

Abstract
Sec14p promotes the energy-independent transfer of either phosphatidylinositol (PtdIns) or phosphatidylcholine (PtdCho) between lipid bilayers in vitro and represents the major PtdIns/PtdCho transfer protein in the budding yeast Saccharomyces cerevisiae. Herein, we employ multi-frequency high-field electron paramagnetic resonance (EPR) to analyze the electrostatic and hydrogen-bonding microenvironments for series of doxyl-labeled PtdCho molecules bound by Sec14p in a soluble protein-PtdCho complex. A structurally similar compound, 5-doxyl stearic acid dissolved in a series of solvents, was used for experimental calibration. The experiments yielded two-component rigid limit 130- and 220-GHz EPR spectra with excellent resolution in the gx region. Those components were assigned to hydrogen-bonded and nonhydrogen-bonded nitroxide species. Partially resolved 130-GHz EPR spectra from n-doxyl-PtdCho bound to Sec14p were analyzed using this two-component model and allowed quantification of two parameters. First, the fraction of hydrogen-bonded nitroxide species for each n-doxyl-PtdCho was calculated. Second, the proticity profile along the phospholipid-binding cavity of Sec14p was characterized. The data suggest the polarity gradient inside the Sec14p cavity is a significant contributor to the driving molecular forces for extracting a phospholipid from the bilayer. Finally, the enhanced g-factor resolution of EPR at 130 and 220 GHz provides researchers with a spectroscopic tool to deconvolute two major contributions to the x-component of the nitroxide g-matrix: hydrogen-bond formation and local electrostatic effects.
INTRODUCTION
Nitroxide site-directed spin labeling, in combination with electron paramagnetic resonance (EPR) spectroscopy, represents a powerful biophysical tool for studying the structure and dynamics of macromolecular systems. Site-directed mutagenesis methods now permit the introduction of nitroxide labels at predetermined sites in a protein, thereby rendering the EPR technology applicable to a variety of water-soluble and membrane-associated proteins of arbitrary molecular weight (1–6). The power of this technology is founded on the exquisite sensitivity of stable nitroxide radical EPR spectra to rotational diffusion, magnetic interactions with other electronic spins, and solvent effects on nitroxide magnetic parameters. The sensitivity of the spin label to these parameters allows for deductions on the nature of the local environment that surrounds the nitroxide moiety. In this way, EPR approaches can yield considerable information even in systems for which there are no available crystal structures.
Both the local electrical field and the formation of hydrogen bonds between the oxygen atom of nitroxide and solvent molecules contribute to solvent effects observed for continuous wave (CW) EPR spectra of nitroxides. These interactions are recorded as changes in the nitrogen hyperfine coupling A tensorial and g-matrix components (7–13). For nitroxide EPR spectra in a fast motional regime, these solvent effects are assessed from the isotropic magnetic parameters Aiso and giso (7,8). This approach is applicable to small nitroxide probes partitioning between phases of different polarity and is widely employed for examining phase behavior of phospholipid bilayers by X-band (9.5 GHz) and W-band (95 GHz) EPR (9,14–16). Even if the fast motion condition is not satisfied for nitroxide EPR spectra, the isotropic magnetic parameters can still be evaluated from (partially) averaged components of magnetic tensors (17). If both Aiso and giso are determined in the course of such measurements, an empirical linear correlation plot for giso versus Aiso measured for a specific nitroxide in a set of solvents can be used to deduce polarity of the local environment surrounding the probe. Alternatively, solvent effects can be evaluated from anisotropic magnetic parameters measured from rigid limit nitroxide EPR spectra. The component of the electronic g-matrix directed along the NO bond, gx, and the component of nitrogen hyperfine coupling A tensor directed along the nitroxide 2pz-orbital, Az, are the most sensitive indicators of local electric fields and hydrogen-bond formation to the nitroxide moiety (10,18,19). A linear correlation between those two nitroxide magnetic parameters has been reported for a wide range of solvents of various polarities (10,19).
The sensitivity of nitroxide magnetic parameters to solvent effects has been previously used to establish and characterize polarity gradients across phospholipid bilayers. Those studies employed a series of stearic acids or, alternatively, phospholipids labeled with a nitroxide at a specific carbon of the acyl chain (8,17,20). Linear correlation between nitroxide anisotropic magnetic parameters measured from rigid limit 95-GHz EPR spectra was used to probe conformational changes in the bacteriorhodopsin channel (11,12). A study of local polarity of spin-labeled side chains in azurin using high-resolution EPR spectroscopy at 275 GHz was also recently reported (13).
Several theoretical studies have been devoted to understanding the basis of solvent effects on nitroxide magnetic parameters. Specifically, shifts between the isotropic nitrogen hyperfine coupling constant, Aiso, and the local charge on the nitroxide have been calibrated (21). These effects were analyzed within a standard Hückel theoretical framework coupled with continuum model approximations of aqueous solvents. More detailed studies of electric field effects on the electronic g-matrix indicate that anisotropic magnetic parameters are exceptionally sensitive to both the magnitude and to the orientation of the electric field (22). Results of ab initio calculations, utilizing an intermediate level Rayleigh-Schrödinger perturbation theory, predicted the electrical field effect on g-matrices of nitroxide radicals that agreed well with experimental results (23).
Effects of hydrogen bonding between the nitroxide moiety and the solvent were also evaluated from quantum mechanical calculations. Results obtained using restricted open-shell Hartree-Fock (ROHF) methods with atomic mean-field approximations demonstrate that hydrogen bonding reduces the gx component of the nitroxide spectrum (24). Semiempirical molecular orbital methods were also used to calculate the shift in gx resulting from formation of a single hydrogen bond with the water molecule. The shift is predicted to be Δgx ≈ 4 × 10−4 for a single hydrogen bond formed (18). Changes of this magnitude in the g-factor component are readily detected in high-resolution EPR spectra measured at high magnetic fields (≥95 GHz).
Herein, we report on high-field (HF) EPR experiments to analyze local polarity and proticity of the interior of the phospholipid-binding pocket of the major yeast phosphatidylinositol/phosphatidylcholine (PtdIns/PtdCho) transfer protein Sec14p. Sec14p is the prototypical member of the large eukaryotic protein Sec14 superfamily and is operationally defined by its ability to efficiently promote the energy-independent transfer of either PtdIns or PtdCho between membrane bilayers in vitro (25,26). A large body of genetic and biochemical evidence demonstrates that Sec14p effects an essential integration of phospholipid metabolism with the membrane-trafficking activity of yeast Golgi membranes. In this regard, the ability of Sec14p to rapidly exchange bound phospholipid with phospholipid monomers that reside in stable membrane bilayers is presumed to be important for Sec14p function in cells. How Sec14p harnesses such a robust phospholipid exchange activity to biological function remains unclear, as there are several distinct mechanistic possibilities for how it may do so. Some of these potential mechanisms do not require bona fide transfer of phospholipids between intracellular membranes.
The available Sec14p crystal structure identifies a novel fold that forms a hydrophobic pocket of sufficient volume to accommodate a single molecule of PtdIns or PtdCho (27). The crystallizing unit is an “open” Sec14p conformer that is interpreted to represent an apo-Sec14p form that exists only transiently on the membrane surface as an intermediate in the phospholipid exchange reaction (26). How Sec14p, and other members of the Sec14 superfamily, bind phospholipids remains to be determined as there is no structural information available that discusses this specific issue. This is an important and unanswered question, as it is unclear how Sec14p and its homologs overcome the thermodynamic barriers inherent in phospholipid exchange reactions. Moreover, it remains unclear how Sec14p employs its individual phospholipid-binding activities to execute biological function.
To gain further insight on the molecular mechanism of phospholipid binding within the Sec14p ligand-binding cavity, we employed HF EPR at 130 and 220 GHz to probe the local electrostatic and hydrogen-bonding environment for a series of spin-labeled n-doxyl-PtdCho molecules incorporated into the Sec14p phospholipid-binding pocket. Using these approaches, we tested the hypothesis that a polarity gradient exists within the Sec14p phospholipid-binding pocket and provides a hydrophobic matching to accommodate the phospholipid molecule. We also hypothesize that the presence of hydrogen-bond donors along the ligand-binding cavity lowers the enthalpic barrier in phospholipid exchange reactions. Within the context of these studies, we also took advantage of the opportunity to investigate whether the individual contributions of hydrogen-bond formation and electrostatic effects to the x-component of the nitroxide g-matrix, gx, could be resolved and evaluated in high magnetic field EPR experiments.
MATERIALS AND METHODS
Expression and purification of Sec14p
Recombinant His6-Sec14p was recovered from Escherichia coli strain KK2186 [Δ(lac-pro) supE thi strA sbcB-15 endA/F′(traD36 lacIQ lacZΔ15)] using two rounds of affinity purification with Talon Sepharose resin (Clontech Laboratories, Mountain View, CA) as affinity matrix and a linear imidazole gradient (0–200 mM) for protein elution. Isopropyl β-thiogalactoside (1 mM final concentration) inducible expression of the His6-Sec14p was driven by a pQE31-based plasmid (Qiagen, Hilden, Germany). His6-Sec14p purity exceeded 95% as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and purified protein was highly active for PtdCho transfer.
Preparation of EPR samples
PtdCho and 1-acyl-2-(n-(4,4-dimethyloxazolidine-N-oxyl) stearoyl)-sn-glycero-3-phosphocholine (n-doxyl-PtdCho) with the nitroxide spin label positioned at n = 5, 7, 10, 12, and 16 were purchased from Avanti Polar Lipids (Alabaster, AL) as chloroform solutions and used without further purification. Organic solvents for calibrating HF EPR spectra for polarity and hydrogen-bonding effects included methanol, ethanol, isopropanol, 1-propanol, 1-butanol, heptane, 2,2,2-triflouroethanol (TFE), tetrahydrofuran (THF), dichloromethane, acetic acid—all purchased from Sigma-Aldrich (St. Louis, MO) and 2-methyl-tetrahydrofuran, purchased from Alfa Aesar (Ward Hill, MA). All solvents were analytical grade or higher and were used as received. Stearic acid with nitroxide label positioned at n = 5, 7, 10, 12, and 16 (n-doxyl-SA) was purchased from Tokyo Chemical Industry America (Portland, OR). Methyl 5-doxyl-stearate was purchased from Acros Organics (Geel, Belgium). To prevent aggregation, solutions of n-doxyl-SA were typically prepared at concentrations below 1 mM, i.e., below the critical micellar concentration. Solutions of 5-doxyl-PtdCho were prepared at a 0.1 mM concentration.
Multilamellar aqueous dispersions of n-doxyl-PtdCho (20% by weight) were prepared as described (16). To load Sec14p with n-doxyl-PtdCho, a 95 μM protein solution was mixed with small molar excess of 100% spin-labeled multilamellar liposomes and incubated at room temperature for at least 2 h. The binding was monitored at room temperature using X-band EPR until no further changes in the signal amplitude were observed. This assay is based on the property of isolated nitroxide spins to exhibit much narrower EPR spectra than the spectra from 100% n-doxyl-PtdCho liposomes that are broadened by spin exchange and/or dipolar interactions.
EPR spectroscopy
HF EPR spectra were acquired with a 130-GHz EPR spectrometer constructed and installed at the Argonne National Laboratory (Argonne, IL). Field-swept echo-detected EPR spectra of spin-labeled samples were recorded at T = 25 K using a two-pulse sequence. Typically, two pulses of 50 and 100 ns in length were separated by a 350-ns delay and the repetition rate was set to 500 Hz. Detecting low-temperature rigid-limit HF EPR spectra in echo-detected field-swept mode holds several advantages over the conventional CW detection scheme. First, the pulse scheme eliminates signals originating from any remaining spin-labeled n-doxyl-PtdCho bilayers (i.e., phospholipids aggregated in a solution and not bound to Sec14p) because these molecules have much shorter phase memory relaxation time, T2, due to strong magnetic interactions with the closely positioned neighboring spins. Second, low-temperature echo-detected HF EPR spectra do not have microwave phase distortions, due to rapid-passage effects and/or admixture of dispersion signal. The absence of these distortions allows for more accurate line shape and g-factor analysis.
For 130-GHz EPR, spin-labeled samples were drawn by capillary action into clear fused quartz tubes (inner diameter = 0.5 mm, outer diameter = 0.6 mm; VitroCom, Mountain Lakes, NJ), sealed with a Critoseal clay (purchased from Fisher Scientific, Hampton, NH) and loaded into precooled cryostat at 25 K. Temperature of the HF EPR resonator and the sample was controlled by an ITC-4 temperature controller coupled to a flow cryostat (all supplied by Oxfords Instruments, Concord, MA).
CW EPR spectra at 220 GHz were recorded using a transmission mode spectrometer constructed and installed at the National High Magnetic Field Laboratory (Tallahassee, FL). The instrument was a transmission-type device in which waves are propagated in cylindrical light pipes (28). The microwave source was a Gunn oscillator operating at 110 ± 3 GHz. Frequency higher by a factor of 2 was obtained using a Schottky diode-based multiplier and appropriate high-pass filter. A superconducting magnet (Oxford Instruments) capable of reaching a field of 17 T was employed. Teflon sample caps were filled with 0.1-ml sample solutions and loaded directly into a flow cryostat precooled to 100 K (supplied by Oxfords Instruments). Spectra were recorded at a frequency of 224 GHz with a field modulation frequency of 26.35 KHz. The temperature of experiments was 5 K.
RESULTS AND DISCUSSION
EPR spectra of n-doxyl-PtdCho molecules bound to Sec14p
Rigid limit echo-detected 130-GHz EPR spectra obtained from a series of Sec14p-bound n-doxyl-PtdCho complexes (n = 5, 7, 10, 12, and 16) are shown in Fig. 1. The spectra are aligned along a magnetic field axis assuming that the value of gz is the same for all the spectra and is equal to that of a free electron (ge = 2.002319). Previous experimental and theoretical studies established that the z-component of the electronic g-matrix is unaffected by solvents. Specifically, the difference in gz for the common spin label MTSL ((1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methy)-methanethiosulfonate) dissolved in toluene versus water is only Δgz ≈ 1 × 10−5 (19). This g-factor shift is within the experimental error of the measurements reported here. To avoid problems associated with magnetic field drift and cross-calibration of two different spectrometers, we utilized the position of the central gz nitrogen hyperfine coupling component as an internal g-factor marker. The field position of the central gz nitrogen hyperfine coupling component was determined by least-squares fitting of the peak to a Lorentzian function. The data in Fig. 1 illustrate the sensitivity of 130-GHz rigid limit EPR spectra to polarity effects for the Sec14p-bound n-doxyl-PtdCho complexes. Systematic changes in spectral line shape, primarily at the magnetic field positions corresponding to the x-component of the electronic g-matrix (low-field) and in nitrogen hyperfine splitting of the z-component, are clearly observed as a function of position of the nitroxide label along the PtdCho acyl chain. In the following sections, we relate these spectral changes with the polarity gradient inside the Sec14p phospholipid-binding cavity.
FIGURE 1.

Rigid limit 130-GHz echo-detected EPR spectra from n-doxyl-PtdCho-Sec14p complex at T = 25 K.
The z-component of the nitrogen hyperfine coupling tensor, Az, was measured directly from a characteristic splitting in the HF region of the spectra (Table 1). The values for Az recorded for Sec14p-bound 5-doxyl- and 7-doxyl-PtdCho were found to be equivalent (Az = 34.7 ± 0.25 G). These values report a similar polarity of the microenvironment at positions C5 and C7 of the PtdCho sn-2 acyl chain within the Sec14p-binding cavity. Values for Az decrease measurably for Sec14p-bound 10-doxyl-PtdCho (Az = 33.85 ± 0.25 G) and 12-doxyl-PtdCho (Az = 34.15 ± 0.25 G), indicating a decrease in local polarity of the spin label environment within the Sec14p-binding cavity as the nitroxide is moved ever farther toward the headgroup-distal end of the sn-2 acyl chain. For 16-doxyl-PtdCho, a sharp increase in Az to 35.17 ± 0.25 G was measured. The small peak observed between the mI = +1 and mI = 0 nitrogen hyperfine coupling components of the spectrum from 7-doxyl-PtdCho was assigned to an impurity, most likely protein-bound Mn2+, and was ignored in the analysis. As the nitroxide label is moved from position C5 to C16 of the sn-2 acyl chain, both the position and the shape of the gx portion (low-field region) of the spectrum undergo noticeable changes (Fig. 1). First, the magnetic field position of the maximum of spectral amplitude progressively shifts to a lower field. Such a shift also reports a progressive decrease in the local polarity of the nitroxide environment and is consistent with data obtained for Az. Second, the shape of the x-component is also affected. Although the gx components of the rigid limit spectra from 5- and 16-doxyl-PtdCho appear as single peaks, the spectra from 7-, 10-, and 12-doxyl-PtdCho may be interpreted as the presence of two unresolved components with varied relative intensities.
TABLE 1.
Magnetic parameters of n-doxyl-PtdCho-Sec14p complexes as measured by 130-GHz EPR at T = 25 K
| N | gx(0) | gx(1) | Az, G |
|---|---|---|---|
| 5 | 2.00940 ± 0.00004 | 2.00892 ± 0.00004 | 34.70 ± 0.25 |
| 7 | 2.00940 ± 0.00004 | 2.00892 ± 0.00004 | 34.70 ± 0.25 |
| 10 | 2.00941 ± 0.00004 | 2.00894 ± 0.00004 | 33.85 ± 0.25 |
| 12 | 2.00944 ± 0.00004 | 2.00893 ± 0.00004 | 34.15 ± 0.25 |
| 16 | 2.00880 ± 0.00004 | 35.17 ± 0.25 |
The x-components of g-tensor are calculated from the fit of the low-field spectral component by a superposition of two Voight functions and assuming gz = gz,e = 2.002319 for all spectra.
The nature of these line shape changes was further investigated by carrying out calibration experiments with a series of pure solvents. Previously, the only solvent calibrations available were for the magnetic parameters of small probes; e.g., Tempo (2,2,6,6-tetramethylpiperidine-1-oxyl) (9) or a cysteine labeled with MTSL and its derivatives (11,12,19). The doxyl-nitroxide spin-labeled PtdCho derivatives employed in this study have rather different molecular structures and, therefore, are expected to exhibit different solvent effects. Previously, a correlation between gx and Az for n-doxyl-PtdCho was reported based on a series of rigid limit 95-GHz EPR spectra from spin-labeled dimyristoyl-PtdCho (DMPC) bilayers containing 40 mol % cholesterol (n was varied from 4 to 9) (17) and from dipalmitoyl-PtdCho (DPPC) bilayers (n = 5, 7, 10, 12, 16) (20). Although such correlations are useful for analyzing relative changes in polarity and water penetration across phospholipid bilayers, no attempts have been made to relate the observed magnetic parameter with those corresponding to pure solvents. Thus, we carried out such magnetic parameter calibrations in a series of protic and aprotic solvents with known dielectric characteristics. In this fashion, we acquired a set of empirical references to relate local polarity and proticity in protein systems.
High-field EPR measurements of solvent effect on magnetic tensors of n-doxyl stearic acid
Typically, a series of solvents of different polarity is employed for calibration of nitroxide magnetic parameters for solvent effects. Unfortunately, such a calibration cannot be carried out for spin-labeled lipids because of very limited solubility of these molecules in polar solvents. However, other structurally similar doxyl-containing spin probes, like n-doxyl stearic acids (n-doxyl-SA) and methyl-n-doxyl-stearates, are known to exhibit somewhat better solubility in polar solvents. We chose n-doxyl-SAs for calibration experiments since these spin probes are routinely used in biophysical studies of phospholipid bilayers and are reasonable mimics for n-doxyl-PtdCho.
Fig. 2 shows echo-detected rigid limit 130-GHz EPR spectra from 5-doxyl-SA dissolved in a series of protic and aprotic solvents. Spectra from aprotic solvents THF, 2-methyl-THF, toluene, and heptane appear as a single component. By contrast, most other spectra obtained for 5-doxyl-SA in protic solvents clearly show an additional spitting in the gx region, indicating the presence of at least two spectral components. The only protic solvent exhibiting a single component is TFE, the solvent of the highest polarity and the strongest hydrogen-bond donor in the series of solvents studied. To confirm that the splitting observed in the gx region is indeed a superposition of two components with different g-factors, we obtained better resolved rigid limit spectra from 5-doxyl-SA at 220 GHz in selected solvents, including THF, ethanol, 1-propanol, and isopropanol (Fig. 3). Again, a single-component 220-GHz spectrum was observed in THF. For three other solvents the two-component spectra were better resolved at 220 GHz, confirming our initial interpretation of the 130-GHz spectra. Note that only the gx component exhibited additional splitting and no extra splitting was detected in the gy and gz components—an expected result on the basis of the lower sensitivity of the latter parameters to solvent effects. Unfortunately, the gain in spectral resolution obtained at 220 GHz was accompanied by a loss in sensitivity: we failed to obtain spectra from n-doxyl-PtdCho-Sec14p complexes at this EPR frequency. Thus, we utilized high-resolution EPR at 220 GHz primarily for confirming two-component assignments and calibration of solvent effects on gx.
FIGURE 2.

Echo-detected rigid limit 130-GHz EPR spectra from 5-doxyl-SA dissolved in a series of protic and aprotic solvents of various polarities at T = 25 K.
FIGURE 3.

Rigid limit 220-GHz EPR spectra from 5-doxyl-SA dissolved in a series of protic and aprotic solvents at T = 5 K.
We have observed that all HF EPR spectra in aprotic solvents exhibited only a low-field gx component, whereas spectra collected in the presence of alcohols that are strong hydrogen-bond donors yielded two component spectra. Consequently, there is a strong correlation between the appearance of the higher field component in the gx region with the ability of solvents to form hydrogen bonds. On this basis, HF EPR spectra exhibiting multiple components in the gx region were tentatively assigned as follows. The component characterized by a higher gx, gx(0), was assigned to nitroxides that are not engaged in hydrogen bonds with solvent molecules. Reciprocally, the component with a lower gx, gx(1), was assigned to nitroxide engaged in a single hydrogen bond (Fig. 2).
To demonstrate that formation of the hydrogen bond is a result of nitroxide-solvent interaction and not a formation of an intramolecular hydrogen bond between the nitroxide moiety and carboxylic groups, we obtained 220-GHz EPR spectra from n-doxyl-SA ester (n = 5, 6, 12, and 16) in ethanol. For all n-doxyl-SA ester samples, two component spectra were observed with gx(0) and gx(1) equal to those measured for 5-doxyl-SA in ethanol (data not shown). We also obtained a 130-GHz EPR spectrum from 5-doxyl-PtdCho in isopropanol (Fig. 1, bottom spectrum).
Modeling of n-doxyl-SA EPR spectra
Spectra from 5-doxyl-SA in all aprotic solvents and in the strongest protic solvent TFE were least-squares fitted with a single component powder pattern model. For two component spectra, all nitroxide magnetic parameters were set to be the same with the exception of gx. An example of the two-component fit for a 130-GHz EPR spectrum from 5-doxyl-SA in butanol is shown in Fig. 4. Although the least-squares simulations produced an overall satisfactory fit, in some cases the rigid limit model failed to faithfully reproduce the relative intensities of the spectral components. One such example is illustrated in Fig. 4. This is expected as the relative intensities of principal axis components of echo-detected EPR spectra could deviate from a normal powder pattern as a result of anisotropic electronic relaxation effects.
FIGURE 4.

Echo-detected 130-GHz EPR spectrum from 5-doxyl-SA in butanol at T = 25 K is superimposed with the result of the fit using the powder pattern model assuming two components. For the two component spectra all nitroxide magnetic parameters were set to be the same with the exception of gx.
To circumvent uncertainties introduced by anisotropic electronic relaxation effects, only the gx region of the 5-doxyl-SA HF EPR spectra was simulated. Furthermore, the simulations of gx(0) and gx(1) components were simplified by modeling this spectral region as a superposition of two Voigt functions (a convolution of Lorentzian and Gaussian functions). The position, intensity, and line widths of each of the components were adjusted during the fitting. An example of such a fit is shown in Fig. 5 for the 5-doxyl-SA spectrum in isopropanol. This empirical fitting approach was also verified by simulation of a modeled powder pattern spectrum (not shown). The correlation between 5-doxyl-SA EPR parameters—gx(0) or gx(1) and Az—in diverse solvents is shown in Fig. 6. Parameters are also listed in Table 2. Those data demonstrate that gx(0), the g-factor of the component corresponding to nonhydrogen-bonded nitroxide radical in protic solvents, is essentially the same as gx for a single-component EPR spectra observed in aprotic solvents. This observation further confirms our initial assignment. Our data further show that gx(0) has a very weak correlation with Az (the slope 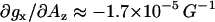 ). For the spectral component assigned to a nitroxide forming a single hydrogen bond, gx(1), the slope
). For the spectral component assigned to a nitroxide forming a single hydrogen bond, gx(1), the slope 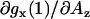 is somewhat higher (≈−8 × 10−5 G−1). It is interesting to note that the slope
is somewhat higher (≈−8 × 10−5 G−1). It is interesting to note that the slope 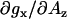 for the hydrogen-bonded and nonhydrogen-bonded components experimentally measured here are smaller than the respective slopes predicted for the limiting protic case (fully hydrogen-bonded MTSL label) and the limiting aprotic cases (various spin-labeled positions in bacteriorhodopsin) (18). Some of these deviations could be attributed to differences in nitroxide structure.
for the hydrogen-bonded and nonhydrogen-bonded components experimentally measured here are smaller than the respective slopes predicted for the limiting protic case (fully hydrogen-bonded MTSL label) and the limiting aprotic cases (various spin-labeled positions in bacteriorhodopsin) (18). Some of these deviations could be attributed to differences in nitroxide structure.
FIGURE 5.

Low-field region of echo-detected 130-GHz EPR spectrum from 5-doxyl-SA in isopropanol (solid line) is superimposed with simulated spectra (Voight function) corresponding to nonhydrogen-bonded (dash-dot line) and hydrogen-bonded (dash line) components. Residual, the difference between the experimental spectrum and the two simulated components, is shown at the bottom as a solid line.
FIGURE 6.

Correlation between doxyl spin label magnetic parameters—gx versus Az—measured from 130-GHz echo-detected rigid limit EPR spectra at 25 K. Results of calibration experiments for 5-doxyl-SA in isotropic solvents are shown by circles. Squares represent parameters for n-doxyl-PtdCho bound to Sec14p: (a) 5-doxyl-PtdCho, (b) 7-doxyl-PtdCho, (c) 10-doxyl-PtdCho, (d) 12-doxyl-PtdCho, and (e) 16-doxyl-PtdCho. Solid symbols correspond to a hydrogen-bonded spectral component, and open symbols represent the nonhydrogen-bonded component. Triangles represent parameters for 5-doxyl-PtdCho in isopropanol. Solvents used in the experiment are 1), heptane, 2,) THF, 3), 2-methyl-THF, 4), toluene, 5), n-butanol, 6), isopropanol, 7), ethanol, 8), methanol, 9), acetic acid, and 10), TFE.
TABLE 2.
Magnetic parameters of 5-doxyl-PtdCho in various solvents as measured by 130-GHz EPR at T = 25 K
| Solvent | gx(0) | gx(1) | Az, G |
|---|---|---|---|
| Ethanol | 2.00927 ± 0.00004 | 2.00863 ± 0.00004 | 34.80 ± 0.25 |
| THE | 2.00927 ± 0.00004 | 2.00849 ± 0.00004 | 36.58 ± 0.25 |
| Isopropanol | 2.00933 ± 0.00004 | 2.00867 ± 0.00004 | 34.79 ± 0.25 |
| Methanol | 2.00929 ± 0.00004 | 2.00868 ± 0.00004 | 35.44 ± 0.25 |
| Butanol | 2.00918 ± 0.00004 | 2.00863 ± 0.00004 | 34.62 ± 0.25 |
| Acetic acid | 2.00921 ± 0.00004 | 2.00857 ± 0.00004 | 35.77 ± 0.25 |
| Toluene | 2.00928 ± 0.00004 | 33.50 ± 0.25 | |
| THF | 2.00941 ± 0.00004 | 33.38 ± 0.25 | |
| Heptane | 2.00926 ± 0.00004 | 33.25 ± 0.25 | |
| 2-methyl-THF | 33.50 ± 0.25 |
The x-components of g-tensor are calculated assuming gz = gz,e = 2.002319 for all spectra.
From our calibration measurements, the difference between gx(1) and gx(0), Δgx, varies from 6.0 × 10−4 to 6.6 × 10−4 depending upon solvent. This g-factor shift reflects formation of a single hydrogen bond between the oxygen atom of the doxyl radical and the OH group of solvent molecules. The shift is larger than the Δgx ≈ 4 × 10−4 value previously predicted by semiempirical calculations for the MTSL nitroxide (18). The magnitude of Δgx measured in course of calibration experiments presented here is, however, in excellent agreement with other published data for the doxyl label. Indeed, the shift in average gx for n-doxyl-PtdCho between the outer and inner regions of lipid bilayer measured with lower resolution at 94 GHz was found to be 6.4 × 10−4 or 6.2 × 10−4 for DMPC bilayers containing 5 or 40 mol % cholesterol, respectively (17). Such a shift was attributed to the effect of water molecules present in the interfacial, but not in the hydrocarbon, regions of the bilayer (17). Our calibration data demonstrate that Δgx observed in the latter phospholipid bilayer experiments is almost entirely due to hydrogen-bonding effects with solvent molecules that, in the case of hydrated phospholipid bilayers, could only be water.
In principle, the magnetic parameters of nitroxides could be affected by proximity to polar chemical groups and interactions of the latter with solvent molecules. This leaves open the possibility that the solvent calibration profile for each of the n-doxyl-SA could exhibit unique features. We investigated whether the position of the doxyl moiety along the hydrocarbon chain influences the magnetic parameters of the rigid limit EPR spectra by measuring 130-GHz echo-detected EPR spectra from 5-, 7-, 10-, 12-, and 16-doxyl-SA in ethanol. For all n-doxyl-SA species tested, well-resolved two-component spectra were observed (not shown). Within the accuracy of our measurements, we found that the magnetic parameters of the nitroxide were independent of the position of the doxyl moiety. We conclude that the polar headgroup of stearic acid exerts no significant effect on doxyl-solvent interactions under the conditions employed. These experiments also demonstrate that long-range substitutions in doxyl-label structure, relative to the position of NO group, do not affect the nitroxide electronic g-matrix and nitrogen hyperfine coupling tensor A.
We also obtained a 130-GHz spectrum from 5-doxyl-PtdCho in isopropanol (bottom trace on Fig. 1). The observed well-resolved two-component spectrum is characterized by gx(0), gx(1), and Az, which are essentially the same as measured for 5-doxyl-SA in isopropanol, confirming that 5-doxyl-SA can be used as a model compound in calibration experiments. It is interesting to note that the fraction of the hydrogen-bonded component in the 5-doxyl-PtdCho spectrum in isopropanol is smaller than the fraction of hydrogen-bonded species formed by 5-doxyl-SA in the same solvent. We attribute this to steric protection of the nitroxide moiety by two acyl chains of the lipid molecule. We hypothesize that local arrangement of the acyl chains around the nitroxide partially excludes rather large isopropanol molecules from the vicinity of the NO group, preventing formation of a hydrogen bond. For a lipid molecule with two chains this effect would be more pronounced than for a stearic acid molecule that has only one chain. Such differential accessibility of the solvent molecules to a nitroxide moiety could result in differences in the fractions of the hydrogen-bonded nitroxide species recorded in these experiments.
Overall, the calibration experiments presented here indicate that increased g-factor resolution at magnetic fields corresponding to 130-GHz and higher resolve EPR signals arising from hydrogen-bonded and nonhydrogen-bonded nitroxide species that coexist in protic solvents. We also conclude that hydrogen-bond formation manifests itself primarily as a shift in the gx component. For 5-doxyl-SA the magnitude of the gx shift for nonhydrogen-bonded species is approximately equivalent for all aprotic and protic solvents studied. This parameter has a rather weak correlation with Az. The gx of the hydrogen-bonded component decreases in protic solvents that are strong hydrogen-bond donors (e.g., TFE). However, the slope 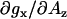 is smaller than what other studies using structurally distinct nitroxide-labeled probes would predict.
is smaller than what other studies using structurally distinct nitroxide-labeled probes would predict.
Although spectral components corresponding to nitroxides in the presence and absence of hydrogen-bond interactions are at least partially resolved in the gx region, the spectral features corresponding to Az overlap completely. This overlap prevents unambiguous determination of the magnitudes of the Az(0) and Az(1) components. Experimentally determined Az values, therefore, represent some averaged form of Az(0) and Az(1). Caution must be exercised when interpreting such “averaged” values. A narrower line width of one of the overlapping signals would result in larger peak amplitude, and this feature would therefore dominate the fitting procedure. In such a case a fitting procedure would report on the narrow component almost exclusively, even if two species contribute equally to the spectrum. Thus, it is anticipated that two solvent effects—formation of hydrogen bond and polarizing field of surrounding polar molecules—contribute to experimentally observed Az.
Polarity and hydrogen-bonding profile of Sec14p phospholipid-binding cavity
Solvent calibration experiments for n-doxyl-SA in a series of protic and aprotic solvents indicate that the main effect on gx arises from formation of a hydrogen bond and that gx(0) and gx(1) assigned to each of the components exhibit very little solvent dependence. Based on this observation, we simulated unresolved 130-GHz echo-detected EPR spectra from n-doxyl-PtdCho-Sec14p complexes as a superposition of two Voigt functions. In Fig. 7, we depict a low-field region of an experimental EPR spectrum from 10-doxyl-PtdCho superimposed with simulated spectra corresponding to a nonhydrogen-bonded component (dash-dot line) and hydrogen-bonded component (dash line). The residual between the experimental spectrum and the two simulated components is shown at the bottom (solid line). This model fits experimental spectra for all positions of n-doxyl-PtdCho bound to Sec14p exceptionally well. Magnetic parameters for both components are shown in Fig. 6 as open squares for gx(0) (nonhydrogen-bonded) components and as solid squares for gx(1) (hydrogen-bonded) components. The g-factor anisotropy, measured as (gx(0) − ge), was equal to 7.07 × 10−3 for position C5. This value increased slightly along the chain reaching 7.12 × 10−3 for position C16. Such a variation is within the accuracy of our experiment (∼4 × 10−5). The gx of the second peak is also nearly constant for the labels attached at positions C5, C7, C10, and C12 with (gx(1) − gz,e) varying within the experimental error from 6.58 × 10−3 to 6.61 × 10−3. For position C16, however, the (gx(1) − gz,e) was determined to be 6.48 × 10−3, a value significantly lower than for all other positions.
FIGURE 7.

Low-field region of echo-detected 130-GHz EPR spectrum from 10-doxyl-PtdCh bound to Sec14p superimposed with simulated spectra (Voigt function) corresponding to nonhydrogen-bonded component (dash-dot line) and hydrogen-bonded component (dash line). Residual, the difference between experimental spectrum and two simulated components, is shown at the bottom as a solid line.
Experimental data suggest that for all spin-labeled lipids bound to Sec14p, the doxyl moiety coexists of two forms: one that engages in hydrogen bonds and one that does not. The magnitude of the gx component for nonhydrogen-bonded label in protein is close to that observed for n-doxyl-SA solubilized in aprotic solvents such as heptane. The gx of the hydrogen-bonded component, however, deviates measurably from the values of gx(1) established in our calibration experiments for n-doxyl-SA incorporated into protic solvents. To account for this observation, we consider two possible hydrogen-bond donors available within the Sec14p phospholipid-binding cavity itself. These include i), water molecules present within the Sec14p hydrophobic cavity, and ii), Sec14p residues. With regard to the former possibility, the apo-Sec14p crystal structure reports an anhydrous phospholipid-binding pocket (27). Although it is formally possible that water molecules are dragged into the Sec14p hydrophobic cavity during the phospholipid exchange reaction (i.e., the phospholipid ligand itself is likely incorporated in a hydrated form) and the trapped water subsequently interacts with polar protein residues, there is no evidence that this actually occurs. At this point, we favor the latter interpretation that the nitroxide moiety interacts with protein side chains or the Sec14p protein backbone itself.
Regardless of whether water or Sec14p residues interact with the spin label, the geometry of the hydrogen bond may be distorted because of steric restrictions on mutual orientations. We posit that one of the results of such a restriction would be an energetically less favorable geometry of the hydrogen bond compared with the unrestrained hydrogen bond formed between the free label and molecules in an isotropic solvent. In this scenario, restriction would result in a weaker hydrogen bond; i.e., the one that should yield a smaller shift in Δgx when compared with a stronger hydrogen bond formed in an isotropic solvent with no steric restrictions. We propose that the doxyl label at positions C5, C7, C10, and C12 forms a hydrogen bond that is subject to some form of steric restriction and, as a result, the gx(1) value observed for the nitroxide at those positions is higher than is observed in protic solvents. It is interesting to note here that the line width of the hydrogen-bonded component for all the positions of doxyl moiety along the lipid chain is broader than for the spectral component arising from the nonhydrogen-bonded label. This could be a consequence of an unresolved hyperfine interaction with the bond-forming hydrogen atom or could indicate a more heterogeneous microenvironment of the nitroxide label in the hydrogen-bonded state. At position 16, however, the gx(1) value is very close to one observed in alcohols. We suggest that the nitroxide label at this position either engages in a hydrogen bond with a water molecule or experiences fewer steric restrictions.
Notwithstanding whether the hydrogen-bond formation directly involves Sec14p residues or water molecules within the Sec14p hydrophobic pocket, the relative fraction of the nitroxide spin label in each of those forms can be estimated by calculating the double integrals of the corresponding spectral components. The fractional engagement of each individual nitroxide in hydrogen-bond formation can be used as a reporter of the local “proticity” of the spin-label environment. This relationship is illustrated in Fig. 8. At positions C5 and C7, the nitroxide probe exists primarily in the hydrogen-bonded form, with corresponding fractions 0.90 ± 0.05 and 0.80 ± 0.07. At position C10, the nitroxide probe exhibits a much smaller fraction in the hydrogen-bonded form (0.30 ± 0.09), thereby characterizing the corresponding local Sec14p environment as “mostly aprotic”. As the nitroxide label is positioned farther along the chain in the headgroup distal direction, the fraction of the hydrogen-bonded form increases again to 0.45 ± 0.17 for position C12 and 1.00 for position C16.
FIGURE 8.

Approximate fraction of hydrogen-bonded nitroxide label as a function of the label position along the chain of n-doxyl-PtdCho bound to Sec14p.
Results of our HF EPR experiments described here provide a basis for comparing the local environment experienced by Sec14p-bound PdtCho relative to a PtdCho molecule resident in a phospholipid bilayer. These two systems can be treated thermodynamically as the final and the initial states in the phospholipid exchange pathway between membrane bilayer and Sec14p. Previously, the polarity gradient across DPPC bilayers was resolved by 250-GHz EPR using n-doxyl-PtdCho (n = 5, 7, 12, and 16) incorporated into the liposomes at 1 mol % concentration (20). Those studies show that the z-component of the nitrogen hyperfine coupling tensor A, Az, decreased from 35.0 ± 0.2 G for the doxyl nitroxide at position C5 to 34.5 ± 0.2 G for position C7, with a further decrease to 34.0 ± 0.2 G at position C12 and 33.5 ± 0.2 G at position C16. The Az values for positions C5–C12 for n-doxyl-PtdCho incorporated into a phospholipid bilayer are very similar to those we report for the PtdCho-Sec14p complex, indicating that the local polarity profiles of these regions of the Ptdcho molecule are essentially equivalent in these two systems.
The local polarity at the distal end of the PtdCho molecule, however, differs substantially. In the center of the bilayer the nitroxide label at C16 reports a far more aprotic environment than we measure for the PtdCho-Sec14p complex. Although in Earl et al. (20), the gx spectral feature was treated as a single component, this parameter can still be used to evaluate the difference (gx − gz), which is equivalent to the parameter (gx − ge) we report herein. At positions C5 and C7 the (gx − gz) for DPPC bilayer is 6.57 × 10−3 ± 0.02 × 10−3 and 6.61 × 10−3 ± 0.02 × 10−3, respectively, indicating that the nitroxide moieties are mostly in the hydrogen-bonded forms. For position C16, the measured value (gx − gz) is 7.17 × 10−3 ± 0.02 × 10−3, and this value is similar to those we record for the nonhydrogen-bonded form of the nitroxide. This interpretation is in agreement with conclusions from other studies that water molecules penetrate as deep as position C7 in DMPC bilayer systems that contain 40% cholesterol and that, in such systems, nitroxide labels located at positions C4–C7 engage in hydrogen bonds with those water molecules (17).
In sum, comparisons of magnetic parameters for Sec14p-bound n-doxyl-PtdCho to those of n-doxyl-PtdCho incorporated into DMPC bilayers indicate that the local polarities of the C5–C10 region of PtdCho are very similar in both systems and that region C5–C7 is exposed to potential hydrogen-bond donors in both the Sec14p hydrophobic cavity and in a genuine membrane bilayer. The environment of the distal end of PtdCho, however, is strikingly different in the Sec14p-bound versus bilayer condition. When PtdCho is incorporated into a bilayer, position C16 resides in a nonpolar/aprotic environment. When PtdCho is Sec14p bound, that region of the PtdCho molecule is exposed to a significantly more polar/protic environment. The collective data indicate that the phospholipid-binding cavity of Sec14p provides a polarity profile along the bound PtdCho molecule that closely matches that found in membrane bilayers. This polarity match provides a rationale for why a PtdCho molecule will efficiently partition from a bilayer into the Sec14p phospholipid-binding pocket and vice versa. The distal end of Sec14p-bound PtdCho is a striking exception to this trend, however, as the C16 position resides in a protic environment. The precise basis for this puzzling C16 microenvironment and what functional implications such a curious arrangement holds await high-resolution structural studies.
SUMMARY
We describe the polarity and proticity profile inside the phospholipid-binding pocket of Sec14p as determined by HF EPR. In a series of calibration experiments, the effects of protic and aprotic solvents of various polarity on rigid limit 130-GHz EPR spectra from 5-doxyl-SA were investigated. We show that, at magnetic fields corresponding to frequencies of 130 GHz and above, the spectral features arising from hydrogen-bonded and nonhydrogen-bonded forms of nitroxide become clearly resolved. In protic solvents, two-component HF EPR spectra were observed with excellent resolution in the gx region of the spectra. Least-squares simulation allowed us to estimate that formation of a single hydrogen-bond shifts the x-component of the electronic g-matrix, gx, of the doxyl label by −6.0 × 10−4 to −6.6 × 10−4 in a manner that depends on solvent. Partially resolved 130-GHz EPR spectra from n-doxyl-PtdCho bound to Sec14p were analyzed using this two-component model. We demonstrate that the fraction of the hydrogen-bonded form of the nitroxide label can be determined from spectral simulations and then be used to characterize the proticity profile along the phospholipid-binding channel. Specifically, HF EPR experiments provide evidence of a more polar and highly protic microenvironment at positions C5 and C7 of the sn-2 acyl chain with the nitroxide probe existing almost completely in the hydrogen-bonded form. Local polarity, characterized by the z-component of nitrogen hyperfine coupling tensor A, Az, decreases along the lipid chain and is accompanied by an increase in the fraction of nitroxide in a nonhydrogen-bonded state. Position C16, however, is located in a polar environment in a completely hydrogen-bonded state. Finally, our experiments further suggest that the protein environment sterically restricts the architecture of hydrogen bonds formed by nitroxide labels incorporated in the Sec14p cavity. These steric restrictions are observed as deviations in the gx component of the electronic g-matrix from those recorded in isotropic bulk solutions.
Acknowledgments
The work at NCSU was supported by National Science Foundation Grant No. MCB-0451510 with partial support from American Chemical Society PRF No. 40771-G4 both to T.I.S. V.A.B., G.S., and M.M.R. were supported by National Institutes of Health grant GM44530 awarded to V.A.B. G.S. was also supported by a Postdoctoral Training Grant from the Deutsche Forschungsgemeinschaft. The work at ANL was supported by the U.S. Dept. of Energy, Office of Basic Energy Sciences, Division of Chemical Sciences, Geosciences, and Biosciences, under Contract W-31-109-Eng-38, and partially by National Institutes of Health 1R01GM072897. A portion of this work was performed at the National High Magnetic Field Laboratory (NHMFL), which is supported by the National Science Foundation Cooperative Agreement No. DMR-0084173 and by the state of Florida. T.I.S. thanks the National High Magnetic Field Laboratory visiting scientist program for financial support.
References
- 1.Hubbell, W. L., D. S. Cafiso, and C. Altenbach. 2000. Identifying conformational changes with site-directed spin labeling. Nat. Strict. Biol. 7:735–739. [DOI] [PubMed] [Google Scholar]
- 2.Hubbell, W. L., L. A. Gross, R. Langen, and M. A. Lietzow. 1998. Recent advances in site-directed spin labeling of proteins. Curr. Opin. Struct. Biol. 8:779–783. [DOI] [PubMed] [Google Scholar]
- 3.Feix, J. B., and C. S. Klug. 1998. Spin labeling: the next millennium. In Biological Magnetic Resonance, Vol. 14. L. J. Berliner, editor. Plenum Press, NY. 251-281.
- 4.Columbus, L., and W. L. Hubbell. 2002. A new spin on protein dynamics. Trends Biochem. Sci. 27:288–295. [DOI] [PubMed] [Google Scholar]
- 5.Cafiso, D. S. 2002. Peptide-membrane interactions determined using site-directed spin labeling. Peptide-Lipid Interactions. 52:3-29. [Google Scholar]
- 6.Malmberg, N. J., and J. J. Falke. 2005. Use of EPR power saturation to analyze the membrane-docking geometries of peripheral proteins: applications to C2 domains. Annu. Rev. Biophys. Biomol. Struct. 34:71–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kawamura, T., S. Matsunami, and T. Yonezawa. 1967. Solvent effects on the g-values of di-t-butyl nitric oxide. Bull. Chem. So. Jpn. 40:1111–1115. [Google Scholar]
- 8.Griffith, O. H., P. J. Dehlinger, and S. P. Van. 1974. Shape of the hydrophobic barrier of phospholipid bilayers (evidence for water penetration in biological membranes). J. Membr. Biol. 15:159–192. [DOI] [PubMed] [Google Scholar]
- 9.Smirnov, A. I., and T. I. Smirnova. 2001. Resolving domains of interdigitated phospholipid membranes with 95 GHz spin labeling EPR. Appl. Magn. Reson. 21:453–467. [Google Scholar]
- 10.Ondar, M. A., O. Ya. Grinberg, A. A. Dubinskii, and Ya. S. Lebedev. 1985. Study of the effect of the medium on the magnetic-resonance parameters of nitroxyl radicals by high-resolution EPR spectroscopy. Sov. J. Chem. Phys. 3:781–792. [Google Scholar]
- 11.Wegener, C., A. Savitsky, M. Pfeiffer, K. Möbius, and H.-J. Steinhoff. 2001. High-field EPR-detected shifts of magnetic tensor components of spin label side chains reveal protein conformational changes: the proton entrance channel of bacteriorhodopsin. Appl. Magn. Reson. 21:441–452. [Google Scholar]
- 12.Steinhoff, H.-J., A. Savitsky, C. Wegener, M. Pfeiffer, M. Plato, and K. Möbius. 2000. High-field EPR studies of the structure and conformational changes of site-directed spin labeled bacteriorhodopsin. Biochim. Biophys. Acta. 1457:253–262. [DOI] [PubMed] [Google Scholar]
- 13.Finiguerra, M. G., H. Blok, M. Ubbink, and M. Huber. 2006. High field (275 GHz) spin-label EPR for high resolution polarity determination in proteins. J. Mag. Res. 180:197–202. [DOI] [PubMed] [Google Scholar]
- 14.Shimshick, E. J., and H. M. McConnell. 1973. Lateral phase separation in phospholipid membranes. Biochemistry. 12:2351–2360. [DOI] [PubMed] [Google Scholar]
- 15.Polnaszek, C. F., S. Schreier, K. W. Butler, and I. C. P. Smith. 1978. Analysis of the factors determining the EPR spectra of spin probes that partition between aqueous and lipid phases. J. Am. Chem. Soc. 100:8223–8231. [Google Scholar]
- 16.Smirnov, A. I., T. I. Smirnova, and P. D. Morse II. 1995. Very high frequency electron paramagnetic resonance of 2,2,6,6-tetramethyl-1-piperidinyloxy in 1,2-dipalmitoyl-sn-glycero-3-phosphatidyl-choline liposomes: partitioning and molecular dynamics. Biophys. J. 68:2350–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kurad, D., G. Jeschke, and D. Marsh. 2003. Lipid membrane polarity profiles by high-field EPR. Biophys. J. 85:1025–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Plato, M., H.-J. Steinhoff, C. Wegener, J. T. Törring, A. Savitsky, and K. Möbius. 2002. Molecular orbital study of polarity and hydrogen bonding effects on the g and hyperfine tensors of site directed NO spin labelled bacteriorhodopsin. Mol. Phys. 100:3711–3721. [Google Scholar]
- 19.Owenius, R., M. Engstrom, M. Lindgren, and M. Huber. 2001. Influence of solvent polarity and hydrogen bonding on the EPR parameters of a nitroxide spin label studied by 9-GHz and 95-GHz EPR spectroscopy. J. Phys. Chem. 105:10967–10977. [Google Scholar]
- 20.Earl, K. A., J. K. Moscicki, M. Ge, D. E. Budil, and J. H. Freed. 1994. 250-GHz electron spin resonance studies of polarity gradients along the aliphatic chains in phospholipid membranes. Biophys. J. 66:1213–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schwartz, R. N., M. Peric, S. A. Smith, and B. L. Bales. 1997. Simple test of the effect of an electric field on the 14N-hyperfine coupling constant in nitroxide spin probes. J. Phys. Chem. B. 101:8735–8739. [Google Scholar]
- 22.Gulla, A. F., and D. E. Budil. 2001. Orientation dependence of electric field effects on the g-factor of nitroxides measured by 220 GHz EPR. J. Phys. Chem. B. 105:8056–8063. [Google Scholar]
- 23.Ding, Z., A. F. Gulla, and D. E. Budil. 2001. Ab initio calculations of electric field effects on the g-tensor of a nitroxide radical. J. Chem. Phys. 115:10685–10693. [Google Scholar]
- 24.Engstrom, M., R. Owenius, and O. Vahtras. 2001. Ab initio g-tensor calculations of hydrogen bond effects on a nitroxide spin label. Chem. Phys. Lett. 338:407–413. [Google Scholar]
- 25.Cleves, A. E., T. P. McGee, and V. A. Bankaitis. 1991. Phospholipid transfer proteins: a biological debut. Trends Cell Biol. 1:30–34. [DOI] [PubMed] [Google Scholar]
- 26.Phillips, S. E., P. Vincent, K. Rizzieri, G. Schaaf, E. A. Gaucher, and V. A. Bankaitis. 2006. The diverse biological functions of phosphatidylinositol transfer proteins in eukaryotes. Crit. Rev. Biochem Mol. Biol. 41:21–49. [DOI] [PubMed] [Google Scholar]
- 27.Sha, B., S. E. Phillips, V. A. Bankaitis, and M. Luo. 1998. Crystal structure of the Saccharomyces cerevisiae phosphatidylinositol-transfer protein. Nature. 391:506–510. [DOI] [PubMed] [Google Scholar]
- 28.Hassan, A. K., L. A. Pardi, J. Krzystek, A. Sienkiewicz, P. Goy, M. Rohrer, and L.-C. Brunel. 2000. Ultrawide band multifrequency high-field EMR technique: a methodology for increasing spectroscopic information. J. Magn. Reson. 142:300–312. [DOI] [PubMed] [Google Scholar]