

Abstract
Background & Aims
Approximately half of hepatitis C virus (HCV)-infected patients do not respond to current interferon (IFN)-α combination therapy. To understand IFN-α resistance in vivo, we examined the dynamic responses to both type I and type II IFNs, human IFN (hIFN)- α, -γ, and consensus IFN, in the chimpanzee model.
Methods
Naive and HCV-infected chimpanzees were treated with 3 forms of hIFNs in vivo. Quantitative real-time polymerase chain reaction was performed to evaluate the expression of IFN-stimulated genes (ISGs) in both peripheral blood mononuclear cells and liver to compare the responses to hIFN between naive and infected chimpanzees. The hepatic expression of IFN signaling components and inhibitory regulators including suppressor of cytokine signaling 3 (SOCS3) were assessed. SOCS3 expression was also evaluated in the liver of HCV-infected patients undergoing IFN treatment.
Results
The in vivo responses to all 3 hIFNs were much lower in the HCV-infected chimpanzees than those in the naive chimpanzees. This defect was particularly evident in the liver because induction of hepatic ISGs was barely detectable in the infected animals. Following IFN administration, the expression of SOCS3 was significantly up-regulated, possibly through induction of interleukin-6, in the liver of HCV-infected chimpanzees. HCV-infected humans also showed a differential pattern of hepatic SOCS3 expression in response to IFN that is associated with treatment response.
Conclusions
Our data indicate a predominantly defective hepatic response to IFN in HCV-infected chimpanzees, which is probably mediated through the activation of SOCS3 and may explain the nonresponse of many HCV patients to IFN-based therapy.
Hepatitis C virus (HCV) infection is one of the most common blood-borne chronic infections with an estimated 170 million infected people worldwide. In the United States, approximately 3 million people are chronically infected, and HCV is the leading cause of liver transplantation.1 No HCV vaccine is available to date, and the current antiviral therapy with pegylated interferon (IFN)- α (Peg-IFN-α) and ribavirin is expensive, effective in approximately 50% of patients, and associated with numerous adverse effects.2 Thus, there is a pressing need for improvement of anti-HCV therapy.3 To achieve this goal, it is crucial to understand the mechanisms of HCV clearance and nonresponse to IFN-based therapy in HCV patients.
IFNs are naturally occurring proteins secreted by mammalian cells that play a critical role in control of viral infection and provide a link between innate and adaptive immunity. There are 3 types of IFN: type I IFNs include the 14 nonallelic subtypes of IFN-α subtypes, as well as IFN-β, -ε, -κ, -ω, and -τ, all of which bind to the type I IFN receptor (IFNAR); type II IFN, IFN-γ, binds to the type II IFN receptor; type III IFNs include 3 recently discovered proteins called IFN-γ that bind to a novel type III receptor.4 Consensus IFN-λ (IFN alfacon-1) is a synthetic type I IFN, whose sequence is derived from the consensus sequences of various IFN-α subtypes.5 It has been shown to be more potent than naturally occurring type I IFNs in cell culture models and more effective in clinical trials for the treatment of chronic hepatitis C.6,7
IFN induces an antiviral state in cells by activating the Janus kinase (JAK)-signal transducers and activators of transcription (STAT) pathway.8 Binding of IFN to its receptor activates constitutively associated JAK proteins, which leads to the docking of STAT molecules to the receptor and subsequent STAT phosphorylation. The activated STATs dissociate from the receptor chain and form dimers that translocate to the nucleus to modulate gene transcriptional activity. For the type I IFNs, the interferon-stimulated gene factor 3 (ISGF3) complex, consisting of a STAT1 and STAT2 heterodimer and interferon regulatory factor 9 (IRF9), binds to the interferon-stimulated response element (ISRE). For the type II IFN, IFN-γ, STAT1 homodimers bind directly to the γ-activated site element. Both types of IFNs induce the expression of a large number of ISGs with substantial overlap and set up an antiviral, antiproliferative, and immunoregulatory state in the host cells. IFN-induced antiviral activities have been extensively studied in the HCV replicon system. Both IFN-α and IFN-γ have been shown to inhibit HCV replication,9,10 and type I/II IFN combinations resulted in a synergistic antiviral effect.11,12 However, standard combination therapy with Peg-IFN-α and ribavirin achieves sustained viral clearance in only approximately half of treated patients, and IFN-γ as a single agent appears to be ineffective in small clinical trials.13 HCV appears to have developed strategies to interfere with the IFN effector pathways, leading to non-response in many HCV-infected patients.
The mechanisms by which HCV interferes with IFN signaling and attenuates its antiviral efficacy have not been fully elucidated. Various hypotheses have been proposed.14 Among them, 2 negative regulators, suppressor of cytokine signaling (SOCS) 3 and protein inhibitor of activated STAT (PIAS), have recently been reported to be induced by HCV proteins leading to inhibition of the JAK-STAT signaling. SOCS3 can be induced by the HCV core protein and suppress JAK-STAT signaling to block the IFN-induced formation of ISGF3 in cell culture.15 HCV protein expression in liver cells is associated with activation of PIAS and inhibition of STAT function, possibly augmented by induction of protein phosphatase 2A (PP2A) expression.16
Despite significant advances in understanding of the actions of IFN in the cell culture system, little is known about the inhibition of IFN signaling by HCV and nonresponse to IFN therapy in vivo. The chimpanzee is the only animal model susceptible to HCV infection. In the present study, we conducted a comprehensive assessment of the antiviral effects of both type I and type II IFNs (IFN-α, -γ, and consensus IFN) in chimpanzees and demonstrated a potential mechanism of IFN resistance. To confirm the clinical relevance in human HCV infection, we also evaluated patient samples before and during Peg-IFN therapy.
Materials and Methods
Chimpanzees and Interferons
Six chimpanzees, X0176, X0284, X0101, X0233, X0142, and X0234, were maintained at the Southwest Foundation for Biomedical Research, an Association for Assessment and Accreditation of Laboratory Animal and Care-accredited facility, and the study protocol was approved by the Institutional Animal Care and Use Committee at the Foundation and by the Interagency Animal Model Committee at the National Institutes of Health (NIH). Two chimpanzees, X0142 and X0234, had persistent infection with HCV genotype 1b derived from a homogenous source as described previously,17 whereas others were naive animals. Whole blood samples from 2 chimpanzees, X6394 and X6475 (infected with genotype 1a viruses),18,19 were provided by Dr. Stephen Feinstone at Center for Biologics Evaluation and Research (CBER), the Food and Drug Administration (FDA).
Human IFN-α2a used for the in vitro study was purchased from Fitzgerald Industries International, Inc. (Concord, MA), and the product used for in vivo study was purchased from Roche (Nutley, NJ). IFN-γ1b and consensus IFN (Infergen, IFN alfacon-1) were provided by InterMune, Inc. (Brisbane, CA).
Patient Samples
Patient samples were derived from 2 sources. Treated patients were recruited from the Liver Diseases Unit at the University of North Carolina (UNC). Patients were given an initial dose of 180 μg Peg-IFN-α-2a and underwent liver biopsy 24 hours later. Control samples came from liver biopsy specimens obtained from patients at the Clinical Center of the NIH prior to undergoing antiviral therapy with Peg-IFN and ribavirin. Control patients were selected to match treated patients in terms of age, gender, ethnicity, initial viral load, and degree of histologic disease. Patients signed informed consent, and the protocol was approved by the Institutional Review Board of UNC and the NIH. All patients had genotype 1 HCV infection. Biopsy samples were snap frozen in liquid nitrogen and stored at −80°C. Details of the human study will be published elsewhere.20 Human liver biopsy tissue was handled similarly to chimpanzee liver tissue as described below. Both treated and control patients subsequently underwent a full course of standard antiviral therapy consisting of Peg-IFN-α-2a 180 μg and weight-based ribavirin (1000 mg daily ≤75 kg and 1200 mg >75 kg for 48 weeks). Patients achieving ≥2-log copies/mL decrease in HCV RNA by 4 weeks of therapy were deemed rapid responders (RR), and those with lesser decreases in viral load were designated slow responders (SR).
Peripheral Blood Mononuclear Cells Isolation and IFN Stimulation
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood samples of chimpanzees and healthy human donors (informed consents were obtained) and stimulated with 3 different doses of human IFN (hIFN)-α, -γ, and consensus IFN in RPMI medium with 10% fetal bovine serum (FBS) (Cellgro, Herndon, VA). After incubation for 6 or 24 hours, cells were harvested and subjected to RNA isolation.
Chimpanzee Experiment
Chimpanzees used for in vivo study were treated sequentially with 10 million IU IFN-α, 400 μg IFN-γ, and 30 μg consensus IFN subcutaneously. Briefly, 4 animals (X0101, X0233, X0142, and X0234) were divided into 2 groups, each comprising 1 naive and 1 infected chimpanzee. The experiment was separated into 3 phases. For the first phase, group 1 chimpanzees (X0101 and X0142) were administered with IFN-α and group 2 (X0233 and X0234) with IFN-γ. Blood samples (40 mL each) were collected at 9 time points (pretreatment, 8 hours, 24 hours, 48 hours, 72 hours, 7 days, 14 days, 21 days, and 28 days posttreatment), and liver biopsy samples were collected at 5 time points (pretreatment, 8 hours, 24 hours, 48 hours, and 72 hours posttreatment). All animals were rested for 6 weeks to avoid any residual drug effect. Next, group 1 was treated with IFN-γ and group 2 with IFN-α for the second phase of the study. Similar sample-collecting procedures were performed during treatment up to 4 weeks. The animals were rested for 6 weeks and started on the third and final phase of the study, in which consensus IFN was given to both groups. Because of a nonspecific infection during the rest period after the phase II study, one of the HCV-infected chimpanzees, X0142, was dropped from the phase III study.
Quantification of Serum HCV RNA and Alanine Transaminase
Sera were isolated from serial blood samples of the HCV-infected chimpanzees. HCV RNA was quantified by using the COBAS AMPLICOR HCV MONITOR TEST, v2.0 (Roche Diagnostics, Branchburg, NJ), which has a detection limit of 600 IU/mL (1620 copies/mL). Alanine transaminase (ALT) values were measured by the Laboratory Medicine Department of the Southwest Foundation for Biomedical Research.
TaqMan Real-Time PCR Analysis
Total RNA was prepared from the PBMCs and liver biopsy tissues with RNeasy Mini Kit according to manufacturer’s instructions (Qiagen, Valencia, CA). Complementary DNA (cDNA) was synthesized from total RNA with First-strand cDNA Synthesis System (Marligen Biosciences, Ijamsville, MD). TaqMan real-time PCR analysis was used to quantify the mRNA expression levels of genes of interest. The primers and probes used were Gene Expression Assays (Applied Biosystems, Foster City, CA). Each reaction was performed in duplicate, and all samples were standardized using the internal control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene. Reactions were set up using 12.5 μL TaqMan universal PCR master mix, cDNA template, and 1.25 μL primers and probe mix in a final volume of 25 μL. Reactions were performed on an iCycler iQ Multicolor Real-Time Detection System (Bio-Rad, Hercules, CA) with the following reaction conditions: 95°C for 10 minutes, followed by 40 cycles of 95°C for 20 seconds, 60°C for 1 minute, and additional incubation at 68°C for 10 minutes.
Western Blot Analysis
Liver biopsy tissues were lysed using mammalian tissue lysis/extraction reagent (CelLytic MT; Sigma Chemical Co., St. Louis, MO) containing a Protease Inhibitor Cocktail Tablet (Roche, Indianapolis, IN) and a Protein Phosphatase Inhibitor Set (Upstate Biotechnology, Lake Placid, NY). The collection of supernatant lysates, determination of protein concentration, and Western blot analysis have been described previously.21 Antibodies to STAT1, SOCS1, SOCS3, Src homology region 2-domain phosphatase (SHP)1, and β-actin were from Abcam Inc. (Cambridge, MA). Antibodies to STAT2, STAT3, phosphorylated STAT1 (Y701), and protein phosphatase 2A C subunit (PP2Ac) were from Upstate Biotechnology. Anti-IRF9 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA), and anti-IFNAR chain 2 antibody was from Fitzgerald Industries International Inc. (Concord, MA). The band intensities in the images were quantified by a public analysis program, ImageJ, offered by the NIH (http://rsb.info.nih.gov/ij/).
Serum Cytokine Analysis
Serum cytokines (eotaxin, GM-CSF, IFN-γ, interleukin (IL)-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, interferon-γ-inducible protein (IP) 10, membrane cofactor protein (MCP)-1, macrophage inflammatory protein (MIP)-1α, tumor necrosis factor (TNF)-α, and RANTES) were measured simultaneously using Beadlyte Human 22-Plex Cytokine Detection System (Upstate Biotechnology) according to the manufacturer’s instructions. Briefly, both standard and chimpanzee samples were diluted with human serum diluent, and 50 μL was loaded onto the 96-well filtration plate. Twenty-five-microliter beads coated with target capture antibodies against cytokines were added to each well and incubated overnight at 4°C with low-speed shaking at 300 rpm in the dark. On the next day, after washing, the plate was supplied with 25 μL premixed biotin conjugate antibodies and incubated for 2 hours at room temperature with shaking at 300 rpm. Finally, streptavidin-phycoerythrin was added, and the results were read with Bioplex Luminex System (Bio-Rad Laboratories Inc., Hercules, CA). The data were analyzed using Bio-Plex Manager software v.4.0 with Five-Parameter Logistics curve fitting.
Results
Response of Chimpanzees to hIFNs In Vitro
Before performing the in vivo chimpanzee study, we first determined the ability of chimpanzees to respond to hIFN. PBMCs from 2 naive chimpanzees (X0284 and X0176) and 2 healthy human donors were stimulated with 3 different doses of IFN-α, IFN-γ, and consensus IFN. A panel of previously reported IFN-α or -γ specific ISGs were selected for analysis: myxovirus resistance 1 (MX1), 2,5-oligoadenylate synthetase (2,5-OAS), IFN-induced protein with tetratricopeptide repeats 1 (IFIT1), and IFN-induced protein 15 (IFI15) were used as markers of IFN-α-induced genes, and the IP10, IRF1, and large multifunctional protease (LMP) 2 and LMP7 were selected as IFN-γ-induced genes.22 All ISGs were induced at 6 hours after IFN treatment (data not shown) and reached a peak at 24 hours (Figure 1). The overall gene expression patterns of the ISGs were comparable between human and chimpanzee PBMCs. Consensus IFN was more potent and induced a broader range of ISGs than either IFN-α or -γ. All 3 IFNs induced higher levels of ISG expression in humans than in chimpanzees (2–5 times higher), suggesting that chimpanzees do not respond as well as humans to hIFN in vitro. Based on these data, 10 million IU IFN-α, 400 μg IFN-γ, and 30 μg consensus IFNs were used for in vivo study in chimpanzees. These doses are 3– 4 times higher than the standard doses for the treatment of human subjects (3 million IU IFN-α, 100 μg IFN-γ, and 9 μg consensus IFN) to compensate for the lower efficacy of hIFNs in chimpanzees.
Figure 1.
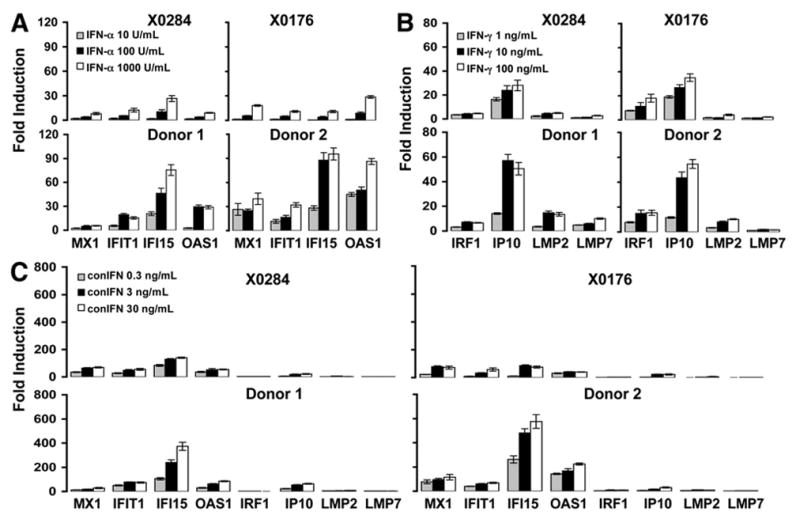
ISG induction of chimpanzee PBMCs to human IFNs in vitro. PBMCs from naive chimpanzees (X0284 and X0176) and healthy human donors (donors 1 and 2) were isolated and stimulated with 3 forms of IFN. (A) IFN-α (10, 100, and 1000 U/mL); (B) IFN-γ (1, 10, and 100 ng/mL); (C) consensus IFN (0.3, 3, and 30 ng/mL). Fold inductions of selected ISG mRNAs were measured by real-time PCR at 24 hours posttreatment. Error bars indicate mean ± SD of 3 experiments. conIFN, consensus IFN.
Impaired Response of HCV-Infected Chimpanzees to hIFN In Vivo
Four chimpanzees (2 naive: X0101 and X0233; 2 infected: X0142 and X0234) were divided into 2 groups in the in vivo study. Each group consisted of 1 naive and 1 infected animal. The experiment was divided into 3 phases: treatment with IFN-α (or IFN-γ), treatment with IFN-γ (or IFN-α), and treatment with consensus IFN, respectively. With this design, each animal received all 3 forms of IFN treatment, which allows for internal control and biologic duplication of the experiment. Consensus IFN was given last because consensus IFN is more potent and induces a broader range of ISGs than either IFN-α or -γ. Similar to the results from the in vitro study, IFN-α and consensus IFN treatment led to the induction of the IFN-α-specific ISGs (MX1, OAS1, IFIT1, and IFI15) in both PBMCs and liver tissues of the chimpanzees in vivo. Likewise, the IFN-γ-specific ISGs (IP10, IRF1, LMP2, and LMP7) were induced by IFN-γ and consensus IFN in vivo. Figure 2 shows the fold inductions of only 4 selected ISGs (MX1, IFI15, IP10, and IRF1), but the other 4 (IFIT1, OAS1, LMP2, and LMP7) behaved similarly. Although PBMCs from both naive and HCV-infected chimpanzees responded to all 3 forms of IFN, reaching a peak at 8 hours posttreatment and returning to basal levels within 48 hours, the levels of ISG induction in PBMCs from HCV-infected chimpanzees were much lower (~4 times) than those in the naive animals (Figure 2A). All 3 forms of IFN induced stronger ISG responses in the liver than in PBMCs of naive chimpanzees (Figure 2B). However, in the HCV-infected animals, little or no hepatic ISG induction was observed. Although hepatic ISG induction was severely blunted in the infected animals, the basal level of ISG expression in the liver were higher than those in naive animals (Figure 2C), suggesting that HCV infection resulted in endogenous IFN production. The higher basal hepatic ISG expression did not account for the failure to respond to exogenous IFN because the absolute levels reached after treatment were still much lower in infected than naive chimpanzees (Figure 2D). These data indicate a deficiency of response, particularly in the liver, to hIFNs in HCV-infected chimpanzees.
Figure 2.
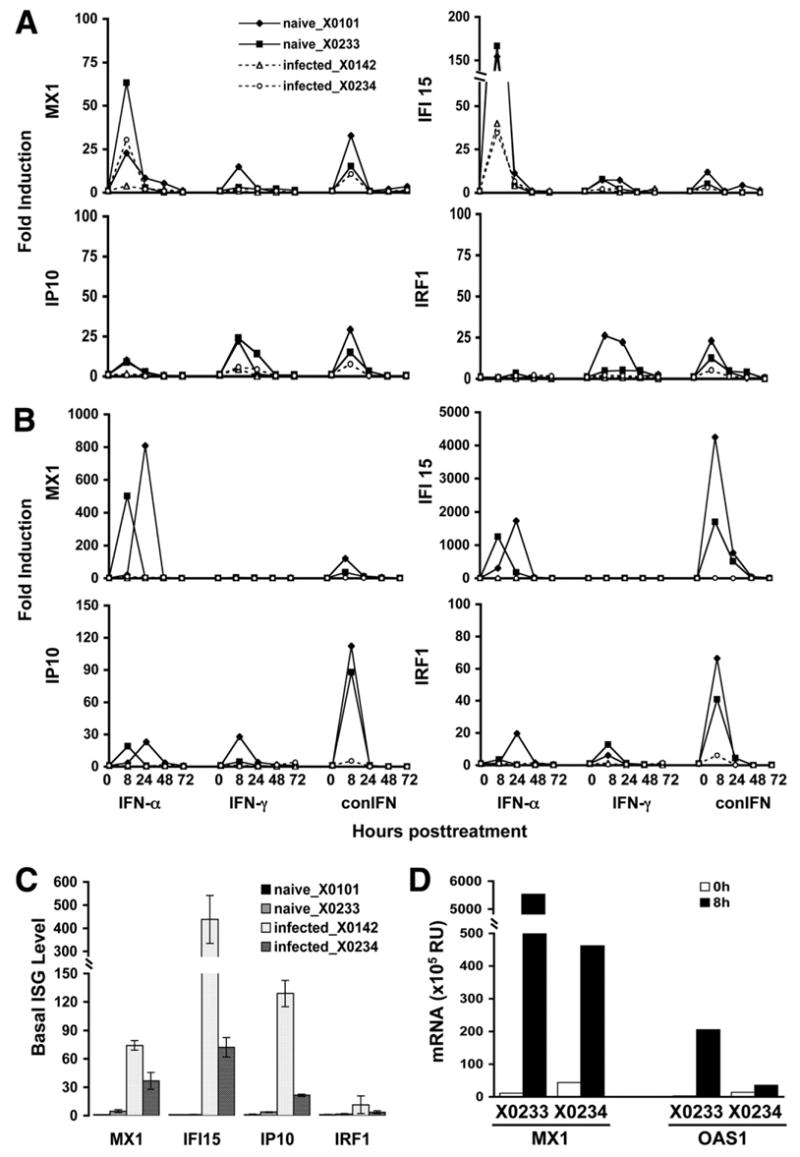
Baseline and induction of ISG in naive and HCV-infected chimpanzees in vivo. Induction of ISG expression in PBMC (A) and liver (B) from naive (X0101 and X0233) and HCV-infected (X0142 and X0234) chimpanzees in vivo. conIFN, consensus IFN. (C) Basal ISG expression in the liver of all animals. The basal ISG levels, after normalized to GAPDH, were adjusted by setting the value of naive chimpanzee (X0101) as 1 for each ISG. (D) The hepatic expression levels of selected ISGs (MX1 and OAS1) in selected animals (X0233 and X0234) following IFN administration (RU; relative unit, defined as copy number normalized to GAPDH).
Effect of IFN on Viral Level of HCV-Infected Chimpanzees
HCV RNA and ALT levels were analyzed from serial serum samples of the 2 chronically HCV-infected chimpanzees (X0142 and X0234) pre- and post-IFN administration. As shown in Figure 3, although chimpanzee X0234 was initially inoculated with the week 2 serum from X0142 (ie, the same viral strain),17 the pattern of infection that ensued differed somewhat between the 2 chimpanzees. The viral load was much higher in X0142 and fluctuated in a range from 104 to 106 copies/mL as compared with a relatively stable viremia (approximately 3 × 104 copies/mL) seen in X0234. Notably, neither IFN-α nor IFN-γ injection resulted in a significant decrease in viral load in either chimpanzee, consistent with the absence of hepatic ISG induction in both animals. However, chimpanzee X0234 did have a transient decrease in viremia in response to IFNs with a reproducible decrease in viral titer of 0.5–1 log copies/mL (IFN-γ < IFN-α < conIFN) at 8–24 hours posttreatment. Interestingly, although the hepatic IFN response in this chimpanzee was blunted compared with naive animals, there was low-level induction of hepatic ISGs in response to IFNs (eg, MX1 and IFI15 induced by IFN-α; IP10 and IRF1 induced by IFN-γ, Figure 2B and 2D). In contrast, chimpanzee X0142 had no hepatic ISG induction after treatment and actually had a transient increase in HCV viremia within 24–48 hours posttreatment, which appeared to coincide with a transient ALT increase in this chimpanzee (data not shown). This effect could be explained by an IFN-induced hepatotoxicity with release of HCV RNA from the injured hepatocytes resulting in a transient rise in viremia.
Figure 3.
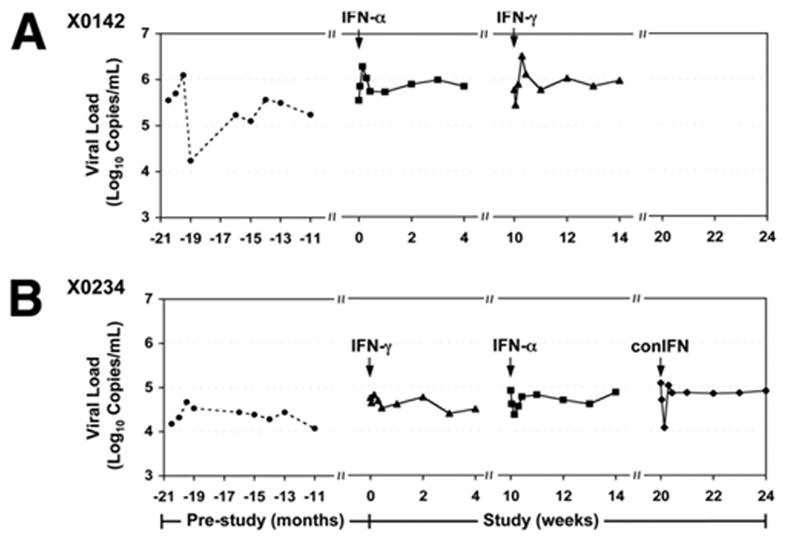
Serum HCV RNA titers of HCV-infected chimpanzees in response to IFN. HCV RNA levels of HCV-infected chimpanzees X0142 (A) and X0234 (B) were monitored for more than 1 year prior to this study. Arrows indicate time points of IFN administration. conIFN, consensus IFN.
Comparable Induction of ISGs in PBMCs From Infected and Naive Chimpanzees Ex Vivo
Because of the apparently different ISG induction in PBMCs between the naive and infected chimpanzees in vivo, we sought to determine whether such a difference persists ex vivo, free of any potential biologic interactions with IFN that may be operational in vivo. Thus, we evaluated the actions of IFNs on PBMCs isolated from naive and infected chimpanzees in vitro. Eight chimpanzees, 4 naive and 4 HCV infected, were studied. The 4 naive animals included the 2 used in the preliminary study to test the response of chimpanzees to hIFNs (X0176 and X0284) and 2 others used in the in vivo study above (X0101 and X0233). The 4 infected animals were the 2 used for the study above (X0142 and X0234) and 2 others infected with a genotype 1a strain H77 (X6394 and X6475).18,19 Figure 4 summarizes the fold inductions of representative ISGs in PBMCs from all 8 chimpanzees at 24 hours post-IFN stimulation. The overall ISG inductions of the HCV-infected animals were comparable with those of the naive animals with the exception of IP10, whose induction by IFN-γ was significantly lower in the infected chimpanzees. However, the difference in IP10 induction of PBMCs by IFN-γ between naive and infected chimpanzees ex vivo was much less than that in vivo (2-to 3-fold vs 4- to 6-fold, respectively). These data suggest that the apparent difference in IFN response occurred mostly in vivo, and, once the PBMCs were removed from the in vivo milieu of the infected animals, they responded to IFN similarly to those from the naive animals.
Figure 4.
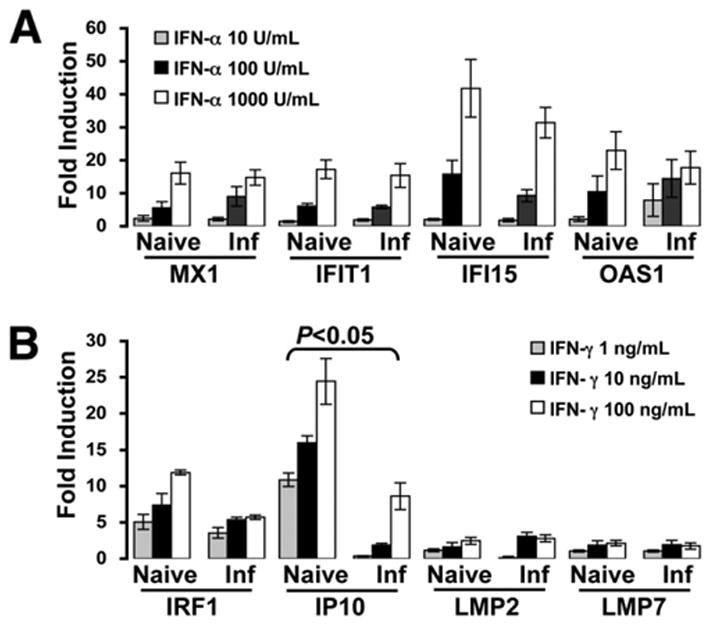
ISG induction of PBMCs from naive and HCV-infected chimpanzees ex vivo. PBMCs from naive (X0176, X0284, X0101, and X0233) and HCV-infected (X0142, X0234, X6394, and X6475) chimpanzees were isolated and stimulated with IFN-α (10, 100, and 1000 U/mL) (A) and IFN-γ (1, 10, and 100 ng/mL) (B), respectively. Fold inductions of selected ISG mRNAs were measured by real-time PCR at 24 hours posttreatment and compared between naive and infected groups. Error bars indicate mean ± SD for the 4 animals in each group. Inf, infected.
Persistent Activation of the JAK-STAT Pathway in the Liver of HCV-Infected Chimpanzees
The lack of hepatic ISG induction after IFN treatment in HCV-infected chimpanzees suggests that the IFN signal transduction pathways are inhibited in the liver. To determine which step of IFN signaling was inhibited, various components involved in the JAK-STAT pathway were evaluated using Western blot analysis from liver biopsy samples of pre- and 8 hours post-IFN-α treatment. The 8-hour time point was chosen to coincide with the peak hepatic ISG expression (Figure 2B). As shown in Figure 5A for the naive chimpanzees, a slight increase of phosphorylated STAT1 was detected with X0101, whereas not with X0233. In these chimpanzees, the JAK-STAT pathway was likely activated by exogenous IFN but quickly returned to baseline by 8 hours. This time course is consistent with the rapid onset and transient nature of JAK-STAT activation by IFN observed in vitro.23 In contrast, all components of the JAK-STAT pathway were activated even prior to IFN treatment in the infected chimpanzees. In addition to a markedly elevated level of phosphorylated STAT1, there was a general increase seen in the total levels of STAT1, STAT2, STAT3, and IRF9 proteins (Figure 5A). This effect was more dramatic in chimpanzee X0142. After IFN treatment, rather than increasing, the phosphorylated STAT1 level actually declined in X0142, which might be due to the activation of negative regulator(s) of IFN signaling. This observation is in keeping with the lack of ISG induction in the liver of these infected animals.
Figure 5.
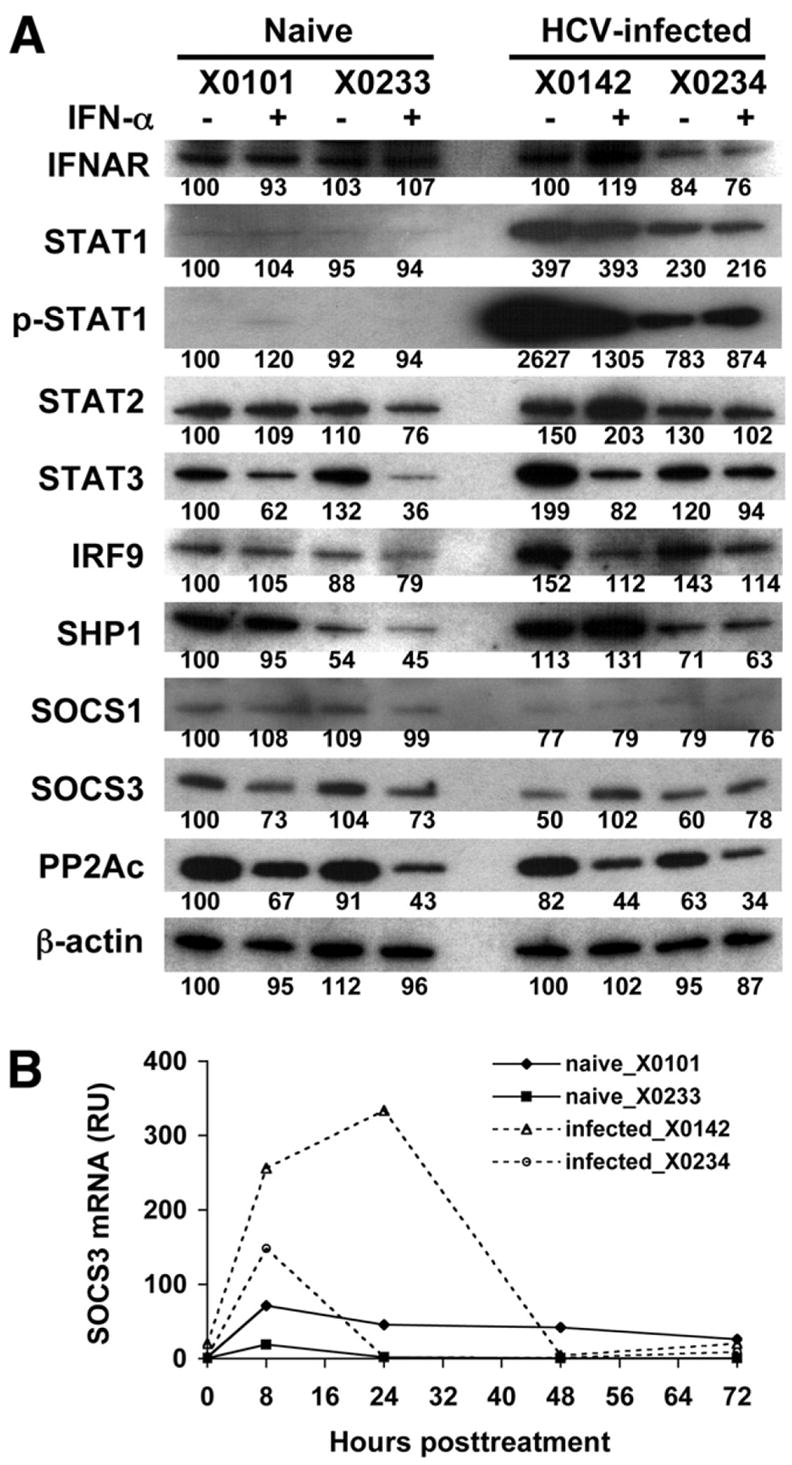
Expression of IFN signaling molecules and inhibitory regulators in the liver of chimpanzees. (A) Fifteen micrograms protein extracts of liver from chimpanzees with or without IFN-α treatment (8 hours) were subjected to Western blot analysis with indicated antibodies. β-actin was used as loading control. Band intensities were quantified with NIH ImageJ software. The relative values for each protein are shown below the bands with the baseline value of naive chimpanzee (X0101) setting as 100. (B) Liver SOCS3 mRNA expression levels measured by real-time PCR (RU; relative unit, defined as copy number normalized to GAPDH).
Enhanced Hepatic SOCS3 Expression in Response to IFN in HCV-Infected Chimpanzees
At least 3 different classes of negative regulators are known to contribute to the inhibition of IFN signaling24: the SHP1, SOCS, and PIAS families. To explore whether these suppressors were responsible for the IFN resistance seen in the HCV-infected chimpanzees, we measured the expression levels of regulatory factors SHP1, SOCS1, SOCS3, and PP2A in liver biopsy samples pre- and post-IFN-α induction (Figure 5A). Among these factors, the SOCS3 protein level increased significantly in HCV-infected but not in naive animals after IFN treatment. This increase was also confirmed at the RNA level by quantitative real-time PCR (Figure 5B). Although the hepatic SOCS3 mRNA level also increased modestly in naive animals, the increase was much more dramatic in HCV-infected animals (X0142 > X0234). These data indicate that the SOCS3 up-regulation may be a potential mechanism for the defective IFN response in HCV-infected chimpanzees.
Induction of IL-6 by IFN in Serum and Liver Tissue of HCV-Infected Chimpanzees
Because SOCS3 can be induced by a variety of cytokines including the proinflammatory cytokine IL-6, we determined the serum IL-6 levels following IFN administration in these chimpanzees. Although the serum IL-6 levels increased in response to IFN in all chimpanzees, the infected chimpanzee X0142 showed a more exaggerated augmentation (Figure 6A). A similar pattern was confirmed by real-time PCR of IL-6 mRNA in the liver. This observation is consistent with the high level of hepatic SOCS3 induction in chimpanzee X0142. In addition to IL-6, we also evaluated a panel of cytokine/chemokine levels in sera of HCV-infected chimpanzees compared with those of naive animals in response to IFN treatment. Among the 22 cytokines/chemokines measured, IP10, MCP-1, MIP-1α, and eotaxin were induced by both type I and type II IFNs and behaved in a pattern similar to the ISG mRNA expression (Figure 6B).
Figure 6.
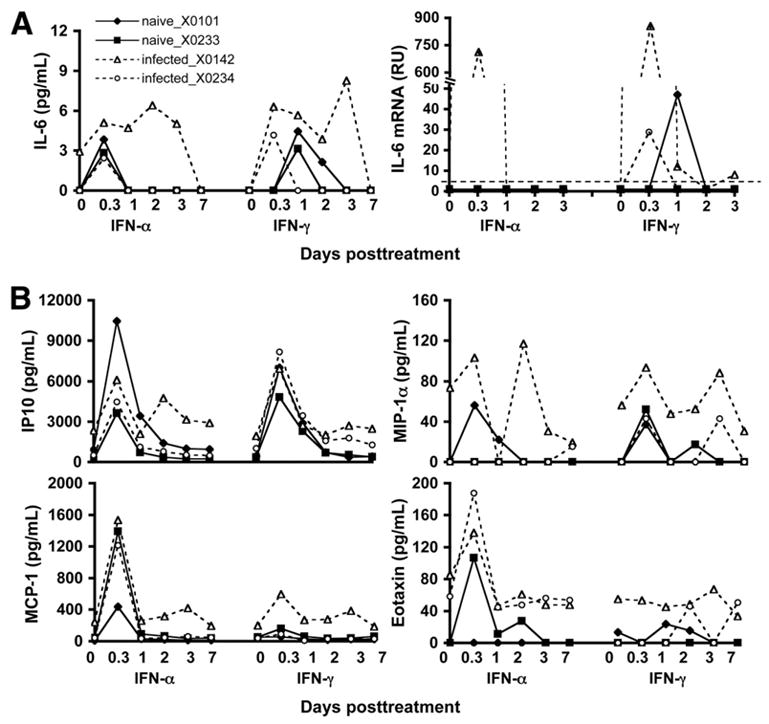
Induction of IL-6 by IFN in the serum and liver of HCV-infected chimpanzees. (A) Serum protein and liver mRNA levels (RU; relative unit, defined as copy number normalized to GAPDH) of IL-6 were determined. The detection limit of IL-6 mRNA level is 5 RU (dotted line) in this assay. (B) A panel of serum cytokines was measured. Selected cytokines (IP10, MIP-1α, MCP-1, and eotaxin) induced by IFN-α are shown.
Hepatic SOCS3 Expression in HCV-Infected Patients Pre- and Post-IFN Treatment
Based on the findings in chimpanzees, we evaluated SOCS3 mRNA expression in HCV-infected patients pre- and post-IFN treatment. Two groups of genotype 1 virus infected-patients were studied: a control group of 21 patients in whom liver biopsies were performed prior to antiviral therapy and a treatment group of 13 patients in whom liver biopsies were performed 24 hours after an initial dose of Peg-IFN-α2a. Two groups were matched for age, gender, and race. All control patients were subsequently treated, and their response to therapy is known. Patients were categorized as rapid responders (RR) or slow responders (SR) depending on whether they achieved a 2-log copies/mL drop in HCV viral titer by 4 weeks of therapy. This early virologic response has been shown to be an accurate predictor of clinical treatment response25 and probably a better marker of the antiviral effect of IFN in vivo because clinical end point is frequently altered by other clinical issues like noncompliance or adverse effects. SOCS3 mRNA expression was evaluated by real-time PCR for all patient samples (Figure 7).
Figure 7.
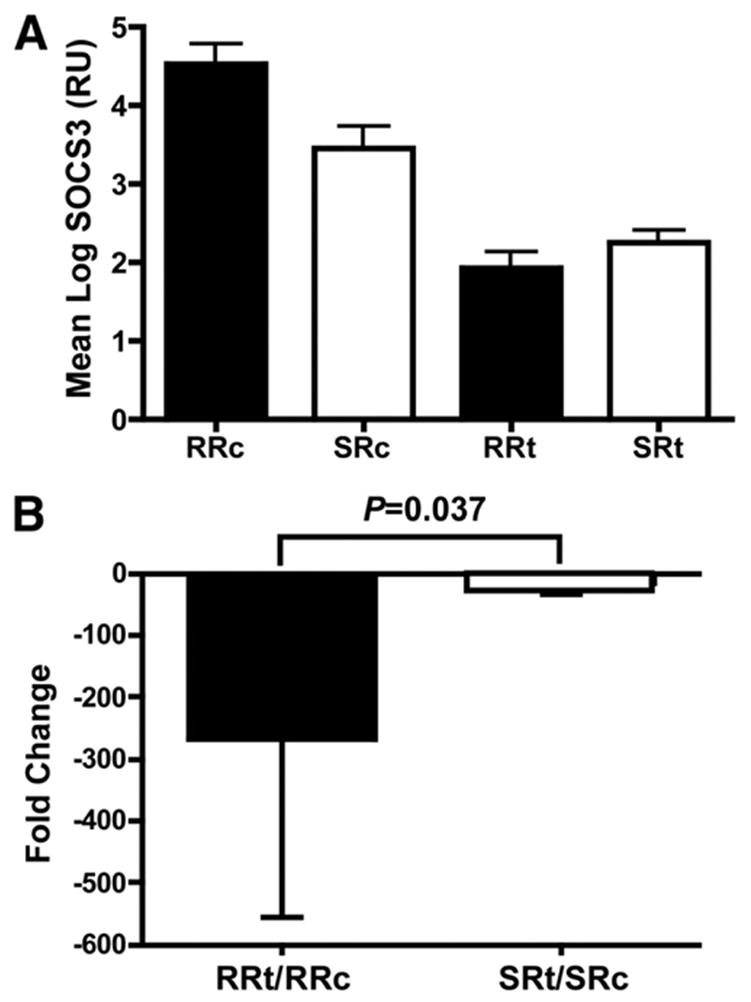
SOCS3 expression in human liver biopsy specimens pre-and post-IFN treatment. (A) The mean SOCS3 expression (RU; relative unit, defined as copy number normalized to GAPDH) is shown for the 2 subgroups of both treatment (t) and control (c) groups as described in the Materials and Methods section, respectively. RR, rapid responder; SR, slow responder. (B) Fold change in SOCS3 expression is calculated by dividing the mean SOCS3 expression of RR or SR in the control group (c) by that of RR or SR in the treatment group (t), respectively; the fold change is then shown as a negative value to account for the fact that SOCS3 expression declined with treatment. Error bars are shown as mean ± SD.
The treatment group had generally lower mean SOCS3 expression than the control patients (Figure 7A). In the control group, patients who were considered as RR when treated later had a higher SOCS3 expression than those as SR, whereas the SOCS3 expression was similar between RR and SR among the treated patients. In light of the inverse trend, the relative change in the RR and SR groups may be more biologically relevant. The fold change of hepatic SOCS3 in response to IFN is determined by taking the control group values as the baseline expression levels for the treatment group according to their treatment response (Figure 7B). Although the SR in the treatment group had a 1.7-fold higher absolute mean SOCS3 expression than that of RR, they had a much less relative change than the RR (22.6-fold vs 268-fold; P = .037).
Discussion
Although new therapies based on small molecule inhibitors of HCV protease and polymerase look promising for HCV infection, it is likely that they will be used in combination with, rather than instead of, IFN-α-based regimens. Therefore, an improved understanding of the mechanisms of action and resistance to IFN in HCV infection will be critical to improve outcomes as well as for the development of new therapeutic approaches. In this study, we have performed an extensive examination of the responses to 3 different forms of IFN in the PBMCs and liver of the chimpanzee, which is the only reliable animal model of HCV infection. We showed that, although less responsive than humans to hIFN, chimpanzees are able to respond to hIFN-α, -γ, and consensus IFN in both PBMCs and liver. The patterns of ISG induction in response to the various IFNs were similar to those previously reported in cell culture,22 chimpanzees,26,27 and humans.12 Although consensus IFN is derived from type I IFNs, it has been shown to induce both type I- and type II-specific ISGs and was shown to be more potent than either IFN-α or -γ in ISG induction. This effect is a result of the higher binding affinity of consensus IFN to IFNAR than IFN-α5,28 and the cross interaction of the signal transduction pathways between the 2 types of IFNs.
Our data are consistent with the recently reported microarray data showing strong ISG induction after IFN-α treatment of naive chimpanzees.29 More importantly, for the first time, we showed that HCV-infected chimpanzees had a blunted response to IFN therapy. Although a lower ISG induction by all 3 forms of IFN was observed in PBMCs, the most notable difference was seen in the liver. Despite high doses of all 3 forms of IFN, little or no hepatic ISG induction was seen in the infected animals. Consistent with the lack of detectable IFN response, there was little or no change in HCV viral load. The basal levels of ISG and phosphorylated STAT1 expression were higher in infected than naive chimpanzees, indicating that HCV infection led to a type I IFN response. This observation is consistent with previous studies in chimpanzees26,27 and humans30,31 and raises the possibility that the ISGs were already maximally induced in infected chimpanzees such that exogenous IFNs did not lead to any further ISG induction. However, the increased basal level in the infected chimpanzees does not account entirely for the reduced ISG induction because the absolute levels of ISGs after IFN treatment were substantially lower in infected animals than those in naive animals (10- to 100-fold lower). This suggests that, in infected chimpanzees, there is interference with ability to respond to IFN. Two possibilities could explain this blunted IFN response. During chronic infection, the continuous but ineffective IFN action leads to a relatively resistant state of the liver to further exposure of IFN. Continuous IFN action could lead to IFN receptor down-regulation, a phenomenon indeed observed in ligand-receptor interaction. We found no decrease in IFNAR in either chimpanzees or humans (data not shown) before or during IFN treatment, consistent with previous in vitro data.32 Alternatively, chronic HCV infection, either through a direct viral mechanism or indirect actions, may activate negative regulator(s) of IFN leading to an IFN resistant state in the liver. This concept is supported by the observation that chronic hepatitis C patients may be more susceptible to other hepatic viral infections.33,34
The observation that hepatic phosphorylated STAT1 showed a paradoxical decline after IFN treatment in the infected animals strongly supports the activation of an IFN-inhibitory pathway, as postulated above. By assessing the status of the various IFN-signaling inhibitors in the liver biopsy samples, we found that, following IFN administration, the expression of SOCS3 was significantly up-regulated. It appears that the induction of SOCS3 expression was much more dramatic at the mRNA (Figure 5B) than the protein level (Figure 5A) for both infected chimpanzees. This difference might be due to the unstable nature of the SOCS proteins and the lack of samples at earlier time points, which made it difficult to compare the 2 levels. SOCS3 acts by interacting with the JAKs, resulting in impaired STAT1 and STAT3 phosphorylation. This leads to reduced STAT1 nuclear translocation, lack of binding to the ISRE, and ultimately decreased ISG expression.24 Being IFN inducible, SOCS3 expression is also induced by IL-6, IL-10, TNF-α, and toll-like receptors 4 and 9.35 A previous study using an HCV transgenic mouse model showed a similar inhibition of JAK-STAT signaling by HCV expression; however, no induction of SOCS1 or SOCS3 was detected.36 On the other hand, HCV core protein has been shown to induce SOCS3 expression in several cell lines, resulting in impaired IFN and specifically STAT1 signaling.15,37,38 In addition, Zhu et al32 also showed that cells harboring HCV replicons resistant to IFN therapy produced higher levels of SOCS3. With silencing of SOCS3, IFN sensitivity was partially restored.32 Our in vivo finding of increased SOCS3 expression and reduced phosphorylated STAT1 after IFN treatment is consistent with the known functional mechanism of this IFN-inhibitory pathway. This pathway could provide a plausible explanation for a direct viral mechanism of IFN resistance in vivo.
If SOCS3 expression were induced by viral proteins, only infected cells would be resistant to IFN. However, our in vivo data suggest a general IFN-resistant state in the infected liver (Figure 2B). Alternatively, activation of IFN inhibitory pathway(s) may occur through the production of soluble factor(s) that act in an autocrine/paracrine fashion in the liver and to a lesser extent PBMCs as they traverse the liver in blood. To examine the possible soluble factor(s), we studied a panel of cytokine/chemokine levels in the serum of HCV-infected and naive chimpanzees in response to IFN treatment. Several cytokines have been reported to be induced by HCV proteins, such as IL-6, IL-8, TNF-α, MCP-1, and RANTES.39–43 Among them, IL-8 has been shown to have anti-IFN activity through inhibition of ISGF3 assembly and interaction with the ISRE. We found no induction of IL-8 by IFN in infected chimpanzees. However, IL-6 was induced in response to IFN in both serum and liver tissue of the infected chimpanzees with a time course similar to SOCS3 induction. Other cytokines known to modulate SOCS3 expression, such as IL-10 and TNF-α, were not induced by IFN in the infected animals. IL-6 mRNA was not detectable in the livers of treated patients in our cohort; however, biopsies were performed 24 hours after IFN treatment compared with 8 hours in the chimpanzees, and, consequently, IL-6 expression may have been missed, particularly given the low levels seen in chimpanzees suggesting that the peak may already have been missed at 8 hours.
Chen et al reported that human nonresponsders to IFN-α-based therapy had a higher expression of numerous ISGs in pretreatment liver biopsy samples as compared with those who achieved a sustained virologic response.44 The opposite pattern was seen with the hepatic SOCS3 expression in our cohort, with ~11-fold higher expression in control patients who responded to treatment (RR group) compared with the nonresponders (SR group). As a negative regulator of IFN, high SOCS3 levels prior to treatment would block ISG expression. With IFN-α treatment, SOCS3 declines, allowing for ISG induction. The degree of ISG induction may be more important than the absolute level of ISG expression for viral clearance. Consequently, the baseline level of SOCS3 expression and the degree to which SOCS3 changes after IFN treatment determine the pretreatment level of ISG expression and the degree to which ISGs can be induced by IFN-α therapy. In our human study, the SOCS3 expression declined much less in the SR than the RR in response to IFN-α. This difference could contribute to a blunted ISG induction in the human nonresponders. Infected chimpanzees, perhaps as an extreme example of the human nonresponders, actually have a rise in hepatic SOCS3 with IFN therapy and therefore exhibit little or no ISG induction and thus no decline in viremia.
Based on these observations, we propose a model (Figure 8) in which HCV infection leads to endogenous IFN production and also to an increased expression of SOCS3, either directly by a viral mechanism or indirectly by the induction of a soluble factor, possibly IL-6. As a consequence of SOCS3 induction and other mechanisms (eg, interaction of HCV NS5A or E2 protein with double-strand RNA-dependent protein kinase [PKR]), the antiviral activity of IFN is impaired, allowing HCV to establish persistent infection. In chimpanzees, IFN treatment leads to further SOCS3 induction, which prevents ISG activation and markedly blunts the IFN response. As described above in humans, SOCS3 declines but much greater in RR than in SR after IFN treatment. This proposed inhibitory pathway is variably operational in HCV-infected humans because of the genetic heterogeneity in the human population, thus accounting for the diverse infection outcomes and treatment responses.
Figure 8.
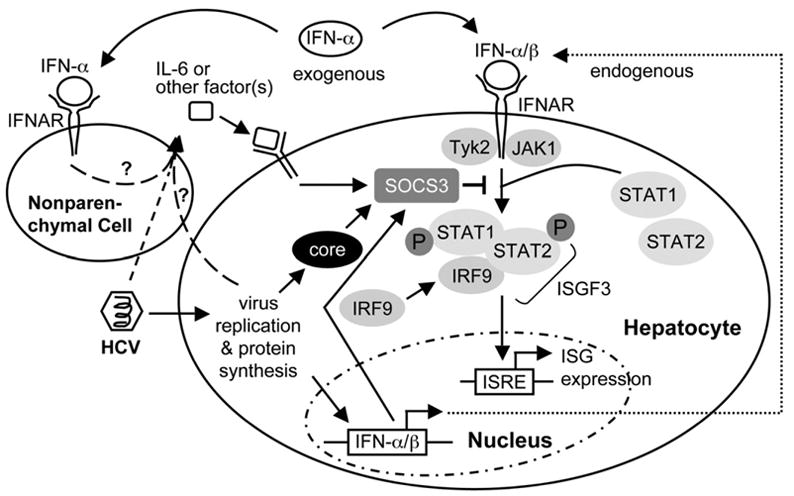
Model of IFN resistance in chronic HCV infection. HCV infection leads to endogenous IFN production and also to increased expression of SOCS3, either directly by a viral protein (eg, core) or indirectly by an IFN inhibitory factor (eg, IL-6 or other soluble factors). SOCS3 can suppress JAK-STAT signaling by blocking the IFN-induced formation of ISGF3, therefore allowing HCV to establish persistent infection. With exogenous IFN treatment, SOCS3 is induced, preventing further ISG activation and markedly blunting the IFN response.
In summary, we have shown that chimpanzees respond to hIFNs with ISG production in both PBMCs and liver. HCV infection leads to the production of type I IFN resulting in sustained activation of the JAK-STAT pathway and ISG induction in the liver. Typical ISG responses were seen in treated naive chimpanzees; however, ISG production was reduced in PBMCs and almost completely abrogated in the liver of HCV-infected animals. SOCS3 expression was markedly increased in the livers of infected chimpanzees after IFN treatment and was associated with a decrease in phosphorylated STAT1. A similar pattern of SOCS3 expression was seen in patients associated with treatment response. This negative regulator of IFN, possibly mediated by the production of IL-6, may be an important mechanism of IFN nonresponse in chronic HCV infection. Further studies to elucidate the biologic importance of this interaction may have crucial implications for the improvement of current HCV therapy.
Acknowledgments
The authors thank Stephen Feinstone and Marian Major (CBER, FDA) for providing the chimpanzee blood samples and the NIH Clinical Center Department of Transfusion Medicine for the virologic testing.
Abbreviations used in this paper
- HCV
hepatitis C virus
- IFN
interferon
- IFI15
IFN-induced protein 15
- IFIT1
interferon-induced protein with tetratricopeptide repeats 1
- IFNAR
interferon-α/β receptor
- IP10
interferon-α-inducible protein 10
- IRF
interferon regulatory factor
- ISG
interferon-induced gene
- ISGF
interferon-stimulated gene factor
- ISRE
interferon-stimulated response element
- LMP
large multifunctional protease
- MCP-1
membrane cofactor protein 1
- MIP-1α
macrophage inflammatory protein 1-α
- MX1
myxovirus resistance 1
- OAS
2,5-oligoadenylate synthetase
- PBMC
peripheral blood mononuclear cell
- PIAS
protein inhibitor of activated STAT
- PP2A
protein phosphatase 2A
- RR
rapid responders
- SHP1
Src homology region 2-domain phosphatase 1
- SOCS
suppressor of cytokine signaling
- SR
slow responders
- STAT
signal transducers and activators of transcription
Footnotes
Supported in part by the Intramural Research Program of the National Institute of Diabetes and Digestive and Kidney Diseases, NIH, and under contract N01-HB-27091 of NHLBI.
References
- 1.Liang TJ, Rehermann B, Seeff LB, Hoofnagle JH. Pathogenesis, natural history, treatment, and prevention of hepatitis C. Ann Intern Med. 2000;132:296–305. doi: 10.7326/0003-4819-132-4-200002150-00008. [DOI] [PubMed] [Google Scholar]
- 2.Feld JJ, Hoofnagle JH. Mechanism of action of interferon and ribavirin in treatment of hepatitis C. Nature. 2005;436:967–972. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 3.Tan SL, He Y, Huang Y, Gale M., Jr Strategies for hepatitis C therapeutic intervention: now and next. Curr Opin Pharmacol. 2004;4:465–470. doi: 10.1016/j.coph.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 4.Ank N, West H, Paludan SR. IFN-λ: novel antiviral cytokines. J Interferon Cytokine Res. 2006;26:373–379. doi: 10.1089/jir.2006.26.373. [DOI] [PubMed] [Google Scholar]
- 5.Blatt LM, Davis JM, Klein SB, Taylor MW. The biologic activity and molecular characterization of a novel synthetic interferon-α species, consensus interferon. J Interferon Cytokine Res. 1996;16:489–499. doi: 10.1089/jir.1996.16.489. [DOI] [PubMed] [Google Scholar]
- 6.Barbaro G, Barbarini G. Consensus interferon for chronic hepatitis C patients with genotype 1 who failed to respond to, or relapsed after, interferon α-2b and ribavirin in combination: an Italian pilot study. Eur J Gastroenterol Hepatol. 2002;14:477–483. doi: 10.1097/00042737-200205000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Melian EB, Plosker GL. Interferon alfacon-1: a review of its pharmacology and therapeutic efficacy in the treatment of chronic hepatitis C. Drugs. 2001;61:1661–1691. doi: 10.2165/00003495-200161110-00009. [DOI] [PubMed] [Google Scholar]
- 8.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 9.Frese M, Schwarzle V, Barth K, Krieger N, Lohmann V, Mihm S, Haller O, Bartenschlager R. Interferon-γ inhibits replication of subgenomic and genomic hepatitis C virus RNAs. Hepatology. 2002;35:694–703. doi: 10.1053/jhep.2002.31770. [DOI] [PubMed] [Google Scholar]
- 10.Guo JT, Bichko VV, Seeger C. Effect of α interferon on the hepatitis C virus replicon. J Virol. 2001;75:8516–8523. doi: 10.1128/JVI.75.18.8516-8523.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larkin J, Jin L, Farmen M, Venable D, Huang Y, Tan SL, Glass JI. Synergistic antiviral activity of human interferon combinations in the hepatitis C virus replicon system. J Interferon Cytokine Res. 2003;23:247–257. doi: 10.1089/107999003321829962. [DOI] [PubMed] [Google Scholar]
- 12.Tan H, Derrick J, Hong J, Sanda C, Grosse WM, Edenberg HJ, Taylor M, Seiwert S, Blatt LM. Global transcriptional profiling demonstrates the combination of type I and type II interferon enhances antiviral and immune responses at clinically relevant doses. J Interferon Cytokine Res. 2005;25:632–649. doi: 10.1089/jir.2005.25.632. [DOI] [PubMed] [Google Scholar]
- 13.Soza A, Heller T, Ghany M, Lutchman G, Jake Liang T, Germain J, Hsu HH, Park Y, Hoofnagle JH. Pilot study of interferon γ for chronic hepatitis C. J Hepatol. 2005;43:67–71. doi: 10.1016/j.jhep.2005.02.023. [DOI] [PubMed] [Google Scholar]
- 14.Gale M, Jr, Foy EM. Evasion of intracellular host defence by hepatitis C virus. Nature. 2005;436:939–945. doi: 10.1038/nature04078. [DOI] [PubMed] [Google Scholar]
- 15.Bode JG, Ludwig S, Ehrhardt C, Albrecht U, Erhardt A, Schaper F, Heinrich PC, Haussinger D. IFN-α antagonistic activity of HCV core protein involves induction of suppressor of cytokine signaling-3. FASEB J. 2003;17:488–490. doi: 10.1096/fj.02-0664fje. [DOI] [PubMed] [Google Scholar]
- 16.Duong FH, Filipowicz M, Tripodi M, La Monica N, Heim MH. Hepatitis C virus inhibits interferon signaling through up-regulation of protein phosphatase 2A. Gastroenterology. 2004;126:263–277. doi: 10.1053/j.gastro.2003.10.076. [DOI] [PubMed] [Google Scholar]
- 17.Thomson M, Nascimbeni M, Gonzales S, Murthy KK, Rehermann B, Liang TJ. Emergence of a distinct pattern of viral mutations in chimpanzees infected with a homogeneous inoculum of hepatitis C virus. Gastroenterology. 2001;121:1226–1233. doi: 10.1053/gast.2001.28669. [DOI] [PubMed] [Google Scholar]
- 18.Major ME, Dahari H, Mihalik K, Puig M, Rice CM, Neumann AU, Feinstone SM. Hepatitis C virus kinetics and host responses associated with disease and outcome of infection in chimpanzees. Hepatology. 2004;39:1709–1720. doi: 10.1002/hep.20239. [DOI] [PubMed] [Google Scholar]
- 19.Logvinoff C, Major ME, Oldach D, Heyward S, Talal A, Balfe P, Feinstone SM, Alter H, Rice CM, McKeating JA. Neutralizing antibody response during acute and chronic hepatitis C virus infection. Proc Natl Acad Sci U S A. 2004;101:10149–10154. doi: 10.1073/pnas.0403519101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feld JJ, Nanda S, Susan P, Schweigler L, Theodore D, Dougherty K, Zacks S, Shrestha R, Liang TJ, Fried MW. Hepatic gene expression profiles during treatment with peginterferon and ribavirin: identifying important molecular pathways for treatment response. Hepatology. 2006;44:315A. doi: 10.1002/hep.21853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Y, Chen XC, Konduri M, Fomina N, Lu J, Jin L, Kolykhalov A, Tan SL. Mechanistic link between the anti-HCV effect of interferon gamma and control of viral replication by a Ras-MAPK signaling cascade. Hepatology. 2006;43:81–90. doi: 10.1002/hep.21011. [DOI] [PubMed] [Google Scholar]
- 22.Der SD, Zhou A, Williams BR, Silverman RH. Identification of genes differentially regulated by interferon α, β, or γ using oligonucleotide arrays. Proc Natl Acad Sci U S A. 1998;95:15623–15628. doi: 10.1073/pnas.95.26.15623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Radaeva S, Jaruga B, Hong F, Kim WH, Fan S, Cai H, Strom S, Liu Y, El-Assal O, Gao B. Interferon-α activates multiple STAT signals and down-regulates c-Met in primary human hepatocytes. Gastroenterology. 2002;122:1020–1034. doi: 10.1053/gast.2002.32388. [DOI] [PubMed] [Google Scholar]
- 24.Yasukawa H, Sasaki A, Yoshimura A. Negative regulation of cytokine signaling pathways. Annu Rev Immunol. 2000;18:143–164. doi: 10.1146/annurev.immunol.18.1.143. [DOI] [PubMed] [Google Scholar]
- 25.Berg T, Sarrazin C, Herrmann E, Hinrichsen H, Gerlach T, Zachoval R, Wiedenmann B, Hopf U, Zeuzem S. Prediction of treatment outcome in patients with chronic hepatitis C: significance of baseline parameters and viral dynamics during therapy. Hepatology. 2003;37:600–609. doi: 10.1053/jhep.2003.50106. [DOI] [PubMed] [Google Scholar]
- 26.Su AI, Pezacki JP, Wodicka L, Brideau AD, Supekova L, Thimme R, Wieland S, Bukh J, Purcell RH, Schultz PG, Chisari FV. Genomic analysis of the host response to hepatitis C virus infection. Proc Natl Acad Sci U S A. 2002;99:15669–15674. doi: 10.1073/pnas.202608199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bigger CB, Guerra B, Brasky KM, Hubbard G, Beard MR, Luxon BA, Lemon SM, Lanford RE. Intrahepatic gene expression during chronic hepatitis C virus infection in chimpanzees. J Virol. 2004;78:13779–13792. doi: 10.1128/JVI.78.24.13779-13792.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein SB, Blatt LM, Taylor MW. Cell surface binding characteristics correlate with consensus type I interferon enhanced activity. J Interferon Cytokine Res. 1996;16:1–6. doi: 10.1089/jir.1996.16.1. [DOI] [PubMed] [Google Scholar]
- 29.Lanford RE, Guerra B, Lee H, Chavez D, Brasky KM, Bigger CB. Genomic response to interferon-α in chimpanzees: implications of rapid down-regulation for hepatitis C kinetics. Hepatology. 2006;43:961–972. doi: 10.1002/hep.21167. [DOI] [PubMed] [Google Scholar]
- 30.Helbig KJ, Lau DT, Semendric L, Harley HA, Beard MR. Analysis of ISG expression in chronic hepatitis C identifies viperin as a potential antiviral effector. Hepatology. 2005;42:702–710. doi: 10.1002/hep.20844. [DOI] [PubMed] [Google Scholar]
- 31.He XS, Ji X, Hale MB, Cheung R, Ahmed A, Guo Y, Nolan GP, Pfeffer LM, Wright TL, Risch N, Tibshirani R, Greenberg HB. Global transcriptional response to interferon is a determinant of HCV treatment outcome and is modified by race. Hepatology. 2006;44:352–359. doi: 10.1002/hep.21267. [DOI] [PubMed] [Google Scholar]
- 32.Zhu H, Nelson DR, Crawford JM, Liu C. Defective Jak-Stat activation in hepatoma cells is associated with hepatitis C viral IFN-α resistance. J Interferon Cytokine Res. 2005;25:528–539. doi: 10.1089/jir.2005.25.528. [DOI] [PubMed] [Google Scholar]
- 33.Vento S, Garofano T, Renzini C, Cainelli F, Casali F, Ghironzi G, Ferraro T, Concia E. Fulminant hepatitis associated with hepatitis A virus superinfection in patients with chronic hepatitis C. N Engl J Med. 1998;338:286–290. doi: 10.1056/NEJM199801293380503. [DOI] [PubMed] [Google Scholar]
- 34.Cacciola I, Pollicino T, Squadrito G, Cerenzia G, Orlando ME, Raimondo G. Occult hepatitis B virus infection in patients with chronic hepatitis C liver disease. N Engl J Med. 1999;341:22–26. doi: 10.1056/NEJM199907013410104. [DOI] [PubMed] [Google Scholar]
- 35.Yoshimura A, Mori H, Ohishi M, Aki D, Hanada T. Negative regulation of cytokine signaling influences inflammation. Curr Opin Immunol. 2003;15:704–708. doi: 10.1016/j.coi.2003.09.004. [DOI] [PubMed] [Google Scholar]
- 36.Blindenbacher A, Duong FH, Hunziker L, Stutvoet ST, Wang X, Terracciano L, Moradpour D, Blum HE, Alonzi T, Tripodi M, La Monica N, Heim MH. Expression of hepatitis C virus proteins inhibits interferon alpha signaling in the liver of transgenic mice. Gastroenterology. 2003;124:1465–1475. doi: 10.1016/s0016-5085(03)00290-7. [DOI] [PubMed] [Google Scholar]
- 37.Kawaguchi T, Yoshida T, Harada M, Hisamoto T, Nagao Y, Ide T, Taniguchi E, Kumemura H, Hanada S, Maeyama M, Baba S, Koga H, Kumashiro R, Ueno T, Ogata H, Yoshimura A, Sata M. Hepatitis C virus down-regulates insulin receptor substrates 1 and 2 through up-regulation of suppressor of cytokine signaling 3. Am J Pathol. 2004;165:1499–1508. doi: 10.1016/S0002-9440(10)63408-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yoshida T, Hanada T, Tokuhisa T, Kosai K, Sata M, Kohara M, Yoshimura A. Activation of STAT3 by the hepatitis C virus core protein leads to cellular transformation. J Exp Med. 2002;196:641–653. doi: 10.1084/jem.20012127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koo BC, McPoland P, Wagoner JP, Kane OJ, Lohmann V, Polyak SJ. Relationships between hepatitis C virus replication and CXCL-8 production in vitro. J Virol. 2006;80:7885–7893. doi: 10.1128/JVI.00519-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polyak SJ, Khabar KS, Rezeiq M, Gretch DR. Elevated levels of interleukin-8 in serum are associated with hepatitis C virus infection and resistance to interferon therapy. J Virol. 2001;75:6209–6211. doi: 10.1128/JVI.75.13.6209-6211.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soo HM, Garzino-Demo A, Hong W, Tan YH, Tan YJ, Goh PY, Lim SG, Lim SP. Expression of a full-length hepatitis C virus cDNA up-regulates the expression of CC chemokines MCP-1 and RANTES. Virology. 2002;303:253–277. doi: 10.1006/viro.2002.1617. [DOI] [PubMed] [Google Scholar]
- 42.Radkowski M, Bednarska A, Horban A, Stanczak J, Wilkinson J, Adair DM, Nowicki M, Rakela J, Laskus T. Infection of primary human macrophages with hepatitis C virus in vitro: induction of tumour necrosis factor-α and interleukin 8. J Gen Virol. 2004;85:47–59. doi: 10.1099/vir.0.19491-0. [DOI] [PubMed] [Google Scholar]
- 43.Basu A, Meyer K, Lai KK, Saito K, Di Bisceglie AM, Grosso LE, Ray RB, Ray R. Microarray analyses and molecular profiling of Stat3 signaling pathway induced by hepatitis C virus core protein in human hepatocytes. Virology. 2006;349:347–358. doi: 10.1016/j.virol.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Borozan I, Feld J, Sun J, Tannis LL, Coltescu C, Heathcote J, Edwards AM, McGilvray ID. Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128:1437–1444. doi: 10.1053/j.gastro.2005.01.059. [DOI] [PubMed] [Google Scholar]