2 release may be unaffected by the protein structures, the diffusion obstacle imposed by the large structure of RyR2s leads to an increase in the probability that “trigger” Ca2+ ions successfully bind to the activating binding sites on the RyR2s, and hence increases ECC gain.
The above results indicate that protein structures are an important factor that influence the dynamics of CICR, and suggest that the location of Ca2+-binding sites on these structures might also affect CICR. To explore this possibility, an alternate set of Ca2+ activation binding sites on the RyR2 were tested in the model. On each subunit, the alternate activation site was located on the surface of domain 1, within ∼3 nm of the RyR2 pore (Fig. 2 A, blue dots). Fig. 7 shows that with the alternate activation sites (dashed line, average of three simulations of 400 dyads each), peak ECC gain is increased nearly twofold compared to the baseline model (solid line). The increase in gain follows from the fact that the LCCs are aligned directly across the dyadic cleft from RyR2s such that each LCC pore is located directly across from a RyR2 pore. Hence, the alternative activation binding sites are located significantly closer to the source of Ca2+ trigger flux than in the control case, increasing the probability that Ca2+ ions encounter and bind activation sites on the RyR2. In a manner similar to that demonstrated earlier for the control RyR2 binding site locations, the RyR2 protein structure plays an important role in funneling the Ca2+ ions to the alternative Ca2+-binding sites. The exclusion of protein structures still leads to decreased ECC gain in the case where RyR2 Ca2+-binding sites have been moved to the alternative locations (e.g., with alternative binding site locations, at 0 mV ECC gain decreases to 14.5 from 19.7 upon removal of protein structures). This finding demonstrates that various locations along the surface of the RyR2s encounter different numbers of Ca2+ ions, and this suggests that RyR2 activity, and hence efficiency of CICR, may vary considerably with changes in the alignment LCCs and RyR2s.
FIGURE 7.

The role of RyR2 Ca2+-binding sites and sarcolemmal surface charges on peak ECC gain. Peak ECC gain as a function of membrane potential for the baseline model (solid line), for the model with Ca2+-binding activation sites relocated near the RyR2 pore (dashed line), and for the model with no membrane surface charges (dash-dotted line).
Previous modeling results (4) have shown that membrane surface charges cause Ca2+ ions to accumulate near the sarcolemma, suggesting a reduction in the ability of LCC Ca2+ influx to trigger CICR. The data of Fig. 6 demonstrate that electric field arising from the membrane surface charges significantly prolongs the latency of Ca2+ release and reduces the probability that a release event occurs (see above). In Fig. 7, peak ECC gain obtained for the baseline model (solid line) is compared to that for a model without membrane surface charges (dash-dotted line, simulation of 400 dyads). At nearly all potentials, ECC gain is significantly increased in the absence of membrane surface charges, indicating that the electric field significantly reduces the efficiency of CICR. As in the case described for the alternate RyR2 activation binding sites above, these results further demonstrate that the efficiency of CICR is correlated with the degree to which Ca2+ ions encounter RyR2 binding sites.
Number of Ca2+ ions in dyad
The peak number of free Ca2+ ions in the dyad has been estimated to be small, ∼1 free Ca2+ ion at a Ca2+ concentration of 10 μM in a dyad of radius 50 nm (2). Fig. 8 demonstrates the contribution of LCCs and RyR2s to the number of free Ca2+ ions in the local vicinity of each channel type within a dyad. The results of Fig. 8 A are obtained from a representative simulation of a model dyad containing only a single LCC in the sarcolemma located at the center of the dyad, where the number of Ca2+ ions within a 50-nm radius of the LCC is shown. The membrane potential is clamped to 0 mV, the initial state of the LCC is open (gray bars at top indicate when the LCC is open), and the initial number of Ca2+ ions is zero. The number of Ca2+ ions is sampled every 0.1 ms (i.e., at 10 kHz) for a duration of 10 ms. In this dyad, the average number of Ca2+ ions within 50 nm of the LCC is ∼0.46 (dashed gray line), however, there is a high degree of variability in the number present at any given instant (mean ± SD ∼1.2, dotted gray line). In this example, the maximum number of Ca2+ ions present is seven, whereas at most times there are zero Ca2+ ions in the dyad. Because each Ca2+ ion corresponds to a Ca2+ concentration of 14.1 μmol/L (within the 50-nm radius of the LCC), the equivalent Ca2+ concentration is effectively changing in large discrete steps as Ca2+ ions enter and exit this volume, reaching a maximum of 98.7 μmol/L corresponding to seven Ca2+ ions. Fig. 8 B shows the average number of Ca2+ ions sampled in the vicinity of an LCC calculated over 1000 independent simulations using the same protocol. At the beginning of the protocol, there is an average of ∼2 Ca2+ ions in the vicinity of the LCC. However, as a result of CDI, LCC open probability decreases and the average number of Ca2+ ions drops to ∼0.5 after ∼3 ms. These results suggest that the number of free Ca2+ ions in the dyad arising from LCC activity is sufficiently small and variable such that their description as a continuously varying Ca2+ concentration may not be adequate.
FIGURE 8.

Counting the number of free Ca2+ ions in the dyad arising from a single LCC or RyR2. In each panel, the number of Ca2+ ions is sampled every 0.1 ms from the volume within a cylinder of 50 nm radius centered at the single LCC or RyR2. (A) Representative simulation of a dyad containing only a single gating LCC (initially open) with membrane potential clamped at 0 mV, showing the number of free Ca2+ ions (black bars), average (dashed gray line), and standard deviation (dotted gray line). The gray bars at the top indicate when the LCC is open. (B) Mean number of free Ca2+ ions based on 1000 dyad simulations of the protocol described in panel A. (C) Representative simulation of a dyad containing only a single gating RyR2 (initially open), showing the number of free Ca2+ ions (black bars), average (dashed gray line), and standard deviation (dotted gray line). The gray bars at the top indicate when the RyR2 is open. (D) Mean number of free Ca2+ ions based on 1000 dyad simulations of the protocol describe in panel C.
Fig. 8 C demonstrates a protocol similar to that described for Fig. 8 A, using a single RyR2 in the JSR membrane. In this example, the RyR2 is initially open (gray bars at top indicate when the RyR2 is open), closes at ∼1.5 ms, reopens at ∼2.5 ms, and then inactivates at ∼7 ms. The average number of Ca2+ ions over the 10-ms simulation is 22.8 (dashed gray line) with mean ± SD of 14.7 (dotted gray line). The number of Ca2+ ions peaks at 49 during Ca2+ release and this corresponds to a concentration of ∼0.7 mM Ca2+, which is similar to the concentration of Ca2+ found in the JSR (52). There is a smaller number of ions (ranging from five to 15) present following RyR2 closure. These ions are present due to the release of Ca2+ ions from buffers and surface charges on the sarcolemmal and the SR membranes and due to diffusion of Ca2+ ions across the boundary of the volume being considered (50-nm radius). With a coefficient of variation of 64%, RyR2 gating produces a high degree of variability in the number of dyadic Ca2+ ions. This is an indication that the LCC-RyR2 signaling involved in CICR, when observed locally at the level of a single dyad, is a rather noisy process. Fig. 8 D shows the average number of Ca2+ ions sampled in the vicinity of a RyR2 calculated over 1000 independent simulations using the same protocol. On average, an open RyR2 introduces a Ca2+ flux that yields ∼31 free Ca2+ ions within 1–2 ms after opening, and the number of Ca2+ ions declines to <20 within 10 ms due to reduced RyR2 open probability following Ca2+ binding to RyR2 inactivation sites.
CICR in the dyad
Panels A and B of Fig. 9 demonstrate fundamental features of a representative Ca2+ release event in the dyad stimulated via a voltage clamp to 0 mV at time zero. Fig. 9 A shows the number of free Ca2+ ions in the dyad resulting from LCC and RyR2 gating, and Fig. 9 B shows the number of LCCs (gray line) and RyR2s (black line) that are open as a function of time during this release event. In this example, two LCCs open at ∼3 ms following the voltage clamp and the Ca2+ influx into the dyad triggers the opening of three RyR2s 1–2 ms later, one of which inactivates after ∼7 ms, the next inactivates after an additional ∼2 ms, and the final one inactivates ∼13 ms after the start of the release event. The temporal shape of the Ca2+ signal in the dyad (Fig. 9 A) is closely correlated to the number of open RyR2s (Fig. 9 B). Fig. 9 C demonstrates an average dyadic Ca2+ signal, as calculated by averaging the number of Ca2+ ions at each point in time for 1000 independent dyad simulations. As would be expected the peak of the average signal is reduced compared to that of an individual dyad due to temporal dispersion of Ca2+ release events and the fact that not all dyads will exhibit a Ca2+ release event. The duration of the average local Ca2+ transient shown in Fig. 9 C is ∼20 ms (at half-maximal amplitude), similar to that measured for Ca2+ spikes in myocytes using confocal imaging techniques (50,53,54). Fig. 9 D demonstrates the snapshot of the spatial distribution of Ca2+ ions in a single dyad (as a function of the distance from the center) during a release event similar to that shown in Fig. 9 A. The Ca2+ ion density was calculated as a function of radial distance from the dyad center as the mean density taken over a 5-ms time window centered at 1.4 ms following the beginning of the release event (a time at which two RyR2s were open). In this case the open RyR2s are centrally located in the dyad, and the density of Ca2+ ions is highest at this location and decreases with increasing radius from the release site.
FIGURE 9.

A CICR event in a single dyad. The response is evoked by a 0-mV voltage clamp step occurring a time 0. (A) The number of free Ca2+ ions in the dyad volume as a function of time. (B) The number of open RyR2s (black solid line) and the number of open LCCs (gray solid line) in the dyad during the release event depicted in panel A. (C) The average number of free Ca2+ ions in the dyad as calculated from 1000 independent dyads subject to the same voltage clamp protocol as the event shown in panel A. (D) Average density (ions nm−3) of Ca2+ ions as a function of radial distance from the center of the dyad at 1.4 ms following a release event. (E) Distribution of the duration of Ca2+ release events. (F) Distribution of the maximum number of RyR2s that open simultaneously during a single Ca2+ release event (bars, left axis) and the mean (± SD) of the maximal number of open LCCs (taken over the 5-ms window preceding each release event) associated with each grouping of release events (solid circles, right axis).
In Fig. 9 E, a distribution of the durations of 317 Ca2+ release events is shown. The average Ca2+ release event duration is 14.7 ± 11.6 ms mean ± SD, which is consistent with typical spark rise time of ∼10 ms (2) and comparable to the major slow component of RyR2 open duration of 13.6 ms measured experimentally by Marx et al. (55). Fig. 9 F (bars) shows the variation in the peak number of open RyR2s among the same set of Ca2+ release events in the model. Overall, ∼36% of the release events were the result of the opening of a single RyR2. However, nearly half of these single-RyR2 events were of duration <5 ms, which may not supply enough Ca2+ release to underlie experimentally detectable Ca2+ sparks since the shortest single-RyR2 Ca2+ spark rise times measured experimentally are ∼4–5 ms (39). If release events <5 ms in duration are excluded, then ∼23% of the release events would be comprised of single-RyR2 events. These features are qualitatively similar to the recent experimental results of Wang et al. (39), where it was found that ∼12% of Ca2+ sparks result from the opening of a single RyR2 and that the typical number of open RyR2s during a spark was two to three. The maximal number of RyR2s that opened during a single release event was seven, in agreement with experiments (39). The number of open RyR2s is predominantly determined by the number of RyR2s that experience Ca2+ binding at all four activation sites simultaneously, which is a function of the distribution and density of Ca2+ ions in the dyadic volume. A closed RyR with all four activation sites bound has high probability of opening before any of the Ca2+ ions unbind. The relatively small number of RyR2s comprising individual release events indicates that the Ca2+ density is not sufficiently high throughout the dyad to bind and activate all RyR2s. This observation is also supported by the results of Fig. 7, showing that ECC gain can be dramatically altered by repositioning the location of RyR2 activation binding sites or altering the electric field. For each grouping of release events (i.e., each bin) in Fig. 9 F, the mean of the maximum number of open LCCs (over the 5-ms window preceding each release event) acting to trigger release is shown (solid circles, ± SD). Regardless of the number of open RyR2s associated with the group of release events, the mean number of triggering LCCs was always in the range of 1.6–2.2, with relatively large standard deviation (up to ∼0.8) in all cases. This finding shows that the number of RyR2s associated with a release event is not correlated with the number of triggering LCCs, which indicates that, in multi-RyR2 release events, the activation of additional RyR2s is likely driven predominantly by Ca2+ arising from earlier RyR2 openings, as is typically assumed due to the positive feedback inherent in CICR.
Ca2+-binding sites are present in the sarcolemmal and JSR membranes of the dyad and have been included in this model as described in Methods. Fig. 10 A shows the ratio of free Ca2+ ions to total Ca2+ ions, calculated as the average ratio over 317 simulated release events occurring in response to a voltage clamp to 0 mV at time zero. This ratio is initially ∼17% (on average), corresponding to the early phase of Ca2+ release. Immediately following release, Ca2+ ions diffuse away from the LCC and RyR2 structures and many encounter and bind to available Ca2+ buffering sites. As a result, the ratio of free Ca2+ ions decreases to an equilibrium value of ∼2%, as can be seen at times >10 ms following Ca2+ release in Fig. 10 A. For the same set of release events, Fig. 10 B shows the average fraction of free Ca2+ ions that are located within 1 nm of the sarcolemmal membrane. Within 1 ms of the beginning of the clamp to 0 mV, and for the entire duration of the clamp, ∼29–30% of the free calcium ions tend to be restricted to the space adjacent to the sarcolemmal membrane as a result of the electric field. This is consistent with the finding that ECC gain is increased in the absence of the electric field (Fig. 7), as the electric field reduces the likelihood that free Ca2+ ions will encounter RyR2 binding sites. The accumulation of Ca2+ ions adjacent to the sarcolemma is qualitatively consistent with the steep gradient in Ca2+ concentration reported adjacent to the sarcolemma in the continuum model of Soeller and Cannell (4).
FIGURE 10.

The effects of Ca2+ buffers and membrane surface charges on free Ca2+ in the dyad. (A) The average ratio of the number of free Ca2+ ions to the total number of Ca2+ ions during CICR is shown as a function of time. (B) The average fraction of free Ca2+ ions located within 1 nm of the sarcolemmal membrane. The responses in both panels are evoked by a 0-mV voltage clamp step occurring at time 0, and the average shown is obtained from 317 release events.
Variability of ECC gain
The data of Figs. 8 and 9 demonstrate that CICR at the level of a single dyad involves a relatively small number of ions and is therefore an inherently noisy process. It is not clear, however, whether the degree of variability in the process of CICR is physiologically relevant. Because the CICR model presented here captures the phenomenon of signaling noise that arises as a result of movement and binding of Ca2+ ions, it can be used to better understand this issue. Fig. 11 demonstrates the degree of variability in measures of ECC gain. Fig. 11 A shows the results of 10 separate simulations of peak ECC gain for voltages in the range from −40 to +50 mV, where each gain curve is calculated for a population of 400 independent dyads. Mean ECC gain (solid black line) and a single standard deviation above and below the mean (gray dashed line) are shown in Fig. 11 B. The variance of peak ECC gain is significant, particularly at voltages between −40 and −20 mV. At potentials more positive than −20 mV, variance is significantly reduced. This follows from that fact that LCC open probability, and hence the probability of triggering a release event in any particular dyad, increases with increasing membrane potential. At low potentials only a very small fraction of the dyads exhibit release events, hence there is a high variability in simulated ECC gain. At higher potentials, the number of dyads (i.e., population size) in which a release event occurs increases. Hence the variability in repeated simulations of ECC gain decreases. A population of 400 dyads represents only ∼3–8% of the number of dyads in a typical cardiac myocyte. Therefore, the variance in ECC gain as measured in a myocyte would be expected to be considerably less than that represented by the standard deviation of gain shown in Fig. 11 B.
FIGURE 11.

Variability of ECC gain. (A) Peak ECC gain as a function of voltage is shown for 10 repeated model simulations based on a population of 400 dyads. (B) Mean peak ECC gain (black solid line) based on the data of panel A is shown along with the mean ± SD (denoted S.D., dashed lines). (C) The distribution of peak ECC gain at −40 mV for the whole myocyte (12,500 dyads) computed using bootstrap replicates. (D) The distribution of peak ECC gain at 0 mV for the whole myocyte computed using bootstrap replicates.
If it is assumed that a single dyad contains four LCCs and 20 RyR2, then a single cardiac myocyte is estimated to contain ∼12,500 dyads (18). Estimating the variance (or standard deviation) of ECC gain measured in a dyad population of this size would require repeated simulations of this large population of dyads, which is extremely computationally demanding and impractical. As an alternative, we use the bootstrap method (56) to estimate the standard deviation of ECC gain based on the data obtained from the simulation of a single population of dyads. The response of 12,500 independent dyads to depolarizing voltage clamp steps to −40 and 0 mV were simulated. From these data, bootstrap samples (each of size 12,500) were generated by resampling (randomly drawing dyads with replacement) from the population of 12,500 simulated dyads. At each voltage, 1000 bootstrap samples were drawn, yielding 1000 bootstrap replicates of ECC gain. Mean and standard deviation of ECC gain are estimated from the set of 1000 replicates at each membrane potential. Fig. 11, C and D, shows the distribution of peak ECC gain estimated for a cardiac myocyte at −40 and 0 mV, respectively, based on the bootstrap replicates of gain. Mean and standard deviation of ECC gain are estimated to be 27.30 ± 2.03 at −40 mV and 11.98 ± 0.25 at 0 mV. As expected, the variance is considerably reduced from that measured in a population of 400 dyads (with mean ± SD of 28.23 ± 7.05 at −40 mV and 11.97 ± 1.32 at 0 mV). These estimates indicate that variance of ECC gain that arises from the stochastic gating of underlying LCCs and RyR2s is expected to be rather small for a whole-cell population of dyads, particularly in the central range of potentials near 0 mV. This variation in gain is smaller than that observed in experiments (51) and therefore may not be easily detectable. The existence of ∼12,500 independent dyads in a single myocyte therefore dramatically reduces the effects of noise inherent in the local triggering events within a dyad that are mediated by tens of Ca2+ ions. ECC gain can therefore be considered a robust and reproducible measurement of CICR at the whole-cell level.
DISCUSSION
In this study, we present a molecularly and structurally detailed model of the cardiac dyad. The model incorporates experimentally determined dyad protein structure and geometry, biophysically detailed Markov channel models for simulation of stochastic channel gating of LCCs and RyR2s, and discrete kinetic models for simulation of stochastic motion of Ca2+ ions in the volume of the dyad in the presence of membrane buffers, ion channel Ca2+ binding sites, and electric potentials. This detailed model of the cardiac dyad allows for the study of the kinetic features of CICR both at the level of the dyad as well as at the level of the whole cell. At the microscopic level, this model allows for a detailed examination of the localized events that are the basis for local control of ECC. The concept of local control embodies the notion that L-type Ca2+ current tightly controls JSR Ca2+ release because elementary, independent JSR Ca2+ release events occur in response to highly localized Ca2+ transients, which are produced by the opening of one or a few LCCs in the vicinity of small clusters of RyR2s within a dyad (57). By increasing the number of dyads to that found in a single myocyte, whole-cell properties such as ECC gain and its variability can be reproduced by the model.
Simulation results suggest that the physical structure of the RyR2, a tetrameric protein with large cytoplasmic domains approaching the LCC pore, may function as a guide by which Ca2+ ions are funneled to and subsequently bind to Ca2+ activation sites within the RyR2. Repeated simulations of ECC gain demonstrated that gain was consistently reduced at all tested membrane potentials when the geometric protein structures of LCCs, RyR2s, and CaM were removed from the dyad (Fig. 5). The volume occupied by these structures constitutes ∼15% of the volume of the dyad. A simple ∼15% reduction of dyad volume (via shortening of dyad height) in the absence of protein structures does not restore ECC gain values to those obtained in the presence of protein structures (Fig. 5). Therefore the difference in ECC gain (nearly twofold greater at 0 mV) in the presence versus absence of protein structures cannot be attributed only to the corresponding change in volume; rather the barrier to Ca2+ diffusion provided by the RyR2 protein structure enhances binding to activation sites. This is further shown by the fact that, in response to a voltage clamp stimulus to 0 mV, the probability of observing a Ca2+ release event is 0.152 in the presence of structures and 0.105 in the absence of structures, demonstrating that protein structures yield a 44% increase in the efficacy of local ECC compared with the absence of protein structures (with all other model properties being identical). It is notable that the mean first latency of Ca2+ release is not significantly different in the presence and absence of structures (Fig. 6, A and B). This suggests that the structure of dyadic proteins increases the likelihood that a Ca2+ ion encounters and binds a RyR2 activation site but may not necessarily reduce the transit time from the LCC pore to the location of the RyR2 binding site.
The role of sarcolemmal surface charges on Ca2+ dynamics in the dyad in this model is consistent with previous predictions from a continuum model of the dyad (4). Fig. 6 demonstrates that in the absence of the electric field that is associated with the surface charges, the probability of triggering JSR Ca2+ release increases approximately twofold and the Ca2+ release latency is reduced from ∼7 to ∼1 ms. Fig. 7 shows that ECC gain is considerably higher in the absence of the electric field. Soeller and Cannell (4) have suggested that the presence of the electric field may reduce CICR by reducing Ca2+ concentration (or the number of Ca2+ ions) sensed by the RyR2s during an LCC opening, and this is precisely what this model predicts. However, Soeller and Cannell have also suggested that the electric field may also augment EC coupling by reducing the rate of decline of dyad Ca2+ concentration following LCC closure (4). Although the role of electric field on the rate of decline of Ca2+ following LCC closure (under conditions where reopening is not allowed) has not been analyzed in this model, the results of Fig. 7 suggest that such an augmentation of ECC does not compensate for the aforementioned reduction in CICR. Future studies will further investigate the role of electric field on the detailed Ca2+ ion distribution in the dyad and on the statistical properties of Ca2+ binding to RyR2s.
The biophysical behavior of the RyR2 model implemented in this study is based on a model originally developed by Stern et al. (38) and the recent findings of Wang et al. (39) regarding the quantal nature of Ca2+ sparks. The model contains a relatively large unitary current (1.24 pA) with typical Ca2+ release events consisting of the opening of only a few (one to seven) RyR2s (Fig. 9 F). In this model, RyR2 open probability is typically in the range of 1–3% (Fig. 12), consistent with the prediction of Wang et al. (39) and previous estimates that ∼2% of simultaneously active RyR2s can account for the amplitude of peak whole-cell SR Ca2+ release flux (2). The small number of RyR2s involved in individual Ca2+ release events indicates that, in this model, these events are not fully regenerative and do not recruit all or even most RyR2s within a single dyad. It remains controversial as to whether this biophysical paradigm is the correct description of the events underlying Ca2+ sparks. Mejia-Alvarez et al. (58) measured unitary RyR2 Ca2+ current under physiological conditions and found it to be two- to threefold smaller than the value of 1.24 pA used in this study. This finding in combination with dyad ultrastructural data (1) and experimental measures of RyR2 and LCC density (17) would suggest that a typical dyad should contain ∼100 RyR2s and ∼10–20 LCCs. The high fraction of release events attributable to opening of a single RyR2 in this model (Fig. 9 F) compared to that observed in experiments (39) may, in part, follow from the assumptions of a relatively high RyR2 unitary current in combination with a smaller number of RyR2s and LCCs per dyad. These assumptions represent a necessary compromise between ultrastructural detail and computational complexity in the development of this model. A recent model of the Ca2+ spark by Sobie et al. (59) simulates gating of clusters of RyR2s and includes mechanisms of coupled gating among RyR2s and dependence of gating on JSR Ca2+ concentration. In their model, RyR2 unitary current is rather small (0.07 pA), open probability is ∼1 during a release event, and nearly all RyR2s (within a single dyad) become activated during Ca2+ release. The distinction between these different paradigms of RyR2 behavior has proven to be difficult to resolve based on experimental findings. The limited regeneration observed in the model presented here arises from the finite probability that all four activation sites on a RyR2 become Ca2+ bound simultaneously given the relatively small number of freely diffusing Ca2+ ions (see Figs. 8 and 9). The finding that protein structures influence properties of Ca2+ release and ECC follows from the nature of Ca2+ release in this paradigm, and may not hold true in a model with strong regenerative Ca2+ release.
FIGURE 12.

Consequences of altering the transit time for Ca2+ in the dyad. The scale factor is applied to the Ca2+ diffusion coefficient, and the open channel flux/permeability of all LCCs and RyR2s. Upper panel shows the fraction of LCCs (solid circles) and RyR2s (open circles) that are open at the time of maximal Ca2+ flux in response to a 0-mV voltage clamp and the lower panel shows the corresponding peak ECC gain (squares). Each data point represents the average of three runs, each consisting of 400 dyads (with the exception of scale factor 5.0, for which only a single run of 400 dyads is shown).
As is the case with any model, some caution is warranted in the interpretation of the results as the dyad model does employ some simplifying assumptions regarding the nature of the interactions between Ca2+ ions and proteins in the dyad. As described in Methods, the positions of Ca2+-binding sites on proteins within the dyad were determined by available experimental data, however, the precise locations of these Ca2+-binding sites remain unclear. In Fig. 7, the role of an alternative set of RyR2 Ca2+ activation site locations on ECC gain was demonstrated. There are many possible combinations of activation and inactivation binding site locations that could be investigated. We chose to illustrate an extreme case where RyR2 activation sites were repositioned within ∼3 nm of the RyR2 pore (see Fig. 2) without altering the locations of the inactivation binding sites, which resulted in increased ECC gain at all potentials. Although the conclusion regarding the ability of dyadic proteins to funnel Ca2+ ions to their binding sites was upheld for this alternative set of RyR2 activation site locations (see above), it is possible that this conclusion may not hold for all combinations of hypothesized Ca2+ binding site locations. There is also uncertainty with respect to placement and orientation of the LCC-tethered CaM and this may have an impact on LCC inactivation. However, this effect is expected to be relatively small, and to have only a minor impact on model ECC gain because the rate-limiting step of LCC inactivation is the conformational change of the protein (i.e., transition from Mode Normal to Mode CDI), which occurs on the timescale of 2–25 ms. Calcium binding to CaM occurs on the timescale of ∼1 μs. Because Ca2+ diffusion is also very fast compared to inactivation, the precise location and orientation of these Ca2+-binding sites would likely not have a major impact on the dynamics of LCC inactivation unless the sites were located at a position to which Ca2+ diffusion was significantly enhanced or obstructed (and there is no evidence to indicate this may be the case). In this study, no attempt has been made to model the behavior of the locally enriched CaM population near LCCs (13) because there is insufficient data describing the mechanism of enrichment. It is not clear how these CaM molecules are restricted to the vicinity of LCCs, whether they remain restricted to the dyad upon Ca2+ binding (see below), how they are arranged and oriented, or how to describe their motion. Based on the results regarding the role of RyR2 structure in funneling Ca2+ ions, an additional ∼25 CaM molecules restricted to within 40 nm of an LCC may act to further hinder Ca2+ diffusion away from the LCC site of entry into the periphery of the dyad (in addition to binding a small number of the Ca2+ ions themselves), and therefore possibly enhance Ca2+ binding to local sites on the RyR2 and LCC. The volume occupied by 25 CaM molecules is ∼38% of the volume of a single RyR2 and may therefore substantially alter and restrict Ca2+ diffusion pathways. However, Mori et al. (13) suggest that these locally enriched CaMs may play a role in the CaM translocation hypothesis, which asserts that these CaMs are driven (out of the dyad) to the nucleus to activate CREB. This further complicates the ability to predict the roles of the CaMs on Ca2+ diffusion because they may not always be present in the dyad and the dynamics of CaM translocation out of and back into the dyad are unknown. The role of the four potentially occupied CaM-binding sites on the RyR2 is even less clear. Four CaMs fill only 6% of the volume of a single RyR2 and would likely have little effect on Ca2+ diffusion alone. However, since the exact locations of Ca2+ binding sites on the RyR2 in relation to CaM binding sites have yet to be determined, it remains unknown as to whether RyR2-bound CaM may influence the availability of Ca2+ binding sites on the RyR2.
Another simplifying assumption of this model is that all dyadic proteins are treated as solid objects, i.e., Ca2+ ions cannot penetrate the space occupied by any protein. In reality, Ca2+ ions may be able to penetrate the folded chains of amino acids that comprise dyadic proteins and may therefore be able to interact with charged residues of these proteins. In addition, these charged residues may alter the electric field in the vicinity of the proteins, and hence may influence the nature of Ca2+ diffusion in the dyad. The addition of these features to the model may influence the conclusions regarding the roles of protein structure and Ca2+-binding site location on CICR. However, accounting for the possibility that Ca2+ ions can penetrate proteins and the role of protein surface charges is beyond the scope of the model presented here, and will be considered in future studies.
As stated above, previous estimates of peak Ca2+ concentration in the dyad range from 100 to 1,000 μM (3,4), which corresponds to 10–100 free Ca2+ ions in the dyad. Simulation results indicate that the number of free Ca2+ ions in the dyad, even during the opening of multiple RyR2s is indeed quite small, on the order of tens of ions. The contribution of individual LCCs and RyR2s to the number of dyadic Ca2+ ions is shown in Fig. 8 and the number of Ca2+ ions that result from local dyadic release events is shown in Fig. 9. The small number of Ca2+ ions that underlie local CICR events contributes noise to LCC-RyR2 signaling within the dyad. This noise is seen in variation of the number of free Ca2+ ions in the dyad under voltage clamp conditions (Fig. 8), by variation of ECC gain measured at any given clamp potential (Fig. 11, A and B) and by variable latency to first opening of a RyR2 in response to LCC opening (Fig. 6). However, the effects of this noise at the cellular level are reduced by the fact that cell behavior reflects the integrated function of a large number (∼12,500) of approximately independent dyads. This reduction of variability is demonstrated by the fact that the standard deviation of ECC gain for a whole-cell population of dyads estimated using bootstrap replicates (Fig. 11, C and D) turns out to be rather low. This result indicates that even though there may be a high degree of noise associated with localized CICR events, ECC gain, as measured at the level of the whole cell, is a robust feature of excitation-contraction coupling.
Although there is still some uncertainty as to the actual level to which Ca2+ rises within the cardiac dyad, it is now well recognized that the number of Ca2+ ions that exist within a single dyad at any given instant is relatively small (2). Detailed studies of Ca2+ diffusion in the dyad have yielded valuable insight into the roles of local buffering, electrodiffusion, and junctional geometry on the ECC process (3,4). Because the methods employed in these studies have assumed that Ca2+ concentration is continuous within the dyad, their results cannot account for stochastic phenomena associated with the small number of Ca2+ ions in the dyad. The model presented in this study is therefore motivated by the need to better understand the role of such phenomena in cardiac ECC. Recently, Koh et al. (60) presented a stochastic model of spark generation in the dyad with results that focus on the temporal properties of localized Ca2+ release. This work further established the fact that the number of Ca2+ ions in the dyad is relatively small (2). Our work extends this prior model in several ways. First, the model used in this study employs descriptions of LCCs and RyR2s that have been validated extensively in prior work and that incorporate voltage-dependent transition rates. Second, in this previous work, motion of Ca2+ ions in the dyad was described by Brownian random-walk diffusion without considering effects of the potential field arising from membrane surface charges. In this study we extend this formulation to include effects of potential fields. Third, the current model incorporates the space-filling structures of membrane proteins. Fourth, we extend analyses to the macroscopic level by investigating the important cellular properties of ECC gain.
The question remains: Does this stochastic model of the dyad reveal qualitative properties of excitation-contraction coupling that are fundamentally different than those demonstrated by deterministic continuum models? One possible way to explore the role of discreteness of Ca2+ ions in this model is to vary the rate at which Ca2+ travels through the dyad. In the limit of very fast Ca2+ transit times, the responses of LCCs and RyR2s will be rate limited by Ca2+-binding and gating kinetics (i.e., channels will not be able to respond to extremely fast fluctuations in free Ca2+ levels). In such a regime, in which channel responses essentially follow the time averaged free Ca2+ signal (i.e., a low-pass filtered Ca2+ signal), Ca2+ concentration becomes a well-defined quantity and the use of a continuum model of Ca2+ diffusion would be justified. Fig. 12 demonstrates the consequences of altering the transit time for Ca2+ in the dyad. The scale factor (with values of 0.1, 0.5, 1.0, 2.0, and 5.0) is applied to the Ca2+ diffusion coefficient, and the open channel flux/permeability of both the LCCs and RyR2s. For example a scale factor of 5.0 means the rate of Ca2+ entry into the dyad via LCCs and RyR2s is fivefold greater and the apparent diffusion coefficient for Ca2+ is fivefold greater (faster). The upper panel of Fig. 12 shows the fraction of channels that are open at the time of maximal Ca2+ flux in response to a 0-mV voltage clamp and the lower panel shows the corresponding peak ECC gain. Each data point represents the average of three runs, each consisting of 400 dyads (with the exception of scale factor 5.0, for which only a single run of 400 dyads was performed). The values in the upper panel can be interpreted as estimates of peak channel open probability in response to a 0-mV voltage clamp. However, it is important to clarify that: a), these values represent the fraction of LCC and RyR2 channels open at a particular instant in time rather than the fraction of channels activated in response to the voltage clamp; and b), the open fraction is calculated over all dyads including those in which Ca2+ was not elevated and no release event occurred. The results demonstrate that peak LCC open probability is in the range of 10–11% for all scale factors, which is expected since peak L-type current is primarily determined by membrane potential (whereas LCC inactivation kinetics are primarily determined by dyadic Ca2+ via CDI). Peak RyR2 open probability is hindered by slowed Ca2+ diffusion and approaches an asymptotic value of ∼2.6% as the scale factor increases. Similarly, peak ECC gain is low (∼4) with a 10-fold reduction in Ca2+ diffusion rate and approach an asymptotic value of ∼13 as the scale factor increases. An important feature of these data is the observation that the baseline model (scale factor 1.0) operates near, but not within the asymptotic regime (with maximal RyR2 open probability of ∼2.2%). This indicates that the discreteness of Ca2+ ions in the dyad may result in RyR2 activation that is less than (by ∼15% in this example) that predicted by an equivalent continuum model. This is a subtle, but potentially significant difference in predicted macroscopic behavior arising from the underlying stochastic simulation of Ca2+ motion in the dyad. Recently, Samoilov et al. (61) showed that stochastic fluctuations (noise) may induce bistable oscillatory behavior in an enzymatic futile cycle (a common type of biochemical reaction mechanism in which a substrate can be converted to product by a forward enzyme and then can be converted back to substrate by a reverse enzyme) that is qualitatively different from what would be predicted using a deterministic model to describe this mechanism. The model presented in this study does not contain an analog to a futile chemical reaction cycle, and therefore, stochastic dynamics of this nature have not been revealed to play a role in ECC. However, such futile cycles may in fact exist in the dyad as a result of kinase and phosphatase activity targeted to LCCs, RyR2s, and other localized proteins. Future work will incorporate these processes in the model, and the stochastic implementation may then potentially reveal emergent macroscopic properties such as bistability, if they exist in the dyad.
The stochastic model of the cardiac dyad presented here provides a step in the direction of better understanding how the events involving a small number of channels and Ca2+ ions in each dyad form the fundamental building blocks of cardiac excitation-contraction coupling. Although the fundamental aspects of LCC and RyR2 function are included in the model description, there are some model limitations that follow from limits in the knowledge of the structure and function of these proteins as based on experiments performed thus far. As discussed above, such limitations include knowledge of the precise location of Ca2+ binding sites on the RyR2 and other dyadic proteins, the position and orientation of the CaM molecule, which is associated with the LCC, and the nature in which mobile CaMs may be locally enriched in the vicinity of each LCC (13). In addition there is limited understanding of the mechanisms of RyR2 regulation by Mg2+, ATP, and other molecules, the position and structure of other proteins that are known to be physically associated with the RyR2 via macromolecular signaling complexes (e.g., kinases, phosphatases, and anchoring proteins), and the mechanistic role of RyR2 interaction and/or phosphorylation on channel gating. As future experiments elucidate and clarify these mechanistic and structural details, they can be incorporated into future versions of the model to improve our understanding of the integrative function of the cardiac dyad.
Acknowledgments
The authors thank Manjuli Sharma and Terence Wagenknecht for providing cryo-EM data for ryanodine receptor structure. A.J.T. and J.L.G. thank Patrick Helm for assistance with image data processing, and Zhong Gao for enlightening discussions. The authors thank the reviewers of this manuscript for insightful suggestions.
This work was supported by National Institutes of Health (RO1 HL60133, RO1 HL61711, P50 HL52307), the Falk Medical Trust, the Whitaker Foundation, and IBM Corporation.
APPENDIX
TABLE 1.
Model parameters
| Dyad | ||
| h | Dyad height | 15 nm |
| w | Dyad width | 200 nm |
| L | Lattice spacing | 1 nm |
| D | Ca2+ diffusion rate (220 μm2 s−1) | 220,000 ms−1 |
| T | Temperature | 309.85 K |
| Ca2+ buffers | ||
| [B-low]SL | Low-affinity Ca2+ buffers on the sarcolemma | 0.5 site nm−2 |
| [B-high]SL | High-affinity Ca2+ buffers on the sarcolemma | 0.05 site nm−2 |
| [B-low]SR | Low-affinity Ca2+ buffers on the SR membrane | 0.05 site nm−2 |
| [B-high]SR | High-affinity Ca2+ buffers on the SR membrane | 0.05 site nm−2 |
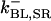 |
Ca2+-unbinding rate, SR low-affinity site | 0.1 ms−1 |
 |
Ca2+-binding rate, SR low-affinity site | 11.5 ms−1 |
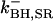 |
Ca2+-unbinding rate, SR high-affinity site | 0.1 ms−1 |
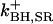 |
Ca2+-binding rate, SR high-affinity site | 115 ms−1 |
 |
Ca2+-unbinding rate, SL low-affinity site | 1.0 ms−1 |
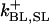 |
Ca2+-binding rate, SL low-affinity site | 115 ms−1 |
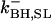 |
Ca2+-unbinding rate, SL high-affinity site | 1.0 ms−1 |
 |
Ca2+-binding rate, SL high-affinity site | 1150 ms−1 |
| L-type Ca2+ channels | ||
| k | Rate constants (see Table 3 and Appendix 1 of Greenstein and Winslow (18) | |
| γ | Rate constant for CDI | |
| with two CaM CDI sites Ca2+-bound | 0.044 ms−1 | |
| with zero or one CaM CDI sites Ca2+-bound | 0.0 ms−1 | |
| kLB | Ca2+-binding rate, CDI binding sites on CaM | 1100 ms−1 |
| kLU | Ca2+-unbinding rate, CDI binding sites on CaM | 0.01 ms−1 |
| Ryanodine receptor | ||
| α | Opening rate | |
| with four activation sites Ca2+-bound | 0.65 ms−1 | |
| with less than four activation sites Ca2+-bound | 0.0 ms−1 | |
| β | Closing rate | 0.06 ms−1 |
| δ | Inactivation rate from open state | |
| with at least one inactivation site Ca2+-bound | 0.075 ms−1 | |
| with zero activation sites Ca2+-bound | 0.0 ms−1 | |
| δ′ | Inactivation rate from closed state | |
| with at least one inactivation site Ca2+-bound | 0.6 ms−1 | |
| with zero activation sites Ca2+-bound | 0.0 ms−1 | |
| γ | Recovery from inactivation (activated) | 0.005 ms−1 |
| γ′ | Recovery from inactivation (deactivated) | 0.005 ms−1 |
| kIO | Ca2+-binding rate, inactivation sites (open state) | 250 ms−1 |
| kIC | Ca2+-binding rate, inactivation sites (closed state) | 450 ms−1 |
| kIU | Ca2+-unbinding rate, inactivation sites | 0.001 ms−1 |
| kAB | Ca2+-binding rate, activation sites | 30,000 ms−1 |
| kAU | Ca2+-unbinding rate, activation sites | 0.333 ms−1 |
Antti J. Tanskanen and Joseph L. Greenstein contributed equally to this work.
References
- 1.Franzini-Armstrong, C., F. Protasi, and V. Ramesh. 1999. Shape, size, and distribution of Ca2+ release units and couplons in skeletal and cardiac muscles. Biophys. J. 77:1528–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bers, D. M. 2001. Excitation-contraction coupling and cardiac contractile force. Kluwer, Boston, MA.
- 3.Langer, G. A., and A. Peskoff. 1996. Calcium concentration and movement in the diadic cleft space of the cardiac ventricular cell. Biophys. J. 70:1169–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Soeller, C., and M. B. Cannell. 1997. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophys. J. 73:97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bhalla, U. S. 2004. Signaling in small subcellular volumes. I. Stochastic and diffusion effects on individual pathways. Biophys. J. 87:733–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tanskanen, A. J., J. L. Greenstein, B. O'Rourke, and R. L. Winslow. 2005. The role of stochastic and modal gating of cardiac L-type Ca2+ channels on early after-depolarizations. Biophys. J. 88:85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu, Z., J. Zhang, P. Li, S. R. W. Chen, and T. Wagenknecht. 2002. Three-dimensional reconstruction of the recombinant type 2 ryanodine receptor and localization of its divergent region 1. J. Biol. Chem. 277:46712–46719. [DOI] [PubMed] [Google Scholar]
- 8.Dulhunty, A. F., and P. Pouliquin. 2003. What we don't know about the structure of ryanodine receptor calcium release channels. Clin. Exp. Pharmacol. Physiol. 30:713–723. [DOI] [PubMed] [Google Scholar]
- 9.Samso, M., T. Wagenknecht, and P. D. Allen. 2005. Internal structure and visualization of transmembrane domains of the RyR1 calcium release channel by cryo-EM. Nat. Struct. Mol. Biol. 12:539–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serysheva, I. I., S. L. Hamilton, W. Chiu, and S. J. Ludtke. 2005. Structure of Ca2+ release channel at 14 Å resolution. J. Mol. Biol. 345:427–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Radermacher, M., T. Wagenknecht, R. Grassucci, J. Frank, M. Inui, C. Chadwick, and S. Fleischer. 1992. Cryo-EM of the native structure of the calcium release channel/ryanodine receptor from sarcoplasmic reticulum. Biophys. J. 61:936–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang, M. C., R. F. Collins, R. C. Ford, N. S. Berrow, A. C. Dolphin, and A. Kitmitto. 2004. The three-dimensional structure of the cardiac L-type voltage-gated calcium channel: comparison with the skeletal muscle form reveals a common architectural motif. J. Biol. Chem. 279:7159–7168. [DOI] [PubMed] [Google Scholar]
- 13.Mori, M. X., M. G. Erickson, and D. T. Yue. 2004. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 304:432–435. [DOI] [PubMed] [Google Scholar]
- 14.Wilson, M. A., and A. T. Brunger. 2000. The 1.0 Å crystal structure of Ca2+-bound calmodulin: an analysis of disorder and implications for functionally relevant plasticity. J. Mol. Biol. 301:1237–1256. [DOI] [PubMed] [Google Scholar]
- 15.Sharma, M. R., L. H. Jeyakumar, S. Fleischer, and T. Wagenknecht. 2000. Three-dimensional structure of ryanodine receptor isoform three in two conformational states as visualized by cryo-electron microscopy. J. Biol. Chem. 275:9485–9491. [DOI] [PubMed] [Google Scholar]
- 16.Wagenknecht, T., M. Radermacher, R. Grassucci, J. Berkowitz, H.-B. Xin, and S. Fleischer. 1997. Locations of calmodulin and FK506-binding protein on the three-dimensional architecture of the skeletal muscle ryanodine receptor. J. Biol. Chem. 272:32463–32471. [DOI] [PubMed] [Google Scholar]
- 17.Bers, D. M., and V. M. Stiffel. 1993. Ratio of ryanodine to dihydropyridine receptors in cardiac and skeletal muscle and implications for E-C coupling. Am. J. Physiol. 264:C1587–C1593. [DOI] [PubMed] [Google Scholar]
- 18.Greenstein, J. L., and R. L. Winslow. 2002. An integrative model of the cardiac ventricular myocyte incorporating local control of Ca2+ release. Biophys. J. 83:2918–2945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takeshima, H., S. Nishimura, T. Matsumoto, H. Ishida, K. Kangawa, N. Minamino, H. Matsuo, M. Ueda, M. Hanaoka, T. Hirose, and S. Numa. 1989. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 339:439–445. [DOI] [PubMed] [Google Scholar]
- 20.Zorzato, F., J. Fujii, K. Otsu, M. Phillips, N. M. Green, F. A. Lai, G. Meissner, and D. H. MacLennan. 1990. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 265:2244–2256. [PubMed] [Google Scholar]
- 21.Chen, S. R., L. Zhang, and D. H. MacLennan. 1992. Characterization of a Ca2+ binding and regulatory site in the Ca2+ release channel (ryanodine receptor) of rabbit skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 267:23318–23326. [PubMed] [Google Scholar]
- 22.Du, G. G., V. K. Khanna, and D. H. MacLennan. 2000. Mutation of divergent region 1 alters caffeine and Ca2+ sensitivity of the skeletal muscle Ca2+ release channel (ryanodine receptor). J. Biol. Chem. 275:11778–11783. [DOI] [PubMed] [Google Scholar]
- 23.Benacquista, B. L., M. R. Sharma, M. Samso, F. Zorzato, S. Treves, and T. Wagenknecht. 2000. Amino acid residues 4425–4621 localized on the three-dimensional structure of the skeletal muscle ryanodine receptor. Biophys. J. 78:1349–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fessenden, J. D., W. Feng, I. N. Pessah, and P. D. Allen. 2004. Mutational analysis of putative calcium binding motifs within the skeletal ryanodine receptor isoform, RyR1. J. Biol. Chem. 279:53028–53035. [DOI] [PubMed] [Google Scholar]
- 25.Li, P., and S. R. Chen. 2001. Molecular basis of Ca2+ activation of the mouse cardiac Ca2+ release channel (ryanodine receptor). J. Gen. Physiol. 118:33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Williams, A. J., D. J. West, and R. Sitsapesan. 2001. Light at the end of the Ca2+-release channel tunnel: structures and mechanisms involved in ion translocation in ryanodine receptor channels. Q. Rev. Biophys. 34:61–104. [DOI] [PubMed] [Google Scholar]
- 27.Du, G. G., and D. H. MacLennan. 1999. Ca2+ inactivation sites are located in the COOH-terminal quarter of recombinant rabbit skeletal muscle Ca2+ release channels (ryanodine receptors). J. Biol. Chem. 274:26120–26126. [DOI] [PubMed] [Google Scholar]
- 28.Sharma, M. R., and T. Wagenknecht. 2004. Cryo-electron microscopy and 3D reconstructions of ryanodine receptors and their interactions with E-C coupling proteins. Basic Appl. Myol. 14:299–306. [Google Scholar]
- 29.Thomas, N. L., F. A. Lai, and C. H. George. 2005. Differential Ca2+ sensitivity of RyR2 mutations reveals distinct mechanisms of channel dysfunction in sudden cardiac death. Biochem. Biophys. Res. Commun. 331:231–238. [DOI] [PubMed] [Google Scholar]
- 30.Tiso, N., D. A. Stephan, A. Nava, A. Baggattin, J. M. Devaney, F. Stanchi, G. Laderet, B. Brahmbhatt, K. Brown, B. Bauce, M. Muriago, C. Basso, et al. 2001. Identification of mutations in the cardiac ryanodine receptor gene in families affected with arrhythmogenic right ventricular cardiomyopathy type 2 (ARVD2). Juman. Mol. Genetics. 10:189–194. [DOI] [PubMed] [Google Scholar]
- 31.Peterson, B., C. DeMaria, J. Adelman, and D. Yue. 1999. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999:549–558. [DOI] [PubMed] [Google Scholar]
- 32.Peterson, B. Z., J. S. Lee, J. G. Mulle, Y. Wang, M. de Leon, and D. T. Yue. 2000. Critical determinants of Ca2+-dependent inactivation within an EF-hand motif of L-type Ca2+ channels. Biophys. J. 78:1906–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuboniwa, H., N. Tjandra, S. Grzesiek, H. Ren, and C. B. Klee. 1995. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 2:768–776. [DOI] [PubMed] [Google Scholar]
- 34.Black, D. J., D. B. Halling, M. D. V., S. E. Pedersen, R. A. Altschuld, and S. L. Hamilton. 2005. Calmodulin interactions with IQ peptides from voltage-dependent calcium channels. Am. J. Physiol. Cell Physiol. 288:C669–C676. [DOI] [PubMed] [Google Scholar]
- 35.Anderson, M. E. 2001. Ca2+-dependent regulation of cardiac L-type Ca2+ channels: is a unifying mechanism at hand? J. Mol. Cell. Cardiol. 33:639–650. [DOI] [PubMed] [Google Scholar]
- 36.Jafri, S., J. J. Rice, and R. L. Winslow. 1998. Cardiac Ca2+ dynamics: the roles of ryanodine receptor adaptation and sarcoplasmic reticulum load. Biophys. J. 74:1149–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rice, J. J., M. S. Jafri, and R. L. Winslow. 1999. Modeling gain and gradedness of Ca2+ release in the functional unit of the cardiac diadic space. Biophys. J. 77:1871–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stern, M. D., L. S. Song, H. Cheng, J. S. Sham, H. T. Yang, K. R. Boheler, and E. Rios. 1999. Local control models of cardiac excitation-contraction coupling. A possible role for allosteric interactions between ryanodine receptors. J. Gen. Physiol. 113:469–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang, S. Q., M. D. Stern, E. Rios, and H. Cheng. 2004. The quantal nature of Ca2+ sparks and in situ operation of the ryanodine receptor array in cardiac cells. Proc. Natl. Acad. Sci. USA. 101:3979–3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zahradnikova, A., I. Zahradnik, I. Gyorke, and S. Gyorke. 1999. Rapid activation of the cardiac ryanodine receptor by submillisecond calcium stimuli. J. Gen. Physiol. 114:787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Josephson, I. R., A. Guia, E. G. Lakatta, and M. D. Stern. 2002. Modulation of the conductance of unitary cardiac L-Type Ca2+ channels by conditioning voltage and divalent ions. Biophys. J. 83:2587–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith, G. D., J. E. Keizer, M. D. Stern, W. J. Lederer, and H. Cheng. 1998. A simple numerical model of calcium spark formation and detection in cardiac myocytes. Biophys. J. 75:15–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Risken, H. 1997. The Fokker-Planck Equation. Springer, Berlin, Germany.
- 44.Wang, H., C. S. Peskin, and T. C. Elston. 2003. A robust numerical algorithm for studying biomolecular transport processes. J. Theor. Biol. 221:491–511. [DOI] [PubMed] [Google Scholar]
- 45.Bortz, A. B., M. H. Kalos, and J. L. Lebowitz. 1975. A new algorithm for Monte Carlo simulation of Ising spin systems. J. Comput. Phys. 17:10–18. [Google Scholar]
- 46.Xing, J., H. Wang, and G. Oster. 2005. From continuum Fokker-Planck models to discrete kinetic models. Biophys. J. 89:1551–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lan, G., and S. X. Sun. 2005. Dynamics of myosin-V processivity. Biophys. J. 88:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Matsumoto, M., and T. Nishimura. 1998. Mersenne twister: a 623-dimensionally equidistributed uniform pseudo-random number generator. ACM Trans. Model. Comput. Simul. 8:3–30. [Google Scholar]
- 49.Nishimura, T. 2000. Tables of 64-bit Mersenne twisters. ACM Trans. Model. Comput. Simul. 10:348–357. [Google Scholar]
- 50.Song, L. S., S. Q. Wang, R. P. Xiao, H. Spurgeon, E. G. Lakatta, and H. Cheng. 2001. β-Adrenergic stimulation synchronizes intracellular Ca2+ release during excitation-contraction coupling in cardiac myocytes. Circ. Res. 88:794–801. [DOI] [PubMed] [Google Scholar]
- 51.Wier, W. G., T. M. Egan, J. R. Lopez-Lopez, and C. W. Balke. 1994. Local control of excitation-contraction coupling in rat heart cells. J. Physiol. 474:463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shannon, T. R., K. S. Ginsburg, and D. M. Bers. 2000. Reverse mode of the sarcoplasmic reticulum calcium pump and load-dependent cytosolic calcium decline in voltage-clamped cardiac ventricular myocytes. Biophys. J. 78:322–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng, H., W. J. Lederer, and M. B. Cannell. 1993. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 262:740–744. [DOI] [PubMed] [Google Scholar]
- 54.Sham, J., L.-S. Song, Y. Chien, L.-H. Deng, M. D. Stern, E. G. Lakatta, and H. Cheng. 1998. Termination of Ca2+ release by a local inactivation of ryanodine receptors in cardiac myocytes. Proc. Natl. Acad. Sci. USA. 95:15096–15101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marx, S. O., J. Gaburjakova, M. Gaburjakova, C. Henrikson, K. Ondrias, and A. R. Marks. 2001. Coupled gating between cardiac calcium release channels (ryanodine receptors). Circ. Res. 88:1151–1158. [DOI] [PubMed] [Google Scholar]
- 56.Efron, B., and R. J. Tibshirani. 1993. An Introduction to the Bootstrap. Chapman & Hall, New York.
- 57.Stern, M. D. 1992. Theory of excitation-contraction coupling in cardiac muscle. Biophys. J. 63:497–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mejia-Alvarez, R., C. Kettlun, E. Rios, M. Stern, and M. Fill. 1999. Unitary Ca2+ current through cardiac ryanodine receptor channels under quasi-physiological ionic conditions. J. Gen. Physiol. 113:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sobie, E. A., K. W. Dilly, J. dos Santos Cruz, W. J. Lederer, and M. S. Jafri. 2002. Termination of cardiac Ca2+ sparks: an investigative mathematical model of calcium-induced calcium release. Biophys. J. 83:59–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koh, X., B. Srinivasan, H. S. Ching, and A. Levchenko. 2006. A 3D Monte Carlo analysis of the role of dyadic space geometry in spark generation. Biophys. J. 90:1999–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samoilov, M., S. Plyasunov, and A. P. Arkin. 2005. Stochastic amplification and signaling in enzymatic futile cycles through noise-induced bistability with oscillations. Proc. Natl. Acad. Sci. USA. 102:2310–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]