

Abstract
Fluorescence resonance energy transfer (FRET) between matched carbocyanine lipid analogs in the plasma membrane outer leaflet of RBL mast cells was used to investigate lateral distributions of lipids and to develop a general method for quantitative measurements of lipid heterogeneity in live cell membranes. FRET measured as fluorescence quenching of long-chain donor probes such as DiO-C18 is greater with long-chain, saturated acceptor probes such as DiI-C16 than with unsaturated or shorter-chain acceptors with the same chromophoric headgroup compared at identical concentrations. FRET measurements between these lipid probes in model membranes support the conclusion that differential donor quenching is not caused by nonideal mixing or spectroscopic differences. Sucrose gradient analysis of plasma membrane-labeled, Triton X-100-lysed cells shows that proximity measured by FRET correlates with the extent of lipid probe partitioning into detergent-resistant membranes. FRET between DiO-C16 and DiI-C16 is sensitive to cholesterol depletion and disruption of liquid order (Lo) by short-chain ceramides, and it is enhanced by cross linking of Lo-associated proteins. Consistent results are obtained when homo-FRET is measured by decreased fluorescence anisotropy of DiI-C16. These results support the existence of nanometer-scale Lo/liquid disorder heterogeneity of lipids in the outer leaflet of the plasma membrane in live cells.
INTRODUCTION
The existence of lateral inhomogeneities or domains in the lipid portion of the plasma membrane is an issue of substantial interest and controversy (1–3). Studies on detergent-resistant membranes (4,5) led to the hypothesis that lateral segregation of liquid ordered (Lo) and liquid disordered (Ld) lipids in the plasma membrane plays an important role in signal transduction, protein sorting, and membrane transport (1,2,6). These cholesterol-dependent ordered membrane domains that selectively contain lipids and proteins are commonly called lipid rafts. However, the difficulty in observing lipid membrane domains at optical resolution has made it challenging to relate biochemical evidence for lipid heterogeneity, such as that from detergent-dependent fractionation, to properties in live cells. Detecting segregated membrane domains in live cells usually requires large-scale cross-linking of lipid raft components and/or low temperature (7–9). Some advanced methods with high spatial (nanometer) and temporal (millisecond) resolution, such as single particle tracking and video microscopy (10–12), nanosecond depolarization homo-FRET (13), and high-resolution electron microscopy (14,15), have provided evidence for membrane domains in intact plasma membranes. However, direct evidence for the existence of lipid rafts in live cells is largely based on measurements of clustering or diffusion of putative raft proteins rather than on measurements of the lipids themselves. A recent exception is the detection of the ordered and disordered lipid phases in live cells with ESR measurements of spin-labeled lipid probes (16).
The ordered state of lipid rafts is based on preferential interactions between cholesterol and phospholipids with long saturated acyl chains such as sphingolipids. Correspondingly, lipid probes with different acyl/alkyl chains are expected to partition differently between ordered domains and disordered regions of the bilayer. In our study, we investigated the lateral distributions of fluorescent lipid probes in the outer leaflet of live cell membranes using FRET between carbocyanine derivatives with differing alkyl chains. Carbocyanines label the outer leaflet of cell membrane with negligible transbilayer flip-flop (17), making them ideal outer leaflet probes. Furthermore, their high photostability, large extinction coefficients, and partition coefficients that strongly favor lipid over aqueous environments (Kp ∼ 103−4 for DiO-C6) (18) make them highly suitable for fluorescence measurements in live cell membranes. FRET is particularly useful for monitoring inhomogeneities in the lateral organization of lipid bilayers on a spatial scale intermediate between the ∼300-nm scale of light microscopy and the nearest neighbor (0.1 nm) scale of Stokes quenching of fluorescent or brominated lipids by spin labels (19,20). Previous measurements of FRET between lipid-anchored fluorescent proteins at the inner leaflet of plasma membranes provided evidence for inhomogeneity on the scale of tens of nanometers (21), illustrating the value of this noninvasive technique for studying membrane domains with dimensions below optical resolution.
Our measurements of FRET between carbocyanine lipid probes in the plasma membrane of live cells provide strong evidence for lateral lipid inhomogeneities that are sensitive to perturbations that enhance or reduce Lo/Ld segregation. These results correlate well with lipid partitioning as measured by detergent resistance. The method we describe provides a novel approach to studying membrane lipid heterogeneity in live cells that is relevant to cell functions.
MATERIALS AND METHODS
Cell labeling with lipid probes
RBL-2H3 cells were maintained in monolayer cultures and harvested with trypsin-EDTA (Life Technologies, Rockville, MD) 3–5 days after passage, as described (22), then resuspended in buffered salt solution (BSS: 135 mM NaCl, 5.0 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, 5.6 mM glucose, 20 mM HEPES, 1 mg/mL BSA) at 5 × 106 cells/mL. Carbocyanine lipid probes or 2-(3-(diphenylhexatrienyl)propanoyl)-1-hexadecanoyl-sn-glycero-3-phosphocholine (DPH-HPC) (Invitrogen, Eugene, OR) in methanol was added to cells at room temperature; cells were mixed immediately and placed on ice for 10 min to facilitate probe incorporation without internalization. Probe concentration varied from 0.2 to 7.0 μM, and solvent was between 0.2% and 1% (v/v). The labeled cells at 4°C were washed twice by centrifugation and resuspended in BSS at 5 × 106 cells/ml. For double labeling with donor and acceptor carbocyanine probes, cells were first labeled with donor, washed, divided, then subsequently labeled or not with acceptor probes to ensure that all the samples have equal amount of donor probe.
The absolute concentrations of donor and acceptor probes incorporated into the plasma membrane were estimated by comparing the fluorescence of cell samples treated with 26 mM octyl glucoside (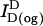 ,
,  , and
, and 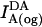 ; see Eqs. 1 and 2) with standard curves. These curves were generated from known amounts of donor and acceptor lipid probes in BSS containing 26 mM octyl glucoside. Approximating the cell surface area as 1000 μm2 (24) and probe area as 4 × 10−7 μm2 /carbocyanine molecule (27), we determined that incubating 5 × 106 cells/ml with 0.2–7.5 μM probes (as described above) yields dye incorporation in the range of 0.02–0.5% (respectively) of the total plasma membrane surface area. This corresponds to a surface density of 103–105 probes/μm2 membrane. As a check on this estimation, we also evaluated the fluorescence of the lipid probes in the supernatant after sedimentation of labeled cells in comparison to a standard curve of lipid probes added to BSS in the absence of cells. Subtracting these values from the total dye incubated yielded consistent results for the amounts of dye incorporated in the plasma membrane.
; see Eqs. 1 and 2) with standard curves. These curves were generated from known amounts of donor and acceptor lipid probes in BSS containing 26 mM octyl glucoside. Approximating the cell surface area as 1000 μm2 (24) and probe area as 4 × 10−7 μm2 /carbocyanine molecule (27), we determined that incubating 5 × 106 cells/ml with 0.2–7.5 μM probes (as described above) yields dye incorporation in the range of 0.02–0.5% (respectively) of the total plasma membrane surface area. This corresponds to a surface density of 103–105 probes/μm2 membrane. As a check on this estimation, we also evaluated the fluorescence of the lipid probes in the supernatant after sedimentation of labeled cells in comparison to a standard curve of lipid probes added to BSS in the absence of cells. Subtracting these values from the total dye incubated yielded consistent results for the amounts of dye incorporated in the plasma membrane.
Liposome preparation
Donor and acceptor carbocyanine lipid probes in methanol were added to chloroform solution of DOPC or DOPC/cholesterol (1:1) (Avanti Polar Lipids, Alabaster, AL), and the mixture was dried under high vacuum at room temperature. The lipid film was redissolved in chloroform, and liposomes were prepared by rapid solvent exchange (23).
Hetero-FRET between lipid probes
FRET between common donor and two different acceptors
Each sample contained 2 ml of a labeled cell suspension at 2 × 106 cells/ml in BSS or a liposome suspension (25 nmol DOPC or 12.5 nmol DOPC + 12.5 nmol cholesterol) in phosphate-buffered saline (PBS). Cells and liposomes were labeled with donors (D) or donors + acceptors (DA), as described above. For FRET measurements, each sample was stirred continuously at 18°C in an acrylic cuvette placed in a thermostatic sample chamber. Donor fluorescence quenching was monitored with excitation and emission wavelengths of 484 nm and 506 nm, respectively. Under these conditions, directly excited emission of the acceptor probes is negligible. For all samples, background fluorescence from unlabeled cells or liposomes and buffers was subtracted. After measurement of cell- or liposome-associated fluorescence, the samples were treated with 26 mM octyl glucoside to solubilize membranes. The intensity of donor fluorescence after octyl glucoside treatment was used as a measure of the absolute amount of donor fluorophore incorporated in D and DA samples. Within a given set, this varied <±5%, primarily because of pipetting variation. Donor fluorescence in D and DA samples were converted to normalized values (FD) by dividing the intensity of each sample before addition of octyl glucoside by the intensity measured after this treatment:
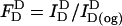 |
(1a) |
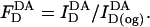 |
(1b) |
A normalized acceptor fluorescence value (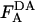 ) was used as a measure of the relative acceptor surface density in the DA samples. For this purpose the acceptor fluorescence (excitation 545 nm, emission 565 nm) after octyl glucoside treatment of each sample,
) was used as a measure of the relative acceptor surface density in the DA samples. For this purpose the acceptor fluorescence (excitation 545 nm, emission 565 nm) after octyl glucoside treatment of each sample,  , was divided by the donor fluorescence (excitation 484 nm, emission 506 nm) of the same sample measured after octyl glucoside treatment,
, was divided by the donor fluorescence (excitation 484 nm, emission 506 nm) of the same sample measured after octyl glucoside treatment, 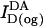 . This conversion corrects for pipetting variation and resulting small differences in cell number in different samples:
. This conversion corrects for pipetting variation and resulting small differences in cell number in different samples:
 |
(1c) |
For experiments with a single donor and two different acceptors (see Figs. 3 and 4), the percentage FRET efficiency was calculated from the steady-state donor emission of each sample:
 |
(2) |
FIGURE 3.

FRET from long-chain, saturated donor DiO-C18 is greater to long-chain, saturated acceptor DiI-C16 (solid circles) than to unsaturated DiI-C18:1Δ9 (open triangles). FRET is measured as donor quenching by different concentrations of acceptors in the plasma membrane of RBL cells at 18°C. For this experiment, cells were labeled with 1 μM DiO-C16 and 0.2–7 μM acceptor probes, and the relative acceptor concentrations (CA) were determined using Eq. 3b. Curves represent the least-squares fits of the experimental data to Eq. 3a to obtain K values. For this set of curves, Q%max= 70.
FIGURE 4.

Long-chain, saturated acceptor DiI-C16 and unsaturated acceptor DiI-C18:1Δ9 show comparable FRET with the long-chain, saturated donor DiO-C18 in single-phase model membranes containing DOPC (A) or DOPC/cholesterol (1:1) (B). Quenching of DiO-C18 fluorescence as a function of acceptors DiI-C16 (solid circles) or DiI-C18:1Δ9 (open triangles) in (A) DOPC and (B) DOPC/cholesterol (1:1) model membranes as fitted by Eq. 3 to obtain K values. For A, Q%max = 78; for B, Q%max = 95.
For experiments with two different donors and a single acceptor (see Fig. 5), it was not possible to normalize in the same way. Aliquots with the same measured number of cells were evaluated, and FRET was calculated according to Eq. 2 except that normalized FD values were replaced by measured ID values.
FIGURE 5.

Long-chain, saturated donor DiO-C16 shows greater FRET than unsaturated donor DiO-C18:2Δ9,12 with long-chain acceptor DiI-C16. Quenching of fluorescence of DiO-C16 (black and light gray bars) and DiO-C18:2Δ9,Δ12 (dark shaded bars) by acceptor DiI-C16. For this experiment, cells were labeled with 0.5–1 μM donor probes. To determine relative concentrations, the donor fluorescence intensity of each sample after octyl glucoside addition ( ) was measured and expressed as a fraction of the highest donor concentration (
) was measured and expressed as a fraction of the highest donor concentration ( ) evaluated. %Q was determined from Eq. 2, except that FD values are replaced by ID values.
) evaluated. %Q was determined from Eq. 2, except that FD values are replaced by ID values.
Fitting of FRET data
For experiments with a single donor and two different acceptors (see Figs. 3 and 4), the FRET data from each set of experiments were separately fitted by a least-squares method to Eq. 3a, where percentage donor quenching, Q% (Eq. 2), is a hyperbolic function of CA, the relative concentration of acceptor in the membrane (Eq. 3b):
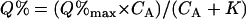 |
(3a) |
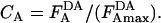 |
(3b) |
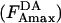 is the highest normalized acceptor intensity measured in a given experiment, in which there is a single donor concentration and multiple acceptor concentrations (represented by
is the highest normalized acceptor intensity measured in a given experiment, in which there is a single donor concentration and multiple acceptor concentrations (represented by 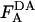 ) for two different acceptors (i.e., the higher
) for two different acceptors (i.e., the higher 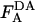 of the two). Note that although a measured CA for each sample cannot exceed the value of 1, extrapolated values can be larger and approach infinity in the limit that the acceptor/donor ratio approaches infinity. The parameter Q%max represents the maximum quenching in this limit. In our standard fitting procedure, we assumed Q%max is the same for both acceptors in a given set of measurements, based on spectral equivalence. However, allowing Q%max to be different resulted in similar values for this parameter and no significant effect on the determined value of the parameter K. The value for K (corresponding to CA at Q% = Q%max/2) represents proximity: a smaller value indicates an average closer distance between donors and acceptors, corresponding to a greater amount of FRET. The K values are relative and can be compared only for a single donor with different (spectrally equivalent) acceptors in the same set of measurements. Ratios of two K values from the same experiment can be compared to ratios of K values from other experiments.
of the two). Note that although a measured CA for each sample cannot exceed the value of 1, extrapolated values can be larger and approach infinity in the limit that the acceptor/donor ratio approaches infinity. The parameter Q%max represents the maximum quenching in this limit. In our standard fitting procedure, we assumed Q%max is the same for both acceptors in a given set of measurements, based on spectral equivalence. However, allowing Q%max to be different resulted in similar values for this parameter and no significant effect on the determined value of the parameter K. The value for K (corresponding to CA at Q% = Q%max/2) represents proximity: a smaller value indicates an average closer distance between donors and acceptors, corresponding to a greater amount of FRET. The K values are relative and can be compared only for a single donor with different (spectrally equivalent) acceptors in the same set of measurements. Ratios of two K values from the same experiment can be compared to ratios of K values from other experiments.
FRET between common acceptor and two different donors
Cells were labeled with either one of the donor probes and washed. Steady-state donor fluorescence was monitored at 18°C as the acceptor probe was added in successive increments. FRET efficiency, measured as donor quenching as a function of acceptor concentration, was quantified according to Eq. 2 except that normalized FD values were replaced by measured ID values as described above.
Sucrose gradient analysis
Sucrose gradient analysis was performed as described previously (25), with a final concentration of 0.04% Triton X-100 (Pierce, Rockford, IL) during lysis of 8 × 106 cells/ml and a 50% sucrose layer in place of the 60% layer. Cells were labeled with various lipid probes as described above to yield probe densities in the range of 104 to 5 × 104 probes/μm2 membrane. Fluorescence emission of isolated fractions was measured in an SLM 8100 fluorescence spectrophotometer (SLM instruments), and background signal caused by buffer alone was subtracted.
Membrane perturbations
Cells were initially labeled with donor, washed, and labeled with varying concentrations of acceptor, as described above. The cells were then subjected to one of the following treatments, and percentage change in FRET efficiency was quantified according to Eq. 4. Before each experiment, the labeled cells were examined in a fluorescence microscope to confirm uniform plasma membrane staining and little or no internalized fluorescence.
 |
(4) |
where steady-state fluorescence values are as follows: ID and 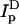 are donor intensities (excitation 484 nm, emission 506 nm) of D-only labeled cells before and after membrane perturbation, respectively; IDA and
are donor intensities (excitation 484 nm, emission 506 nm) of D-only labeled cells before and after membrane perturbation, respectively; IDA and 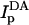 are donor intensities of DA-labeled cells before and after membrane perturbation, respectively.
are donor intensities of DA-labeled cells before and after membrane perturbation, respectively.
Cholesterol depletion with mβCD
Labeled, suspended cells (2–4 × 106 cells/ml) were incubated for 45 min at 4°C in the presence of 10 mM methyl-β-cyclodextrin (mβCD) (Sigma, St. Louis, MO) in BSS, then washed and resuspended in BSS for measurement of 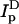 and
and  . For measurement of ID and IDA, cells were incubated in BSS without mβCD under the same conditions. After completion of FRET measurements, mβCD-treated and nonperturbed cells were washed and then incubated with 26 mM octyl glucoside. Comparative measurements of donor and acceptor fluorescence in these two washed samples showed that loss of lipid probes because of cholesterol depletion was <6%.
. For measurement of ID and IDA, cells were incubated in BSS without mβCD under the same conditions. After completion of FRET measurements, mβCD-treated and nonperturbed cells were washed and then incubated with 26 mM octyl glucoside. Comparative measurements of donor and acceptor fluorescence in these two washed samples showed that loss of lipid probes because of cholesterol depletion was <6%.
Addition of filipin III and ceramide
Steady-state fluorescence for D and DA cells was measured before and after addition of perturbants filipin III (Sigma) or C2-ceramide, or dihydro C2-ceramide (Calbiochem, San Diego, CA) in DMSO or ethanol stock solutions. Control experiments showed that the solvents added (<1% v/v total) had negligible effects on the steady-state fluorescence emission.
Large-scale clustering of cell surface molecules
Before labeling with donor and acceptor probes, suspended cells were presensitized by incubation with excess anti-DNP IgE for at least 1 h at 37°C and washed. Labeled, sensitized cells were incubated with 5 μg/ml of OX-7 (mAb specific for Thy-1; PharMingen, San Diego, CA) for 45 min at 4°C and washed, followed by 10 μg/ml of secondary anti-mouse-IgG (Invitrogen, Eugene, OR) at 4°C for 1 h. Alternatively, labeled cells were incubated with 5 μg/ml of biotinylated cholera toxin B subunit (Invitrogen) for 45 min at 4°C and washed, followed by 10 μg/ml of streptavidin for 1 h at 4°C. Steady-state fluorescence was measured for D- and DA-labeled cells before and after the perturbation of cross-linking (Eq. 4).
Homo-FRET
Cell suspensions were labeled with varying concentrations of DiI-C16 and incubated with either mβCD or buffer under identical conditions. The cells were washed and resuspended in BSS. For each experiment, 2 ml of cell suspension (2 × 106 cells/ml) in BSS was stirred continuously in an acrylic cuvette placed in a thermostatic sample chamber. Steady-state fluorescence anisotropy (r) of cholesterol-depleted cells and control cells was measured according to Eq. 5, as described previously (26):
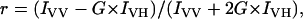 |
(5) |
where IVV, IVH, IHV, and IHH correspond to fluorescence intensities measured with excitation and emission polarizers, respectively, oriented in vertical (V) or horizontal (H) positions; G is the monochromator grating correction factor given by G = IHV/IHH. For all samples, background fluorescence caused by unlabeled cells and buffer was subtracted.
In other experiments, cells were first labeled with DiI-C16, and steady state anisotropy was measured before and after 2,4,6-trinitrobenzene sulfonate (TNBS), C6-ceramide, C2-ceramide, or dihydro C2-ceramide was added in successive increments from stock solutions in ethanol or DMSO. Control experiments showed that the solvents added (<1% v/v total) had negligible effects on anisotropy values.
To normalize concentrations in the homo-FRET experiments, the fluorescence intensity of each sample after addition of octyl glucoside, I(og), was determined. This value was used as a measure of probe surface density and expressed as a fraction of the surface density at the maximal concentration tested ([I(og)]max). Absolute densities of the incorporated probes were estimated by comparison to a standard curve as described above.
RESULTS
Cell surface FRET
To investigate plasma membrane lipid heterogeneity in live cells, we introduced fluorescent carbocyanine lipid analogs into the outer leaflet of RBL cell plasma membranes using a labeling method previously described (27). For FRET experiments, DiO carbocyanine lipid probes served as donors, and DiI carbocyanine lipid probes served as acceptors. Localization of DiO-C16 to the plasma membrane is illustrated in Fig. 1. Carbocyanine probes are particularly advantageous for this study because they remain localized to the plasma membrane at 18°C for extended periods of time and because the fluorophore moieties are structurally homologous to each other. Thus, these FRET donors and acceptors with variable alkyl chains have the potential to undergo differential partitioning into Lo/Ld environments and are spectroscopically similar, allowing direct comparisons.
FIGURE 1.

Confocal micrograph showing plasma membrane labeling of RBL cells by donor DiO-C16 in RBL cells. Average diameter of cells is 10 μm.
In one type of experiment, suspended cells were labeled with the donor probe DiO-C16 and then divided into two aliquots, one of which was then labeled with acceptor probe DiI-C16. As shown in Fig. 2 A, the emission spectrum (excitation 480 nm) of cells labeled with DiO-C16 alone shows a peak at 506 nm, and that for cells labeled with DiO-C16 and DiI-C16 shows decreased fluorescence in the region of the DiO-C16 peak emission and a second peak with a maximum emission at 565 nm. A parallel cell sample containing the same amount of DiI-C16 in the absence of DiO-C16 shows no significant fluorescence at 506 nm and a peak with much less fluorescence emission at 565 nm, indicating that most of the increased fluorescence intensity in the 565 nm peak when DiO-C16 is present is sensitized emission caused by FRET. To evaluate whether donor quenching and sensitized acceptor emission result from proximity of these probes in the plasma membrane, the same cell samples were solubilized in 26 mM octyl glucoside, and emission spectra were again compared. Under these conditions, the spectrum of the sample containing both donor and acceptor is the simple sum of the spectra of samples with donor only or acceptor only (Fig. 2 B), as expected for the absence of FRET.
FIGURE 2.

Spectroscopic evidence for FRET between carbocyanine probes DiO-C16 and DiI-C16 when incorporated into the plasma membrane of live RBL cells. Emission spectra of cells labeled with both donor DiO-C16 and acceptor DiI-C16 (circles), acceptor DiI-C16 alone (triangles), and donor DiO-C16 alone (squares) before (A) and after (B) treatment with 26 mM octyl glucoside. Samples were excited at 480 nm, and fluorescence intensities in A and B are separately normalized to the peak of the donor-only spectra.
We monitored FRET by donor quenching (excitation 480 nm, emission 506 nm), as this provides a more direct, quantitative measurement of transfer efficiency. The relative concentration of the acceptor probe was assessed for each sample at the end of the measurement by adding octyl glucoside to solubilize cellular membranes and measuring the fluorescence intensity of the directly excited acceptor (excitation 545, emission 565 nm; Eq. 1b). Fig. 3 shows an example of an experiment of this design, in which FRET is measured in matched samples between donor DiO-C18 and either DiI-C16 or DiI-C18:1Δ9, which contains a single double bond in each alkyl chain at position 9. In a direct comparison, increasing concentrations of DiI-C16 cause a larger amount of donor quenching (i.e., higher efficiency of energy transfer, Eq. 2) than for equal concentrations of DiI-C18:1Δ9. Assuming the two acceptor probes are spectroscopically identical, these results indicate that the acceptor with saturated alkyl chains has a greater probability of being close to DiO-C18, the donor with saturated alkyl chains, than does the acceptor with unsaturated alkyl chains.
To quantify the differences observed in these FRET experiments, we used a simple model where percentage donor quenching, Q%, is a hyperbolic function of CA, the relative membrane concentration of acceptor (Eq. 3). The parameter Q%max is a hypothetical value corresponding to the limit of infinite acceptor concentration. The parameter K provides information about the tendency of the donors and acceptors to occupy the same region of the membrane at low probe concentrations. K corresponds to the value of CA at which there is half-maximal quenching (Q%max/2), with a smaller value of K indicating closer proximity between donors and acceptors. As shown by the representative experiment in Fig. 3, Eq. 3 provides good fits to the data, and the ratio of K values for DiO-C18/DiI-C18:1Δ9 compared to DiO-C18/DiI-C16 is 2.1, indicating significantly closer proximity of the latter donor/acceptor pair. In multiple experiments (n = 4) with this set of probes, the average K ratio is 2.3 ± 0.3. Similar evaluations were made in experiments with a single donor and two acceptors with different tendencies to occupy Lo/Ld regions. We found that the K ratio for DiO-C18/DiI-C12 compared to DiO-C18/DiI-C16 is 2.0 ± 0.2 (n = 4); the K ratio for DiO-C16/DiI-C18:1Δ9 compared to DiO-C16/DiI-C16 is 2.1 ± 0.3 (n = 3); and the K ratio for DiO-C16/DiI-C12 compared to DiO-C16/DiI-C16 is 1.8 ± 0.2 (n = 3). In all of these comparisons, DiI acceptor probes with longer or more saturated alkyl chains exhibit greater proximity to DiO donor probes with saturated alkyl chains than do DiI acceptor probes with shorter or unsaturated alkyl chains.
To test whether spectroscopic differences between the DiI analogs or nonideal mixing of the carbocyanine probes could contribute to the difference in FRET observed in plasma membrane of cells, we carried out similar FRET experiments in homogeneous liposomes (23) containing either pure DOPC or 1:1 DOPC:cholesterol. According to published phase diagrams, both DOPC and DOPC/cholesterol liposomes are in a single phase (28,29), but the potential exists for suboptical nonideal mixing of the fluorescent lipid probes. Fig. 4 shows that, when evaluated with the common donor DiO-C18, equimolar concentrations of the acceptor DiI-C16 with long, saturated alkyl chains and the acceptor DiI-C18:1Δ9 with unsaturated alkyl chains undergo comparable FRET yielding similar K values (K ratio ∼1) in both DOPC (Fig. 4 A) and DOPC/cholesterol (Fig. 4 B) liposomes. These results support our assumption that the DiI analogs used are spectroscopically identical and have similar proximity to a common donor, DiO-C18, in these single-phase systems. Thus, the difference in FRET between the DiI analogs in live cells can be attributed to the presence of nanoscopic heterogeneities.
In a complementary set of experiments, cells were first labeled with either DiO-C16 or with DiO-C18:2Δ9,12 as donor probes. DiI-C16, the common acceptor, was titrated into the donor-labeled cells as donor fluorescence quenching was monitored. Samples contained the same measured number of cells, and the relative concentration of each donor probe incorporated was determined by measuring fluorescence intensity after solubilization of an aliquot with octyl glucoside. Two samples of DiO-C16-labeled cells were prepared such that their respective concentrations bracketed the concentration of DiO-C18:2Δ9,12 in the third sample. Fig. 5 shows a representative experiment in which these three samples were titrated with DiI-C16. These data show that FRET is greater for acceptor DiI-C16 when the donor contains long, saturated alkyl chains (DiO-C16) than when the donor contains unsaturated alkyl chains (DiO-C18:2Δ9,12). The results from experiments of this type support those described above for comparisons between two acceptors and a single donor. These quantitative differences observed in FRET experiments provide a consistent picture of a heterogeneous distribution of lipids in the plasma membrane. More FRET between lipids with similar alkyl chains implies that the alkyl chain structure plays a significant role in the relative partitioning of these lipid probes in the plasma membrane of living cells.
Sucrose gradient analysis
To compare FRET results between lipid probes on live cells to a common biochemical analysis of lipid raft association, we carried out sucrose gradient fractionation of Triton X-100 (0.04%)-lysed cells that had been labeled with carbocyanine lipid probes. Fig. 6 A shows representative gradient distributions of several DiO and DiI lipid probes used in our FRET measurements. We also compared the phospholipid analog DPH-HPC, which we showed previously to partition nearly equally between Lo and Ld regions of model membranes (26). Within the gradients, fractions 2–7 contain components associated with detergent-resistant Lo regions of the membrane. Solubilized proteins and lipids associated with Ld regions of membranes are found in fractions 10–16 (8,30). In this analysis, carbocyanine lipid probes with longer, saturated alkyl chains (C16, C18) are more abundant in the Lo fractions, whereas probes with a shorter alkyl chain (C12) or unsaturated alkyl chains (C18:1Δ9, C18:2Δ9,12) preferentially fractionate to the Ld region of the gradients. DPH-PC shows a fractionation pattern that is very similar to that for DiO-C18. DPH-PC also shows considerable partitioning into Lo phase in the presence of coexisting fluid phases in giant unilamellar vesicles (T. Baumgart and G. Feigenson, unpublished results) as detected by fluorescence imaging, consistent with other evidence that DPH-PC has significant affinity for ordered phases.
FIGURE 6.

Distributions of the carbocyanine lipid probes from labeled cells after sucrose gradient fractionation of TX-100-lysed cells are consistent with FRET results. (A) Percentage of DiO-C18 (solid circles), DiO-C18:2Δ9,12 (open triangles), DiI-C16 (solid squares), DiI-C18:1Δ9 (open diamonds), DiI-C12 (solid triangles), and DPH-HPC (open hexagons) in different fractions from a representative experiment. (B) Percentage of DiO-C16 and DiI-C16 before (filled circles and squares) and after (open triangles and diamonds) treatment with 10 mM methyl β-cyclodextrin in different fractions. Fractions 2–7 (∼10–20% sucrose) represent the DRMs, whereas fractions 10–16 (∼40% sucrose) contain the solubilized proteins and lipids. Cells were labeled with 1–3 μM carbocyanine probes or 5 μM DPH-HPC.
To evaluate the carbocyanine dye distributions further, labeled cells were depleted of cholesterol using methyl β-cyclodextrin (mβCD), and sucrose gradient fractionation of TX-100-lysed cells was carried out. As shown in Fig. 6 B, both DiO-C16 and DiI-C16 exhibit a lower ratio of Lo-associated probe to Ld probe after cholesterol depletion. Taken together with previous studies on detergent resistance of cellular phospholipids (31) and model membranes (32), these sucrose gradient data support the view from our FRET measurements that carbocyanine lipid analogs with longer, saturated alkyl chains have a greater preference for Lo membrane domains than carbocyanine probes with unsaturated or shorter alkyl chains, which prefer Ld regions.
Changes in FRET caused by membrane perturbations
To characterize further the relationship of FRET between carbocyanine lipid probes to Lo regions in live cells, we monitored the quenching of donor DiO-C16 by acceptor DiI-C16 under different conditions of plasma membrane perturbation. As calculated with Eq. 4 and shown in Fig. 7 A, cells labeled with DiO-C16 and DiI-C16 exhibited an average decrease of 47% for FRET between these lipid probes after depletion of cholesterol with mβCD. Addition of 10 μM filipin III to sequester cholesterol (33,34) caused a decrease of 22%. Short-chain ceramides also have been shown to disrupt the Lo phase of model membranes and decrease the lipid order of plasma membrane vesicles (35,36). Because of its allylic trans double bond, C2-ceramide has a more rigid and bulkier headgroup than dihydro C2-ceramide, with two rigid planes of H-bonded atoms, and the resulting cone-shaped lipid can disrupt the packing of saturated lipids and cholesterol in ordered bilayers. As shown in Fig. 7 A, the FRET efficiency between DiO-C16 and DiI-C16 in the plasma membrane of live cells was found to be reduced by 16% when 32 μM C2-ceramide was added. Dihydro C2-ceramide, which does not affect lipid order (35), caused <5% inhibition of FRET at the same concentration. These results with a variety of perturbants are consistent with the view that enhanced FRET between DiO-C16 and DiI-C16 is dependent on Lo structure in the plasma membrane of live RBL cells.
FIGURE 7.

FRET between DiO-C16 and DiI-C16 in the plasma membrane of live cells is sensitive to membrane perturbations that reduce the Lo membrane structure. Percentage decrease in FRET efficiency (Eq. 4) after treatment with 10 mM methyl-β-cyclodextrin (solid bar), 10 μm filipin III (light gray bar), 32 μM C2 ceramide (dark shaded bar), or 32 μM C2-dihydro ceramide (off-white bar). For these experiments, cells were labeled with 1 μM DiO-C16 and 2.5-3.5 μM DiI-C16.
We examined the sensitivity of FRET between these carbocyanine probes to cross-linking of cell surface proteins. Previously, we showed that cross-linking of IgE receptor complexes and the GPI-linked protein Thy-1 at 4°C induces large-scale coclustering of characteristic lipid raft components on the surface of RBL cells (37). Under conditions of simultaneous cross-linking of these proteins, we find that the FRET efficiency between DiO-C16 and DiI-C16 increases by 35%. Cross-linking of GM1 by biotinylated cholera toxin, followed by streptavidin, causes an increase in FRET of 10% between these carbocyanine lipid probes. These results indicate that large-scale plasma membrane segregation caused by cross-linking of cell surface proteins can increase proximity of these carbocyanine lipid probes in the plasma membrane. From these results, we conclude that FRET between these carbocyanine lipid probes is sensitive to optically detectable heterogeneity in cells caused by large-scale clustering (micrometer) as well as perturbations that are optically undetectable (tens of nanometers).
Homo-FRET
FRET measurements between DiO and DiI lipid analogs require sequential labeling of cells and multiple samples to provide adequate controls for accurate quantification and interpretation. An alternative method for FRET-based detection of lipid probe proximity utilizes fluorescence depolarization of a single type of probe acting as both donor and acceptor, commonly called homo-FRET (13). In these measurements, FRET between probes results in decreased fluorescence anisotropy (Eq. 5). Carbocyanine lipid probes have relatively short lifetimes of ∼1 ns (38), and their fluorescence is highly polarized (39), making them suitable candidates for homo-FRET measurements. Furthermore, their small Stokes shift and overlap between absorption and emission spectra result in efficient homo-FRET over nanometer distances. Thus, we investigated the self-proximity of carbocyanine lipid probes with saturated acyl chains in live cell membranes by measuring the extent of homo-FRET between DiI-C16 molecules.
As shown in Fig. 8 A, we found that steady-state anisotropy of DiI-C16 in the plasma membrane decreases monotonically with increased surface density of this probe, consistent with a FRET-dependent mechanism. DiI-C16 anisotropy is significantly increased by cholesterol depletion (Fig. 8 A), indicating a decrease in probe proximity under these conditions. These results are consistent with results from hetero-FRET measurements with DiO-C16 donors and DiI-C16 acceptors (mβCD effect in Fig. 7). We found that cholesterol depletion does not alter significantly the concentration of cell-associated DiI-C16, as tested by washing the cells before and after mβCD treatment and measuring fluorescence in the presence of octyl glucoside. As shown in Fig. 8 B, collisional quenching of DiI-C16 by membrane-impermeant TNBS (17) causes increased fluorescence anisotropy that is more sensitive to TNBS at higher concentrations of the lipid probe for two different densities of DiI-C16. In this experiment, collisional quenching with TNBS decreases the effective density of fluorescent DiI-C16 at the cell surface, and the observed increase in anisotropy is consistent with an expected decrease in homo-FRET. This increase in anisotropy is more pronounced at higher concentrations of DiI-C16, where more FRET occurs in the absence of a quenching reagent.
FIGURE 8.

Steady-state anisotropy of DiI-C16 incorporated in the plasma membrane of RBL cells is used as a measure of homo-FRET between the lipid probes. Results are averages of replicate samples with standard deviations from a single representative experiment. (A) Concentration-dependent homo-FRET detected by steady-state anisotropy in the plasma membrane of live cells in the absence (solid circles) and presence of (open triangles) methyl-β-cyclodextrin. For these experiments, cells were labeled with 1–6 μM of DiI-C16, and the DiI-C16 concentration is expressed as a fraction of the highest DiI-C16 concentration evaluated. (B) Homo-FRET between DiI-C16 molecules is substantially reduced on treatment with the cell-impermeable quencher TNBS. Cells were labeled with 1.5 μM (sample 1) or 0.5 μM (sample 2) DiI-C16. The amount of DiI-C16 incorporated in the plasma membrane in sample 1 was 2.4 times more than that in sample 2 as evaluated after treatment with octyl glucoside (I(og)). (C) C2-ceramides reduce homo-FRET of DiI-C16-labeled cells (solid circles), whereas C2-dihydroceramide (open triangles) does not. For this experiment, cells were labeled with 6.5 μM DiI-C16.
Similar to results from hetero-FRET measurements, we find that C2-ceramide increases the steady-state anisotropy of DiI-C16 (corresponding to decreased homo-FRET), and dihydro C2-ceramide has no significant effect (Fig. 8 C). These results indicate that disruption of Lo membrane structure by short-chain C2-ceramide (or by cholesterol depletion, Fig. 8 A) decreases the effective probe proximity in the plasma membrane. Thus, the homo-FRET results support the view that segregation of ordered and disordered regions contributes to lateral partitioning of DiI-C16 molecules in the plasma membrane of live cells.
DISCUSSION
Ordered regions of the plasma membranes, often called lipid rafts, are widely recognized to be important in cellular function. Rafts have been notoriously difficult to characterize because they are not distinctive by optical microscopy, and relating these diverse, dynamic structures to biochemical preparations from lysed cells is problematic. We utilized FRET between carbocyanine probes in several different types of experiments to evaluate the relevant lateral organization of lipids in the heterogeneous plasma membrane of live cells. We observed higher FRET efficiency between carbocyanine probes with similar alkyl chain structure, indicating differential partitioning of lipid molecules into segregated plasma membrane domains that correlates with preferential Lo/Ld partitioning (Figs. 3 and 5). The capacity of FRET measurements to discriminate between laterally separated molecules is predicted to be greatest when the dimensions of the separation are one to four times the Ro for a particular donor/acceptor pair (40,41). The Ro for FRET between DiO and DiI carbocyanine probes is ∼5–6 nm (42), making them sensitive to lateral segregation with dimensions of tens of nanometers as has been implicated for lipid rafts by a variety of studies with different experimental approaches (10–15). Our results provide strong evidence that although these carbocyanine derivatives appear homogeneous in the plasma membrane at optical resolution, nanoscopic lateral segregation of these probes occurs, even in unstimulated cells.
With fluorescence microscopy, we recently visualized micrometer-scale Lo/Ld phase separation in giant plasma membrane vesicles that pinch off from RBL cells after cytoskeletal detachment. In these vesicles, endogenous proteins and lipids exhibit preferential Lo/Ld partitioning as can be distinguished by selective probes (43). Consistent with previous studies of detergent-resistant membranes, these results demonstrate that compositionally complex biological membranes have the intrinsic capacity to segregate into Lo/Ld phases. Thus, the submicroscopic heterogeneities detected by our FRET measurements in live cells may represent nanoscopic phase separation or Lo/Ld-based concentration fluctuations in the plasma membrane that are prevented from assuming optically distinctive dimensions in live cells by the underlying cellular cytoskeleton and associated proteins. The temporal resolution of FRET depends on the excited state lifetime of the fluorophores, and, as the carbocyanine probes have lifetimes in the order of nanoseconds (38), it is not yet known whether the heterogeneities detected are transient or stable on longer time scales.
To address whether addition of exogenous lipid probes to the cell membrane causes confounding perturbations, we compared FRET measurements using single donor DiO-C18 on cells and simple liposomes. For cells, FRET to acceptors DiI-C16 and DiI-C18:1Δ9 is distinctive (Fig. 3), whereas for liposomes, the FRET is the same (Fig. 4). These results support our conclusion that heterogeneities detected in cell plasma membranes are not caused by nonideal interactions of probe molecules. We can calculate the surface density of dyes incorporated into the plasma membrane by comparing the fluorescence of washed cells after octyl glucoside treatment with a standard curve. Initial incubations of cells (2 × 106/ml) with 0.2- to 7-μM probes resulted in incorporation of 103–105 dyes/μm2, corresponding 0.02–0.5% of the total lipids of the plasma membrane (based on surface area). Although we cannot rule out some perturbing effects of the probes, we believe these are not substantial because of our use of low concentrations and the liposome results.
RBL-2H3 cells are known to laterally segregate carbocyanine lipid probes into optically visible, micrometer-size domains as a result of IgE-FcɛRI cross-linking at the cell surface or during phagocytosis of micrometer-sized, antigen-coated beads (22,27). Induced clustering at 4°C of a minor cell surface component such as FcɛRI, which comprises ∼1–2% of cell surface proteins, causes dramatic and selective coredistribution of DiI-C16 and other lipid probes present at much higher concentrations (27). Similarly, RBL cells effectively segregate several different membrane proteins and lipids in the plasma membrane during phagocytosis, such that, for example, saturated DiI-C16, but not unsaturated DiI-C18:2Δ9,12, is substantially excluded from the forming phagosomes (22). These results are consistent with the hypothesis that lipid probes such as DiI-C16 preferentially partition into submicrometer membrane domains in the plasma membrane of resting cells and are then gathered together into large, stable domains during cross-linking-induced stimulation or endocytosis. Mukherjee et al. found that DiI derivatives differing solely in the composition of their alkyl chains show differential trafficking after internalization from the cell surface into sorting endosomes (44). The DiI analogs with long and saturated tails preferentially enter the late endocytic pathway, whereas those with shorter or unsaturated chains are recycled via the endocytic recycling compartment, indicating that sorting of lipids is driven in part by their alkyl chain structure.
Perturbation of FRET between DiO-C16 and DiI-C16 by reagents that are known to affect cholesterol content or in other ways disrupt Lo structure provides further evidence for lateral segregation of ordered and disordered regions in live cell membranes. As described above, FRET-detected proximity of probes that have long, saturated alkyl chains provides strong evidence for their preferential partitioning (Figs. 3 and 5). The disruption of the lateral heterogeneity by agents such as mβCD and short-chain ceramides can cause local dilution of the probe molecules and consequently a decrease in FRET efficiency (Fig. 7). The observed increase in FRET between carbocyanine probes with saturated alkyl chains that occurs after cross-linking of lipid raft-associated FcɛRI and GPI-linked Thy-1 (35%, see Results) indicates that such redistributions are accompanied by changes in the spatial distributions of membrane lipids. Thus, these carbocyanine donor/acceptor pairs can be utilized as sensitive probes for detecting such induced membrane reorganization.
Because sucrose gradient analysis of detergent-extracted cells is often used to distinguish ordered (Lo) from disordered (Ld) regions of the plasma membrane, we compared this rough measure of lipid heterogeneity to our FRET measurements in live cells (Fig. 6). Our low-detergent condition for cell lysis (0.04% Triton X-100 for 8 × 106 cells/ml) was shown in previous studies to separate detergent-resistant membranes (Lo) such that the association of cross-linked and of monomeric IgE-FcɛRI are distinguished (25). We find that differential partitioning in the detergent-resistant membrane fractions of the carbocyanine probes based on their alkyl chain structure is consistent with differential partitioning of endogenous lipids (31) and is also consistent with differential partitioning into Lo versus Ld domains in the plasma membranes of live cells as observed with our FRET measurements. Selective partitioning of carbocyanine probes that differ only in their alkyl chain structures strongly supports the view that lipids segregate in the two-dimensional plane of the cell membrane depending on the capacity of their alkyl/acyl chains to pack together into cholesterol-dependent ordered regions. The decrease in association of the long-chain, saturated carbocyanine lipids with detergent-resistant membranes on cholesterol depletion (Fig. 6 B) is also consistent with the FRET measurements of membrane heterogeneity (Fig. 7). The lipid probe DPH-HPC shows approximately the same partitioning as DiO-C18, the carbocyanine probe that associates most preferentially with the DRMs. We showed previously with anisotropy measurements that DPH-HPC partitions roughly equally between Lo and Ld regions in model membranes (26), but it is possible that this partition coefficient is altered in the more complex plasma membrane milieu. Nevertheless, the presence of a large fraction of this probe and the long-chain saturated carbocyanine probes in the detergent-resistant membranes (up to 60–65%) is consistent with previous findings that a substantial fraction of the plasma membrane is ordered (26). Partition coefficients of the carbocyanine probes for Lo/Ld region of plasma membranes are also expected to affect their distributions, but these values are not known.
In a recent study, fluorescence recovery after photobleaching of various membrane proteins at the apical surface of epithelial cells indicated that Lo is the percolating phase, occupying the major fraction of the plasma membrane in this region at 25°C; at 37°C, Ld becomes the percolating phase in this region (45). Recent electron spin resonance measurements in live RBL cells measured over temperatures of 5–37°C (16) are also consistent with the view that a large fraction of the plasma membrane is in an ordered state. These results challenge a common assumption that lipid rafts represent a small fraction of the membrane surface and serve to concentrate associating signaling components. However, current results are consistent with the view that Lo/Ld segregation participates in separating key signaling proteins from each other, regardless of the relative amount of each type of domain (6). For example, we showed recently that segregation of a transmembrane phosphatase in disordered regions of the membrane from the lipid-anchored Lyn kinase in ordered regions of membrane plays an important role in regulating the initial signaling steps triggered by cross-linking IgE-FcɛRI (46).
Homo-FRET between carbocyanine lipid probes provides a complementary approach to evaluate probe clustering based on lateral heterogeneity in live cell membranes (Fig. 8). These experiments require labeling the cells with only a single fluorophore that acts as both donor and acceptor. Homo-FRET is more sensitive to the presence of small clusters and lateral segregations than hetero-FRET because of the high probability of transfer back and forth between the same fluorophore as donor and acceptor. Such back-transfer can occur multiple times and over many nearest-neighbor distances, further reducing the measured anisotropy and increasing the sensitivity. The highly efficient homo-FRET observed with carbocyanine probes such as DiI-C16 in live cell membranes makes them particularly useful for investigating changes in lateral heterogeneity with this approach.
We found our homo-FRET measurements to be sensitive to Lo perturbing agents such as mβCD and short-chain ceramides (Fig. 8, A and C), wholly consistent with results from hetero-FRET experiments (Fig. 7). Regarding the homo-FRET anisotropy experiments, we note that these perturbants could cause changes in membrane viscosity, which could in turn affect the rotational correlation times of probes located there. However, changes in membrane viscosity are not expected to significantly affect the measured anisotropy of carbocyanine probes, which have their transition dipole moment parallel to the plane of the membrane (39), because rotation of these probes around their principal axis, perpendicular to the plane of the membrane, is not expected to be different for a wide range of Lo versus Ld environments. In contrast, our previous anisotropy measurements in model membranes showed that DPH-HPC, with a transition dipole moment along the length of its acyl chains, is very sensitive to ordering caused by cholesterol content (26). Our current results are consistent with these: cholesterol depletion causes a decrease in the homo-FRET of DiI-C16, corresponding to an increase in average distance between the probe molecules. The caveat remains that potential multiple effects of cholesterol depletion and other perturbations on the plasma membrane (and incorporated probes) are not completely known and could cause additional changes in DiI-C16 anisotropy.
Decreasing the concentration of fluorescent probe molecules in the excited state by the chemical quencher TNBS causes attenuation of homo-FRET and consequently increased anisotropy (Fig. 8 B). These results provide additional evidence that DiI-C16 molecules preferentially partition into ordered regions in the cell membrane, such that efficient homo-FRET between the DiI-C16 molecules decreases the steady-state anisotropy. The disruption of the lateral segregation of Lo/Ld phases by cholesterol depletion causes decreased homo-FRET efficiency and recovery of fluorescence anisotropy as a result of increased average distance between the probe molecules (Fig. 8 A). Thus, our homo-FRET and hetero-FRET results consistently support the hypothesis of heterogeneous distribution of lipid molecules based on Lo/Ld phase separation in live cell membranes.
In summary, carbocyanine probes and FRET measurements are shown to be valuable for investigating lateral distributions of lipids in the plasma membrane of live cells. We obtained strong evidence for nanoscopic, laterally segregated, ordered and disordered regions of this membrane. The observed lateral heterogeneity is sensitive to the cholesterol content of the cell membrane as well as to other agents that disrupt the Lo phase such as short-chain ceramides. FRET between the carbocyanine probes is also sensitive to cross-linking of cell surface proteins and thus can be used to probe membrane redistributions in real time that may accompany various signaling processes.
Acknowledgments
We thank Drs. G. Feigenson and T. Baumgart for helpful discussions and F. Heberle for assistance with liposome preparations.
This work was supported by National Institutes of Health grant AI18306 and in part by National Science Foundation-Nanoscale Interdisciplinary Research Team grant DMR-0404195.
References
- 1.Simons, K., and D. Toomre. 2000. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 1:31–39. [DOI] [PubMed] [Google Scholar]
- 2.Edidin, M. 2003. The state of lipid rafts: from model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 32:257–283. [DOI] [PubMed] [Google Scholar]
- 3.Munro, S. 2003. Lipid rafts: elusive or illusive? Cell. 115:377–388. [DOI] [PubMed] [Google Scholar]
- 4.Brown, D. A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- 5.Brown, D. A., and J. K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 68:533–544. [DOI] [PubMed] [Google Scholar]
- 6.Holowka, D., J. A. Gosse, A. T. Hammond, X. Han, P. Sengupta, N. L. Smith, A. Wagenknecht-Wiesner, M. Wu, R. M. Young, and B. Baird. 2005. Lipid segregation and IgE receptor signaling: a decade of progress. Biochim. Biophys. Acta. 1746:252–259. [DOI] [PubMed] [Google Scholar]
- 7.Harder, T., P. Scheiffele, P. Verkade, and K. Simons. 1998. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 141:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheets, E. D., D. Holowka, and B. Baird. 1999. Critical role for cholesterol in Lyn-mediated tyrosine phosphorylation of FcepsilonRI and their association with detergent-resistant membranes. J. Cell Biol. 145:877–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Janes, P. W., S. C. Ley, and A. I. Magee. 1999. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol. 147:447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pralle, A., P. Keller, E. L. Florin, K. Simons, and J. K. Horber. 2000. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells J. Cell Biol. 148:997–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subczynski, W. K., and A. Kusumi. 2003. Dynamics of raft molecules in the cell and artificial membranes: approaches by pulse EPR spin labeling and single molecule optical microscopy. Biochim. Biophys. Acta. 1610:231–243. [DOI] [PubMed] [Google Scholar]
- 12.Kusumi, A., C. Nakada, K. Ritchie, K. Murase, K. Suzuki, H. Murakoshi, R. S. Kasai, J. Kondo, and T. Fujiwara. 2005. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 34:351–378. [DOI] [PubMed] [Google Scholar]
- 13.Sharma, P., R. Verma, R. C. Sarasji, Ira, K. Gousset, G. Krishnamoorthy, M. Rao, and S. Mayor. 2004. Nanoscale organization of multiple GPI-anchored proteins in living cell membranes. Cell. 116:577–589. [DOI] [PubMed] [Google Scholar]
- 14.Prior, I. A., C. Muncke, R. G. Parton, and J. F. Hancock. 2003. Direct visualization of Ras proteins in spatially distinct cell surface microdomains. J. Cell Biol. 160:165–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilson, B. S., S. L. Steinberg, K. Liederman, J. R. Pfeiffer, Z. Surviladze, J. Zhang, L. E. Samelson, L. Yang, J. M. Kotula, and J. M. Oliver. 2004. Markers for detergent-resistant lipid rafts occupy distinct and dynamic domains in native membranes. Mol. Biol. Cell. 15:2580–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Swamy, M. J., L. Ciani, M. Ge, D. Holowka, B. Baird, and J. H. Freed. 2006. Coexisting domains in the plasma membranes of live cells characterized by spin-label ESR spectroscopy. Biophys. J. 90:4452–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolf, D. E. 1985. Determination of the sidedness of carbocyanine dye labeling of membranes. Biochemistry. 24:582–586. [DOI] [PubMed] [Google Scholar]
- 18.Castanho, M. A. R. B., and S. Lopes. 2004. Does aliphatic chain length influence carbocyanines' orientation in supported lipid multilayers? J. Fluoresc. 14:281–287. [DOI] [PubMed] [Google Scholar]
- 19.London, E., and G. W. Feigenson. 1981. Fluorescence quenching in model membranes. 1. Characterization of quenching caused by a spin-labeled phospholipid. Biochemistry. 20:1932–1938. [DOI] [PubMed] [Google Scholar]
- 20.Silvius, J. R., D. del Giudice, and M. Lafleur. 1996. Cholesterol at different bilayer concentrations can promote or antagonize lateral segregation of phospholipids of differing acyl chain length. Biochemistry. 35:15198–15208. [DOI] [PubMed] [Google Scholar]
- 21.Zacharias, D. A., J. D. Violin, A. C. Newton, and R. Y. Tsien. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 296:913–916. [DOI] [PubMed] [Google Scholar]
- 22.Pierini, L., D. A. Holowka, and B. Baird. 1996. Fc epsilon RI-mediated association of 6-micron beads with RBL-2H3 mast cells results in exclusion of signaling proteins from the forming phagosome and abrogation of normal downstream signaling. J. Cell Biol. 134:1427–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Buboltz, J. T., and G. W. Feigenson. 2001. A novel strategy for the preparation of liposomes: rapid solvent exchange. Biochim. Biophys. Acta. 1417:232–245. [DOI] [PubMed] [Google Scholar]
- 24.Metzger, H. 1978. The IgE-mast cell system as aparadigm for the study of antibody mechanisms. Immunol. Rev. 41:186–199. [DOI] [PubMed] [Google Scholar]
- 25.Field, K. A., D. Holowka, and B. Baird. 1999. Structural aspects of the association of FcepsilonRI with detergent-resistant membranes. J. Biol. Chem. 274:1753–1758. [DOI] [PubMed] [Google Scholar]
- 26.Gidwani, A., D. Holowka, and B. Baird. 2001. Fluorescence anisotropy measurements of lipid order in plasma membranes and lipid rafts from RBL-2H3 mast cells. Biochemistry. 40:12422–12429. [DOI] [PubMed] [Google Scholar]
- 27.Thomas, J. L., D. Holowka, B. Baird, and W. W. Webb. 1994. Large-scale co-aggregation of fluorescent lipid probes with cell surface proteins. J. Cell Bio. 125:795–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Veatch, S. L., and S. L. Keller. 2003. Separation of liquid phases in giant vesicles of ternary mixtures of phospholipids and cholesterol. Biophys. J. 85:3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lentz, B. R., D. A. Barrow, and M. Hoechli. 1980. Cholesterol-phosphatidylcholine interactions in multilamellar vesicles. Bochemistry. 19:1943–1954. [DOI] [PubMed] [Google Scholar]
- 30.Field, K. A., D. Holowka, and B. Baird. 1997. Compartmentalized activation of the high affinity immunoglobulin E receptor within membrane domains. J. Biol. Chem. 272:4276–4280. [DOI] [PubMed] [Google Scholar]
- 31.Fridriksson, E. K., P. A. Shipkova, E. D. Sheets, D. Holowka, B. Baird, and F. W. McLafferty. 1999. Quantitative analysis of phospholipids in functionally important membrane domains from RBL-2H3 mast cells using tandem high-resolution mass spectrometry. Biochemistry. 38:8056–8063. [DOI] [PubMed] [Google Scholar]
- 32.Schroeder, R., E. London, and D. Brown. 1994. Interactions between saturated acyl chains confer detergent resistance on lipids and glycosylphosphatidylinositol (GPI)-anchored proteins: GPI-anchored proteins in liposomes and cells show similar behavior. Proc. Natl. Acad. Sci. USA. 91:12130–12134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Robinson, J. M., and M. J. Karnovsky. 1980. Evaluation of the polyene antibiotic filipin as a cytochemical probe for membrane cholesterol. J. Histochem. Cytochem. 28:161–168. [DOI] [PubMed] [Google Scholar]
- 34.Bolard, J. 1986. How do the polyene macrolide antibiotics affect the cellular membrane properties? Biochim. Biophys. Acta. 864:257–304. [DOI] [PubMed] [Google Scholar]
- 35.Simon, C. G., and A. R. Gear. 1998. Membrane-destabilizing properties of C2-ceramide may be responsible for its ability to inhibit platelet aggregation. Biochemistry. 37:2059–2069. [DOI] [PubMed] [Google Scholar]
- 36.Gidwani, A., H. A. Brown, D. Holowka, and B. Baird. 2003. Disruption of lipid order by short-chain ceramides correlates with inhibition of phospholipase D and downstream signaling by FcepsilonRI. J. Cell Sci. 116:3177–3187. [DOI] [PubMed] [Google Scholar]
- 37.Holowka, D., E. D. Sheets, and B. Baird. 2000. Interactions between Fc(epsilon)RI and lipid raft components are regulated by the actin cytoskeleton. J. Cell Sci. 113:1009–1101. [DOI] [PubMed] [Google Scholar]
- 38.Packard, B. S., and D. E. Wolf. 1985. Fluorescence lifetimes of carbocyanine lipid analogues in phospholipid bilayers. Biochemistry. 24:5176–5181. [DOI] [PubMed] [Google Scholar]
- 39.Wolf, D. E. 1988. Probing the lateral organization and dynamics of membranes. In Spectroscopic Membrane Probes, Vol. 1. L. M. Loew, editor. CRC Press, Boca Raton, FL. 193–220.
- 40.Wolber, P. K., and B. S. Hudson. 1979. An analytic solution to the Forster energy transfer problem in two dimensions. Biophys. J. 28:197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dewey, T. G., and G. G. Hammes. 1980. Calculation on fluorescence resonance energy transfer on surfaces. Biophys. J. 32:1023–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Feigenson, G. W., and J. T. Buboltz. 2001. Ternary phase diagram of dipalmitoyl-PC/dilauroyl-PC/cholesterol: nanoscopic domain formation driven by cholesterol. Biophys. J. 80:2775–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baumgart, T., A. Hammond, P. Sengupta, S. Hess, D. Holowka, B. Baird, and W. W. Webb. 2006. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc. Natl. Acad. Sci. USA. 104:3165–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukherjee, S., T. T. Soe, and F. R. Maxfield. 1999. Endocytic sorting of lipid analogues differing solely in the chemistry of their hydrophobic tails. J. Cell Bio. 144:1271–1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meder, D., M. J. Moreno, P. Verkade, W. L. Vaz, and K. Simons. 2006. Phase coexistence and connectivity in the apical membrane of polarized epithelial cells. Proc. Natl. Acad. Sci. USA. 103:329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Young, R. M., X. Zheng, D. Holowka, and B. Baird. 2005. Reconstitution of regulated phosphorylation of FcepsilonRI by a lipid raft-excluded protein-tyrosine phosphatase. J. Biol. Chem. 280:1230–1235. [DOI] [PubMed] [Google Scholar]