

Abstract
Mass spectrometry has played an integral role in the identification of proteins and their post-translational modifications (PTM). However, analysis of some PTMs, such as phosphorylation, sulfonation, and glycosylation, is difficult with collision-activated dissociation (CAD) since the modification is labile and preferentially lost over peptide backbone fragmentation, resulting in little to no peptide sequence information. The presence of multiple basic residues also makes peptides exceptionally difficult to sequence by conventional CAD mass spectrometry. Here we review the utility of electron transfer dissociation (ETD) mass spectrometry for sequence analysis of post-translationally modified and/or highly basic peptides. Phosphorylated, sulfonated, glycosylated, nitrosylated, disulfide bonded, methylated, acetylated, and highly basic peptides have been analyzed by CAD and ETD mass spectrometry. CAD fragmentation typically produced spectra showing limited peptide backbone fragmentation. However, when these peptides were fragmented using ETD, peptide backbone fragmentation produced a complete or almost complete series of ions and thus extensive peptide sequence information. In addition, labile PTMs remained intact. These examples illustrate the utility of ETD as an advantageous tool in proteomic research by readily identifying peptides resistant to analysis by CAD. A further benefit is the ability to analyze larger, non-tryptic peptides, allowing for the detection of multiple PTMs within the context of one another.
Keywords: electron transfer dissociation (ETD) mass spectrometry, labile post translational modification (PTM), phosphorylation, sulfonation, glycosylation, nitrosylation, disulfide bond, proteomics
Introduction
Transcription of DNA to RNA and subsequent translation of RNA into protein does not necessarily result in a final protein product. A variety of covalent protein modifications can take place during or after protein synthesis. Protein modifications result from the chemical modification of amino acid side chains as well as the amine and carboxy terminus of the protein. Modifications on eukaryotic proteins tend to be widespread. These modifications are needed for normal cellular function, while alterations in the regulation of these modifications can lead to diseases such as Alzheimer Disease (AD) [1], cancer [2] and erectile dysfunction [3].
Protein modifications can enhance/decrease protein activity, allow interaction with other proteins, localize protein to a specific place in the cell, and affect folding. For example, post-translational modifications (PTM) such as phosphorylation, acetylation, and methylation are used as chemical switches to activate/inactive the histone regulation of gene transcription, DNA replication, and DNA damage repair [4]. In the ubiquitin-proteasome pathway, phosphorylation [5, 6] and O-linked N-acetylglucosamine (O-GlcNAc) modifications [7, 8] play a major role in the degradation process of intracellular proteins. Tyrosine sulfonation has been implicated in the mediation of inflammation, leukocyte adhesion, chemokine receptor signaling, and modulation of the blood coagulation cascade. However, O-sulfonation of serine and threonine has only been recently described and implicated in protein assembly and signal transduction [9]. S-nitrosylation is the covalent attachment of a nitrogen monoxide group to the thiol side chain of cysteine. S-nitrosylation is a broad based PTM with roles in cellular signal transduction, regulation of redox state, and regulation of enzyme activity (reviewed in [10]).
Identifying such PTMs is essential as it leads to insights into the function and role of the protein in the biological system. The analysis of proteins and their post-translational modifications is particularly important for the study of many diseases including cancer, diabetes, cardiovascular disease, and neurodegenerative disorders such as Alzheimer Disease.
A common technique to analyze protein(s) by mass spectrometry is to digest the proteins into smaller peptides with an enzyme, separate the peptides by reverse phase chromatography, and introduce the peptides into a mass spectrometer by electrospray ionization (ESI) to obtain sequence information by tandem mass spectrometry (MS/MS). The enzyme typically utilized to digest proteins into smaller peptides is trypsin, which cleaves C-terminal to Arg or Lys. This produces a complex mixture of peptides containing one basic amino acid residue per peptide. These peptides ionize via ESI to low charge states (generally +3 or less, with the +1 and +2 charge states most favored), which are optimal for collision-activated dissociation (CAD) mass spectrometry.
Low-energy CAD tandem mass spectrometry has been, by far, the most common method used to dissociate peptide ions for subsequent sequence analysis. Typically, the preferred sites of cleavage, in a gas-phase peptide ion, are the amide bonds of the peptide backbone. Upon collisional activation, the amide bond of the peptide backbone will fragment to produce, ideally, a homologous series of b and y-type fragment ions (Figure 1) (please refer to [11, 12] for a more detailed description of the mechanism of peptide fragmentation). Successful peptide sequence analysis, regardless of the dissociation method, requires the production of fragment ions corresponding to a complete or near-complete set of the possible backbone bond cleavages of the precursor.
Figure 1.
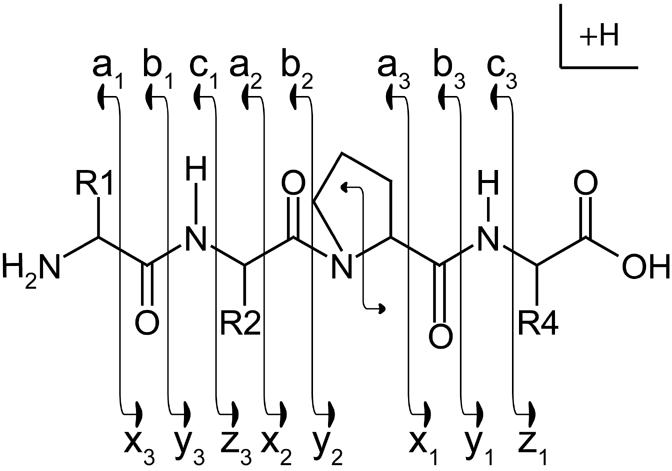
Roepstorff Nomenclature Scheme
Illustration of fragment ions formed from the backbone cleavage of protonated peptides. Fragment ions retaining the positive charge on the amino terminus are termed a-, b-, or c-type ions. Fragment ions retaining the positive charge on the carboxy terminus are termed x-, y-, or z-type ions.
Depending on the protein sequence, sometimes trypsin can generate peptides that are too small or too large for analysis. Missed cleavages due to post translational modifications or other effects, can generate larger tryptic peptides containing more than one basic amino acid. The presence of multiple basic residues prevents random protonation along the peptide backbone and upon collisional activation, directs the backbone bond dissociation to specific sites and therefore often inhibits the production of a sufficiently diverse set of sequence ions to enable sequence analysis. In this regard, CAD is most effective for short, low-charged (tryptic) peptides. This widely used approach to identify proteins in order to understand their biological function limits investigators to the analysis of small peptides (approximately 20 residues or less). This can also prevent the detection of multiple PTMs within the context of one another and the understanding of the interaction of various PTMs with each other.
While CAD MS/MS is a very powerful technique, it does impose some limitations on investigators in their efforts to identify proteins and understand their biological function. Due to splice variation during transcription and post translational modifications, proteins often are present in biological systems in multiple forms and these different forms have different biological functions or activity. The biological activity and function of any protein is substantially determined by its sequence and structure including the aggregate of its PTMs. The functional effect of particular PTM at a particular amino acid may well depend on the type, presence or absence of other PTMs at other relatively remote sites on the protein. Digestion of proteins into peptides inherently makes it problematic to observe correlated PTMs that are not proximate when more than one variant of the protein is in the original sample. PTMs may only be jointly observed when they occur in close enough proximity such that there is not an intervening site for enzymatic cleavage. Otherwise, the PTMs will be retained on different peptides and thus cannot be associated as jointly belonging to any particular variant of the protein. The peptide type and size limitations (tryptic peptides of less than ∼20 residues) effectively imposed by using CAD generally allows only the type and sites of modifications to be identified without much information regarding the relationship between them. Only modifications very closely located to each other can be observed simultaneously.
Further, chemical modifications (or PTMs) of certain residues of the peptide can alter, or, redirect the sites of preferred cleavage during CAD (often cleaving the modifying moiety, leaving the peptide backbone intact). For example, peptide ions containing phosphorylated serine (Ser) or threonine (Thr) residues, the phosphate group competes with the peptide backbone as the preferred site of cleavage. Collisional-activation of these peptides readily displaces phosphoric acid from the peptide, and frequently results in the peptide’s backbone bonds remaining intact. The resultant spectra, because of the preferential loss of the labile PTM over peptide backbone cleavage, tend to contain minimal peptide sequence information and may not allow for unambiguous peptide sequence assignment. The yield of such product ions with the phosphoric acid moiety intact can be very low.
While phosphorylation of serine and threonine residues is arguably the most well-known and widely documented of "MS/MS labile" chemical modifications, other PTMs such as glycosylation [13], sulfonation [13], and nitrosylation [14] are also firmly seated in this category . Loss of these labile PTMs during CAD tandem mass spectrometry, regardless of the mechanism that defines these preferential cleavages or of the specifics of the CAD implementation, results in product ion spectra that are frequently devoid of consecutive peptide backbone cleavage product ions that retain the labile PTM moiety. Since sequential peptide backbone fragments are necessary if peptide sequence identification is to be successfully performed, thus results in reduced success rates in sequence determination.
Even with this limitation, the methodology of proteolytic digestion followed by CAD MS/MS has successfully provided sequence identifications (including PTM locations) of thousands of peptides with labile PTMs. Many thousands of peptides with labile PTMs have had enough sequence information in their CAD spectrum to determine their amino acid sequence and location of PTM. For example, 2,000 phosphorylated peptides were identified from the nuclear fraction of HeLa cells [15]. However, it has been estimated that >100,000 potential phosphorylation sites occur in the human proteome, and only a few thousand are currently known [16].
An alternative method for peptide dissociation (electron capture dissociation, ECD) was introduced by McLafferty and co-workers eight years ago [17]. In that work, low energy electrons were reacted with peptide cations in the magnetic field of a Fourier transform ion cyclotron resonance mass spectrometer (FT-ICR-MS). The reaction resulted in the attachment of electrons to the protonated peptides producing peptide cations containing an additional electron. The odd-electron peptide then undergoes rearrangement with subsequent dissociation. Peptide backbone cleavage by ECD is relatively indifferent to either peptide sequence or length. Unlike CAD, ECD does not cleave chemical modifications from the peptide, but rather induces random breakage of the peptide backbone. With ECD, labile modifications such as phosphorylation [18, 19], N- and O-glycosylation [20, 21], sulfonation [22], as well as other labile PTMs remain intact. However, ECD requires an FT-ICR mass spectrometer, instruments that are not readily available to many in the proteomics field.
Electron transfer dissociation (ETD) is a new method to fragment peptides that utilizes ion/ion chemistry [12, 23-25]. ETD fragments peptides by transferring an electron from a radical anion to a protonated peptide. This induces fragmentation of the peptide backbone, causing cleavage of the Cα-N bond (Figure 2) just as ECD does. This creates complementary c and z-type ions instead of the typical b and y-type ions observed in CAD (Figure 1). Although the exact mechanism of ECD and ETD are debated, ETD preserves PTMs that are labile by CAD and sequence information on the peptide can be obtained. ETD uses a RF quadrupole ion trapping device instead of an FT-ICR-MS for ion trapping and detection [12]. RF ion trap mass spectrometers are low-cost, low-maintenance, and widely accessible as compared to the FT-ICR-MS. In this review, we illustrate that ETD fragmentation produces many more product ions amenable to peptide interpretation than the CAD fragmentation methodology.
Figure 2.

ETD fragmentation scheme
Fragmentation scheme of a multiply protonated peptide after reaction with a low energy electron to produce c- and z-type ions [12] Copyright (2004) National Academy of Sciences, USA. Reprinted with permission.
Phosphorylation
Phosphorylation regulates many critical cellular processes including cell cycle progression, signal transduction, cell migration, gene expression, and apoptosis [26-28]. Phosphate is added in an ATP dependent manner to the side chain of Ser, Thr, or Tyr amino acids in proteins via protein kinases. The most common method to identify these sites of phosphorylation is by tandem mass spectrometry. As described above, when phosphorylated peptides are fragmented by CAD, a single peak corresponding to the loss of phosphoric acid (H3PO4 - loss of 98 Da) from the parent mass is what is typically observed (Reviewed in [29]).
More than a thousand phosphorylated peptides have been analyzed in our laboratory from a variety of samples by both CAD and ETD on an ion trap mass spectrometer ([12, 23, 30], manuscripts in preparation, and unpublished data). Many have demonstrated that phosphorylated peptides primarily show a strong neutral loss of phosphoric acid by CAD. However, we observe that phosphorylated peptides fragment completely along the peptide backbone by ETD. As an example, the peptide ERpSLpSRER from an immobilized metal affinity chromatography (IMAC) enriched tryptic digest of human nuclear proteins analyzed by CAD and ETD during a data-dependent nHPLC-μESI-MS/MS analysis is shown in Figure 3 [12].
Figure 3.

Comparison of CAD vs. ETD spectrum of a phosphorylated peptide
Consecutive single-scan CAD vs. ETD mass spectrum comparison of phosphorylated peptides generated from a tryptic digest of human nuclear proteins recorded during a data-dependent analysis (nHPLC-μESI-MS/MS). All peptides were converted to methyl esters and enriched for phosphorylated peptides by immobilized metal affinity chromatography before analysis. (A) CAD spectrum dominated by fragment ions corresponding to the loss of phosphoric acid and either methanol or water. (B) ETD spectrum containing a near complete series of c- and z-type product ions. Note that the spectrum is devoid of fragment ions corresponding to the loss of phosphoric acid [12] Copyright (2004) National Academy of Sciences, USA. Reprinted with permission.
The CAD spectrum of (M+3H)+3 at m/z 412 of this doubly phosphorylated peptide is shown in Figure 3A. The majority of the signal in this CAD spectrum corresponds to the loss of one and two phosphoric acid molecules. Additional losses of water or methanol are also seen. Minimal peptide backbone fragmentation is observed. Conversely, the ETD spectrum of the same peptide, shown in Figure 3B, reveals peptide backbone cleavage generating a near complete c and z-type ion series which is more than sufficient to identify the peptide. In the ETD spectrum, no loss of phosphoric acid is observed.
Sulfonation
O-sulfonation has been implicated in protein assembly and signal transduction [9] and is an important PTM to study as little is known about it. Analysis of sulfonated peptides by CAD often results in the loss of SO3 [9]. An example of a sulfonated peptide, GRLGsSRAGR, (Mikesh et al., unpublished data) fragmented by CAD is shown in Figure 4A. The spectrum contains one major ion at m/z 310 corresponding to the neutral loss of SO3 from the (M+3H)+3 precursor ion at m/z 337. As seen in the figure, no other product ions are detectable in the spectrum. When the spectrum is magnified by a factor of 50, the only observable product ion is the loss of NH3 from m/z 310 and possibly y +27 as shown in Figure 4B. When GRLGsSRAGR was fragmented by ETD, a complete c and z-type ion series was observed with no detectable loss of SO3 from the precursor or product ions (Figure 4C). The peptide MGPRRRsSRKPE with carboxylic acids converted to methyl esters, shows similar behavior (data not shown). To differentiate the isobaric modifications, sulfonation from phosphorylation, our ETD modified LTQ can perform sequential CAD-ETD analysis. These two modifications can usually be differentiated the by their different CAD neutral loss patterns (80 amu vs. 98 amu).
Figure 4.
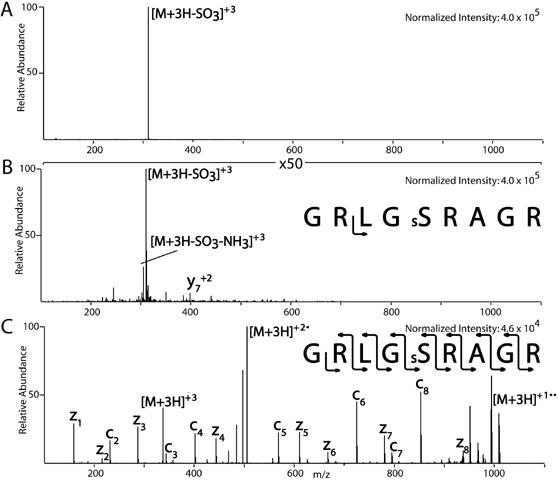
Comparison of CAD vs. ETD spectrum of a sulfonated peptide
Sulfonation of peptides was achieved by reacting the peptide with 5% chlorosulfonic acid in neat trifluoroacetic acid (TFA) for 20 minutes at room temperature. The reaction was terminated by the addition of water and was purified by RP-HPLC. Mass spectrometry analysis before and after sulfonation confirmed reaction. Sulfonated peptides (1pmol/ul) were infused at a flow rate of 60 nl/min into a ThermoElectron LTQ ion trap mass spectrometer modified to perform ETD. (A) The CAD spectrum of the illustrated sulfonated peptide contains one major ion corresponding to the neutral loss of SO3 from the (M+3H)+3 precursor ion. (B) Magnification of the spectrum shown in A by 50X. (C) Fragmentation of the sulfonated peptide by ETD where a complete c and z-type ion series was observed with no detectable loss of SO3 from the precursor ion or the peptide backbone.
O- and N-linked Glycosylation
Addition of a single O-GlcNAc moiety to serine or threonine residues is proposed to act as a dynamic modification reciprocal to phosphorylation. O-GlcNAc is thought to regulate protein stability, subcellular localization and protein-protein interactions as well as modify transcription factors [31]. However, currently, only 80-100 O-GlcNAc modified proteins have been identified [32, 33]. This may be due to the labile nature of the O-GlcNAc modification and the limitations of the analytical methodologies currently available. Improved methodologies for the identification of O-linked glycosylation, such as ETD, would facilitate a more global understanding of the regulatory role of this PTM.
We demonstrate that O-linked glycosylation is retained by ETD during fragmentation (Mikesh et al., unpublished data). Using synthetic O-GlcNAc modified peptides (gift from Gerald Hart, Johns Hopkins University) such as, KKFELLPgTPPLSPSRR, the (M+4H)+4 ion at m/z 518 was dissociated. In Figure 5A, the CAD spectrum from the O-GlcNAc modified peptide shows ions at m/z 204, which corresponds to the O-GlcNAc oxonium ion, and the corresponding charge reduced (M+3H)+3 product ion with a loss of 203 amu (GlcNAc) at m/z 623. The rest of the CAD spectrum is composed of a total of three b and y-type ions (b6 +2, b14 +2, and y10 +2), none of which are more than 20% of the abundance of the (M+3H-GlcNAc)+3 product ion. In the ETD spectrum of KKFELLPgTPPLSPSRR shown in Figure 5B, an almost complete c and z-type ion series for the peptide is observed. The only c and z-type ions not observed corresponds to bonds adjacent to a proline residue. In ETD, because the amine bond is broken corresponding to c and z-type ions, this peptide is still intact due to the N-C ring structure of proline (see Figure 1). In the ETD spectrum of this synthetic peptide, there is no observable loss of GlcNAc.
Figure 5.
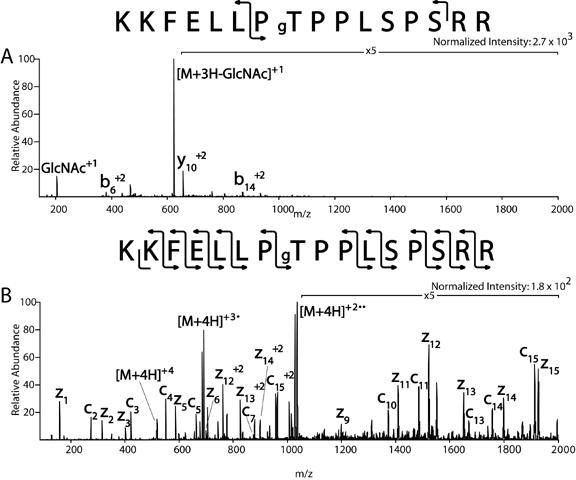
Comparison of CAD vs. ETD spectrum of O-GlcNAc containing peptide
O-GlcNAc containing peptides (1pmol/ul) donated by Gerry Hart were infused at a flow rate of 60 nl/min into a ThermoElectron LTQ ion trap mass spectrometer modified to perform ETD. (A) The CAD spectrum of a synthetic O-GlcNAc modified peptide illustrates the loss of an O-GlcNAc oxoniom ion at m/z 204 and the corresponding charge reduced (M+3H)+3 product ion with a loss of 203 (GlcNAc). (B) The ETD spectrum of the O-GlcNAc modified peptide illustrates an almost complete c and z-type ion series. The only c and z-type ions not observed corresponds to bonds adjacent to a proline residue. In the ETD spectrum of this synthetic peptide, there is no observable loss of GlcNAc.
In a study to identify sites of protein post translational modifications in paxillin, ETD was used to identify a site of glycosylation from a tryptic digest of this protein. The ETD spectrum from a 55 amino acid tryptic peptide provided the necessary fragment ions to assign Ser 74 of paxillin as being modified by 203 amu, corresponding to the mass of GlcNAc [34]. The CAD spectrum of this peptide produced highly charged fragment ions with a loss of 203 amu. Neither the site of modification nor the peptide sequence could be readily identified by CAD.
In addition to O-linked glycosylation, N-linked glycosylation is an important and common post translational modification. N-glycosylation is the attachment of polysaccharide chains to the amide nitrogen of the asparagine side chain. This modification is typically found in membrane and secreted proteins and can be required for proper protein folding. Glycosylation may also be involved in cell adhesion, embryonic development, immune function, and cell division. N-linked glycopeptide analysis using typical low-energy CAD is difficult as it generally yields only glycan, but not peptide, structural information. The observation of oxonium ions such as m/z 204 (HexNAc), 163 (Hexose), 292 (sialic acid), and 366 (Hexose-HexNAc) can identify these peptides as glycopeptides (for a more detailed review on the analysis of glycoproteins by mass spectrometry see [35]).
Hogan et al. reports the application of ETD on a 3D ion trap to an N-linked glycopeptide from a fractionated tryptic digest of the Erythina cristagalli (coral tree) lectin. The average mass of the corresponding glycopeptide is 3002 Da with the following known glycan structure Manα3(Manα6)(Xylβ2)Manβ4GlcNAcβ4(Fucα3)GlcNAc [36] (Figure 6). The CAD spectrum for this triply charged glycopeptide ion contains information about the glycan structure, however, there is no fragmentation of the peptide backbone (Figure 6A). In contrast, the ETD spectrum of this glycopeptide shows multiple z-type ions corresponding to the dissociation of the peptide backbone (Figure 6B). Again, no loss of the glycan structure was observed (glycan structure fragments in the spectrum are thought to arise during resonance ejection of the triply and doubly charged ions) [37]. Although a near complete z-type ion series was observed, the complementary c-type ions normally produced by ETD are missing from this spectrum. The authors note this may be a characteristic of the glycopeptide selected for study or due to the structural nature of the large sugar moiety [37]. Others have also reported that gas-phase protein conformation can affect the generation/observation of fragment ions using ECD [38].
Figure 6.
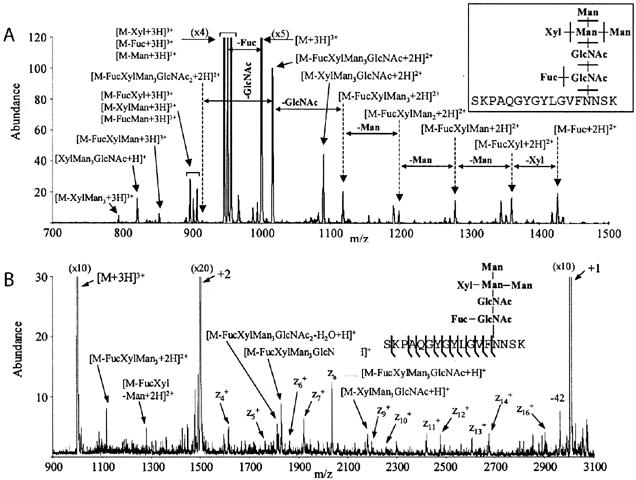
Comparison of CAD vs. ETD spectrum of an N-linked glycosylated peptide
Spectrum of the N-linked glycopeptide generated from a tryptic digest of the Erythina cristagalli (coral tree) lectin. The average mass of the corresponding glycopeptide is 3002 Da with the following N-linked glycan structure Manα3(Manα6)(Xylβ2)Manβ4GlcNAcβ4(Fucα3)GlcNAc. (A) CAD spectrum of the (M+3H)+3 glycopeptide ion. Note fragmentation is of the glycan structure. (B) ETD spectrum of the (M+3H)+3 glycopeptide. Sulfur dioxide was used as the anion. Note that most fragmentation occurs along the peptide backbone [37] Copyright (2005) American Chemical Society. Reprinted with permission.
Nitrosylation
Nitrosylation is a very labile PTM, making analysis difficult [14, 39]. We analyzed nitrosylated bovine insulin beta chain as a model of this type of PTM (Mikesh et al., unpublished data). The majority of the signal in the CAD spectrum of the (M+5H)+5 of FVNQHLnCGSHLVEALYLVnCGERGFFYTPKA corresponds to the neutral loss of both NO groups on the cysteine residues (M+5H-2NO)+5. Minimal peptide backbone fragmentation is obtained as only a few product ions are observed above 5% relative abundance (y y +2, -NO b +2 +2 13 , -NO b +2 16 , 17-NO , and b24-NO ) (Figure 7A). In the ETD spectrum of the same peptide, the following charge reduced (electron transfer without fragmentation) species with and without losses of NO are observed: (M+4H-NO)+4(may also be z ), (M+3H)+3, (M+3H-NO)+3, (M+3H-2NO)+3, (M+2H)+27 ,(M+2H-NO)+2, and (M+2H-2NO)+2. The loss of NO from the charged reduced species may be acting as its own proton transfer reagent directing mostly charge reduction of the nitrosylated insulin as opposed to fragmentation. However, 6-7 low level (2% or less of the largest ion in the spectrum) c and z-type ions are observed (Figure 7B). Three of these c-type ions demonstrate the retention of NO on the insulin product ions after ETD.
Figure 7.
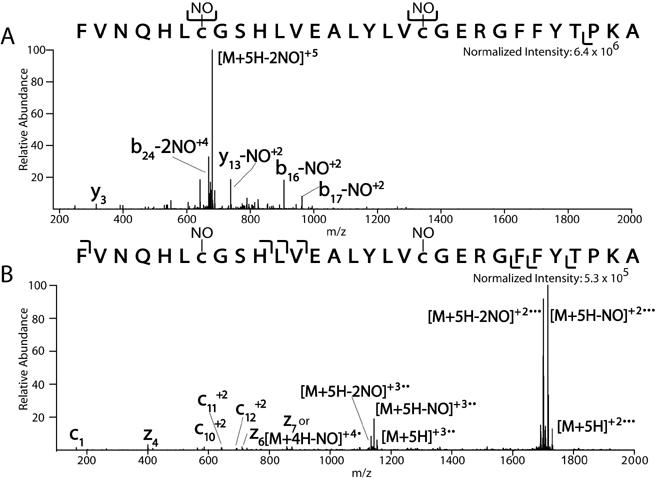
Comparison of CAD vs. ETD spectrum of a nitrosylated peptide
The beta chain of insulin containing two cysteine residues was S-nitrosylated using S-Nitroso-N-acetylpenicillamine (SNAP). The resulting nitrosylated polypeptide was analyzed by nanoflow RP-HPLC interfaced to a ThermoElectron LTQ ion trap mass spectrometer modified to perform ETD. (A) CAD spectrum of the (M+5H)+5 nitrosylated insulin beta chain is mostly composed of the neutral loss of both NO groups on the cysteine residues of insulin. Minimal peptide backbone fragmentation is observed. (B) ETD spectrum of nitrosylated insulin beta chain illustrates a charge reduced (electron transfer without fragmentation) species with and without losses of NO. However, a few low level c and z-type ions demonstrate the retention of NO on some product ions after ETD.
Disulfide Linkage
Another common post-translational modification important to protein folding, structure, and function, is the disulfide linkage of two cysteine residues in proteins/peptides. Disulfide bonds are not typically fragmented by CAD [40], but it has been previously shown that disulfide bonds can be broken by ECD [41] and ETD [42]. In Chrisman et al., two polypeptide chains held together by disulfide bonds were analyzed by ETD in a three-dimensional quadrupole ion trap mass spectrometer using SO -2· as the reagent anion. When the intra chain disulfide containing peptide, Arg8-conopressin G (Cys-Phe-Ile-Arg-Asn-Cys-Pro-Arg) is digested with trypsin, it produces an alpha chain composed of the first half of the peptide, Cys-Phe-Ile-Arg now linked by an inter chain disulfide bond to the beta chain composed of the second half of the peptide, Asn-Cys-Pro-Arg. The major ETD products from this trypsin digested peptide are the alpha and beta chain product ions resulting from the cleavage of the disulfide bond. Although there are c- and z-type ions resulting in the cleavage of the peptide backbone as well, the disulfide bond appears to be cleaved preferentially over the peptide backbone [42]. With disulfide bonded peptides, it is also suspected that noncovalent bonding between the chains may hold the two peptide chains together after disulfide bond cleavage [43].
Modified Peptides Containing Multiple Basic Residues
In addition to labile PTMs, ETD is useful for sequence analysis of peptides with multiple basic residues such as the N-terminal tail (approximately amino acids 1-50) of histones (involved in chromatin mediated processes, i.e. gene regulation), small nuclear ribonucleoprotein polypeptides (SMD2 or SMD3) (involved in RNA binding), or certain peptides associated with the major histocompatibility complex (related to immune system recognition/function).
For example, PTMs on histones are difficult to analyze by CAD tandem mass spectrometry for a variety of reasons. The N-terminal tail of histones contain 30% basic residues. Because of the abundant number of Arg and Lys residues in histones, most of the peptide fragments generated from a trypsin digest would be too small for analysis. An advantage of ETD is that it is not restricted to low charged peptides generated from trypsin digests. With ETD, a variety of enzymes that produce potentially longer peptides with higher charge states can be utilized to digest biological samples.
As an example of the fragmentation pattern of modified peptides containing multiple basic amino acids, synthetic peptides from histone H3, TARKSTGGKAPRKQL, (amino acid 6-20 or 6-19) with a variety of PTMs were analyzed (Mikesh et al., unpublished data) (Table 1). Results from one of the histone H3 peptides is illustrated in Figure 8, however, results are typical for all histone peptides analyzed, independent of their modification status. The CAD spectrum from the (M+4H)+4 molecule from histone H3 (6-20), TAR(acK)STGGKAPRKQL, is illustrated in Figure 8A. The spectrum consists of a product ion corresponding to the neutral loss of water (M+4H-2H O)+42 . Limited peptide backbone fragmentation is obtained as only nine product ions are observed (b5, b +3, y , y , y +2, y +310 , y +2, y +3 +2 14 5 6 9 11 13 , and a5-NH3 ). In the corresponding ETD spectra, an almost complete c and z-type ion series for the peptides are observed (Figure 8B). The only fragment ions not observed are those N-terminal to proline (due to the ring structure of proline), z-type ions where there is no amino acid residue to be protonated at the C-terminus (z1), and c1.
Table 1.
| Sequence of Modified Human Histone H3 Synthetic Peptides (6-20) or (6-19) |
|---|
| TAR(acK)STGGKAPRKQL |
| TAR(acK)STGG(triK)APRKQL |
| TAR(diK)STGG(triK)APRKQL |
| TAR(triK)pSTGGKAPRKQL |
| TAR(diK)pSTGG(acK)APRKQ |
tri=trimethylation
di=dimethylation
ac=acetylation
p=phosphorylated
bold indicates data shown (Figure 8)
Figure 8.
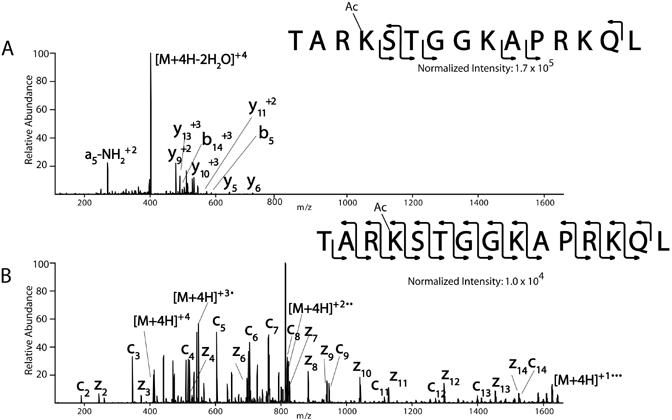
Comparison of CAD vs. ETD spectrum of histone H3 peptides
Human histone H3 peptides (amino acids 6-20 or 6-19) were synthesized on an AMS 422 multiple peptide synthesizer (Gilson Medical Electronics, Middletown, WI) by standard solid-phase 9-fluorenylmethoxycarbonyl (Fmoc) chemistry using Wang resins. Novabiochem Fmoc protected amino acids, modified amino acids (phosphorylated, acetylated, and methylated), and resins were used for peptide synthesis. Synthetic histone peptides (1pmol/ul) were infused at a flow rate of 60 nl/min into a ThermoElectron LTQ ion trap mass spectrometer modified to perform ETD. (A) The CAD spectrum from histone H3 (6-20), TAR(acK)STGGKAPRKQL, is illustrated. The spectrum consists of a product ion corresponding to the neutral loss of water with minimal peptide backbone fragmentation. (B) The corresponding ETD spectrum illustrates an almost complete c and z-type ion series for the peptide. The only fragment ions not observed are those N-terminal to proline (due to the ring structure of proline), z-type ions where there is no amino acid residue to be protonated at the C-terminus (z1), and c1.
Larger peptides/proteins
In addition, we have previously shown ETD can be utilized to fragment peptides larger than 20 amino acids as well as whole proteins [30]. In this type of experiment, proteins were gradient eluted into the mass spectrometer where they are isolated and reacted with the radical anion, fluoranthene. Next, the resulting highly charged product ions are reacted with even electron anions of benzoic acid to reduce the various charge states of the c and z-type product ions into predominantly singly charged species, simplifying the spectrum. Proton transfer reduction (PTR) along with ETD is needed to reduce the complexity of the spectra generated from these highly charged (z>10) species. Electron transfer dissociation followed by proton transfer of larger peptides and proteins typically only generate the first 15-40 amino acids at both the N- and C-termini, constrained by the m/z range of the mass spectrometer. The hybridization of ETD capable linear ion traps to other types of mass spectrometers is expected to relieve this m/z restriction. Nonetheless, information from current ETD mass spectrometers is more than sufficient to identify the protein. Shown in Figure 9, is the tandem mass spectrum of ubiquitin (8.5 kDa) generated by sequential ion/ion reactions. Shown in Figure 9A is the ETD spectrum of ubiquitin with several hundred highly charged c and z-type ions. In Figure 9B-9D are the sequential PTR reactions of these product ions for 50ms, 100ms, and 150ms respectively. Illustrated by these figures is the gradual reduction in charge state of the product ions to predominately the doubly and singly charge state (Figure 9D-150ms). This makes spectral interpretation easier and allows for the identification of c and z-type ions corresponding to almost the entire N-terminus (17 amino acids) and C-terminus (16 amino acids). In addition to ubiquitin, cytochrome C (∼12 kDa), several histones (∼13 kDa), and albumin (66 kDa) have also been analyzed by ETD followed by PTR on a chromatographic time scale [30].
Figure 9.
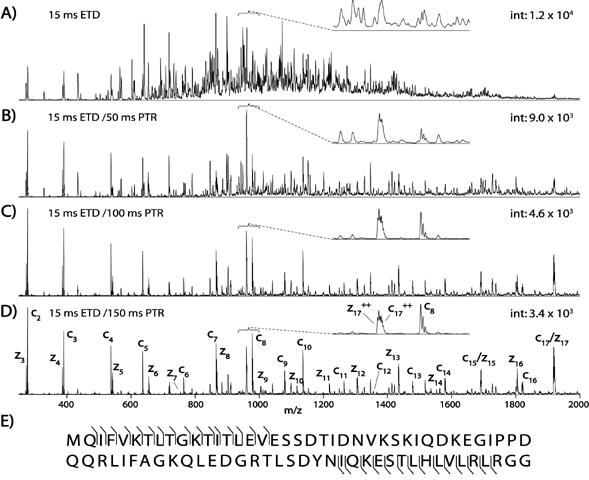
Tandem mass spectrum of ubiquitin generated by sequential ion/ion reactions
(A) Spectrum illustrating whole protein dissociation of ubiquitin, a 8.5 kDa protein (+13, m/z 659), after a 15-ms reaction with the radical anion of fluoranthene. Several hundred highly charged unresolved c- and z-type fragment ions dominate the spectrum. (B-D) Sequestering the entire mixture of highly charged product ions and reacting them with a second anion, deprotonated benzoic acid, simplified this mixture. Note the gradual degradation of multiply charged products when reacted with even-electron anions of benzoic acid for 50 (B), 100 (C), and 150 (D) ms., leaving predominately doubly and singly charged fragments after 150 ms. (E) The resulting sequence coverage considering only singly charged product ions. Each spectrum is the average of ∼50 spectra (∼30-s acquisition), and the y axis indicates the relative ion abundance [30] Copyright (2005) National Academy of Sciences, USA. Reprinted with permission.
ETD followed by PTR allows for the sequence analysis of mixtures of highly modified large peptides. Detecting post translational modifications on the N-terminal tail of histones in context of one another is invaluable information in deciphering the histone code. From Coon et al., histone H3.1 (residue 1-50) from asynchronous human cells was isolated and analyzed on a chromatographic time scale with ETD-PTR mass spectrometry. Shown in Figure 10A is a near complete series of c-type ions of the first ten amino acids of the N-terminus of histone H3.1 demonstrating monomethylation and dimethylation on K4 and K9 respectively. In addition, the z-type ions from the C-terminus indicate K36 is dimethylated. In Figure 10B of histone H3.1 (residue 1-50) is a peptide eluting six seconds later than the peptide shown in Figure 10A, which is composed of a slightly different modification pattern. In the later eluting peptide, the N terminus of this peptide is modified identically to the previous species, however, an m/z shift of 14 U of the z 14 ion confirms a monomethylated K37. A shift in the higher mass z-type ions confirms an unmodified K36 residue [30].
Figure 10.

Sequencing highly modified, large peptides by ETD
Online chromatographic separation of large peptides (residues 1-50 from histone H3.1) followed by automated sequential ion/ion reactions of 15-ms ETD followed by 150-ms PTR. (A) The tandem mass spectrum from early eluting histone H3.1 peptide. Note the c- and z-type ion series allows interpretation of the N- and C-termini and demonstrates that K and K 4 9 are modified with monomethyl and dimethyl groups respectively. (B) The tandem mass spectrum generated from a histone H3.1 peptide eluting 6 seconds later. Here the c-type ion series indicates the N terminus is modified identical to the peptide shown in (A); however, the C terminus is monomethylated at K37, as opposed to being dimethylated at K36. Note the spectrum shown in (B) contains fragment ions derived from both species (co-elution) [30] Copyright (2005) National Academy of Sciences, USA. Reprinted with permission.
Conclusions
This review illustrates that dissociation by ETD produces product ions from peptides containing common post translational modifications, peptides with multiple basic residues, and intact proteins amenable to peptide interpretation. Peptides with labile modifications typically produced an abundant loss of phosphoric acid, SO3, glycan, or NO when dissociated by CAD. However, when phosphorylated, sulfonated and N- and O-glycosylated peptides were dissociated using ETD, fragmentation occurred before the bond with the lowest energy was localized and preferentially broken (i.e. the labile modification). ETD can also readily fragment peptides with multiple basic residues or peptides containing disulfide bonds. In these examples, ETD produced a complete or near complete c and z-ion series. On our ETD mass spectrometer, ETD adds only about a 100msec/scan premium over CAD. Depending on ion abundance (lower femtomole range) and ion injection time, we can obtain about three ETD spectrum/second vs. about four CAD spectrum/second, which is compatible with a chromatographic timescale. For peptides, one microscan per spectrum is typical, while some spectral averaging is desirable for proteins. The application of ETD in proteomic research will be useful in studies involving disease, cancer, drug discovery, as well as characterizing cellular function and signaling. ETD will expand current analysis to include more basic, non-tryptic peptides and proteins which will enable the identification of various post translational modifications within the context of each other as well as potentially identify new protein isoforms [30].
Acknowledgement
We thank Dr. Cynthia Brame for critical review of the manuscript. This work was supported by NIH Grant GM37537 to DFH.
Abbreviations used
- acK
acetylated lysine
- AD
Alzheimer Disease
- CAD
collision activated dissociation
- diK
dimethylated lysine
- ECD
electron capture dissociation
- ESI
electrospray ionization
- ETD
electron transfer dissociation
- FT-ICR-MS
Fourier transform ion cyclotron resonance mass spectrometer
- GlcNAc-N
acetylglucosamine
- IMAC
immobilized metal affinity chromatography
- LT
linear ion trap mass spectrometer
- MS/MS
tandem mass spectrometry
- nC
nitrosylated cysteine
- PTM
post translational modification
- PTR
proton transfer reduction
- sS
sulfonated serine
- pS
phosphorylated serine
- gT-O
GlcNAc modified threonine
- TFA
trifluoroacetic acid
- triK
trimethylated lysine
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101:10804–9. doi: 10.1073/pnas.0400348101. Epub 2004 Jul 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Crane R, Gadea B, Littlepage L, Wu H, Ruderman JV. Aurora A, Meiosis and Mitosis. Biology of the Cell. 2004;96:215–229. doi: 10.1016/j.biolcel.2003.09.008. [DOI] [PubMed] [Google Scholar]
- [3].Musicki B, Kramer MF, Becker RE, Burnett AL. Inactivation of phosphorylated endothelial nitric oxide synthase (Ser-1177) by O-GlcNAc in diabetes-associated erectile dysfunction. PNAS. 2005;102:11870–11875. doi: 10.1073/pnas.0502488102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–5. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- [5].Li N, Lerea KM, Etlinger JD. Phosphorylation of the Proteasome Activator PA28 Is Required for Proteasome Activation. Biochemical and Biophysical Research Communications. 1996;225:855–860. doi: 10.1006/bbrc.1996.1263. [DOI] [PubMed] [Google Scholar]
- [6].Rechsteiner M, Rogers SW. PEST sequences and regulation by proteolysis. Trends in Biochemical Sciences. 1996;21:267–271. [PubMed] [Google Scholar]
- [7].Zhang F, Su K, Yang X, Bowe DB, Paterson AJ, Kudlow JE. O-GlcNAc modification is an endogenous inhibitor of the proteasome. Cell. 2003;115:715–25. doi: 10.1016/s0092-8674(03)00974-7. [DOI] [PubMed] [Google Scholar]
- [8].Han I, Kudlow JE. Reduced O glycosylation of Sp1 is associated with increased proteasome susceptibility. Mol Cell Biol. 1997;17:2550–8. doi: 10.1128/mcb.17.5.2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Medzihradszky KF, Darula Z, Perlson E, Fainzilber M, Chalkley RJ, Ball H, Greenbaum D, Bogyo M, Tyson DR, Bradshaw RA, Burlingame AL. O- sulfonation of serine and threonine: mass spectrometric detection and characterization of a new posttranslational modification in diverse proteins throughout the eukaryotes. Mol Cell Proteomics. 2004;3:429–40. doi: 10.1074/mcp.M300140-MCP200. Epub 2004 Jan 29. [DOI] [PubMed] [Google Scholar]
- [10].Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S- nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–66. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- [11].Wysocki VH, Tsaprailis G, Smith LL, Breci LA. Mobile and localized protons: a framework for understanding peptide dissociation. J Mass Spectrom. 2000;35:1399–406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- [12].Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101:9528–33. doi: 10.1073/pnas.0402700101. Epub 2004 Jun 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mann M, Jensen ON. Proteomic analysis of post-translational modifications. Nat Biotechnol. 2003;21:255–61. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- [14].Mirza UA, Chait BT, Lander HM. Monitoring Reactions of Nitric Oxide with Peptides and Proteins by Electrospray Ionization-Mass Spectrometry. J. Biol. Chem. 1995;270:17185–17188. doi: 10.1074/jbc.270.29.17185. [DOI] [PubMed] [Google Scholar]
- [15].Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP. Large-scale characterization of HeLa cell nuclear phosphoproteins. PNAS. 2004;101:12130–12135. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Morandell S, Stasyk T, Grosstessner-Hain K, Roitinger E, Mechtler K, Bonn GK, Huber LA. Phosphoproteomics strategies for the functional analysis of signal transduction. Proteomics. 2006;6:4047–56. doi: 10.1002/pmic.200600058. [DOI] [PubMed] [Google Scholar]
- [17].Zubarev RA, Kelleher NL, McLafferty FW. ECD of multiply charged protein cations. A non-ergodic process. J Am Chem Soc. 1998;120:3265–3266. [Google Scholar]
- [18].Stensballe A, Jensen ON, Olsen JV, Haselmann KF, Zubarev RA. Electron capture dissociation of singly and multiply phosphorylated peptides. Rapid Commun Mass Spectrom. 2000;14:1793–800. doi: 10.1002/1097-0231(20001015)14:19<1793::AID-RCM95>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- [19].Shi SD, Hemling ME, Carr SA, Horn DM, Lindh I, McLafferty FW. Phosphopeptide/phosphoprotein mapping by electron capture dissociation mass spectrometry. Anal Chem. 2001;73:19–22. doi: 10.1021/ac000703z. [DOI] [PubMed] [Google Scholar]
- [20].Mirgorodskaya E, Roepstorff P, Zubarev RA. Localization of O- glycosylation sites in peptides by electron capture dissociation in a Fourier transform mass spectrometer. Anal Chem. 1999;71:4431–6. doi: 10.1021/ac990578v. [DOI] [PubMed] [Google Scholar]
- [21].Hakansson K, Cooper HJ, Emmett MR, Costello CE, Marshall AG, Nilsson CL. Electron capture dissociation and infrared multiphoton dissociation MS/MS of an N-glycosylated tryptic peptic to yield complementary sequence information. Anal Chem. 2001;73:4530–6. doi: 10.1021/ac0103470. [DOI] [PubMed] [Google Scholar]
- [22].Kelleher NL, Zubarev RA, Bush K, Furie B, Furie BC, McLafferty FW, Walsh CT. Localization of labile posttranslational modifications by electron capture dissociation: the case of gamma-carboxyglutamic acid. Anal Chem. 1999;71:4250–3. doi: 10.1021/ac990684x. [DOI] [PubMed] [Google Scholar]
- [23].Coon JJ, Syka JE, Shabanowitz J, Hunt DF. Tandem mass spectrometry for peptide and protein sequence analysis. Biotechniques. 2005;38:519–521. doi: 10.2144/05384TE01. [DOI] [PubMed] [Google Scholar]
- [24].Coon JJ, Syka JEP, Schwartz JC, Shabanowitz J, Hunt DF. Anion dependence in the partitioning between proton and electron transfer in ion/ion reactions. International Journal of Mass Spectrometry. 2004;236:33–42. [Google Scholar]
- [25].Pitteri SJ, Chrisman PA, Hogan JM, McLuckey SA. Electron transfer ion/ion reactions in a three-dimensional quadrupole ion trap: reactions of doubly and triply protonated peptides with SO2*. Anal Chem. 2005;77:1831–9. doi: 10.1021/ac0483872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Graves JD, Krebs EG. Protein phosphorylation and signal transduction. Pharmacol Ther. 1999;82:111–21. doi: 10.1016/s0163-7258(98)00056-4. [DOI] [PubMed] [Google Scholar]
- [27].Morley SJ, Coldwell MJ, Clemens MJ. Initiation factor modifications in the preapoptotic phase. Cell Death and Differ. 2005;12:571–584. doi: 10.1038/sj.cdd.4401591. [DOI] [PubMed] [Google Scholar]
- [28].Hunter T. Signaling--2000 and Beyond. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- [29].Reinders J, Sickmann A. State-of-the-art in phosphoproteomics. Proteomics. 2005;5:4052–61. doi: 10.1002/pmic.200401289. [DOI] [PubMed] [Google Scholar]
- [30].Coon JJ, Ueberheide B, Syka JEP, Dryhurst DD, Ausio J, Shabanowitz J, Hunt DF. Protein identification using sequential ion/ion reactions and tandem mass spectrometry. PNAS. 2005;102:9463–9468. doi: 10.1073/pnas.0503189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Slawson C, Hart GW. Dynamic interplay between O-GlcNAc and O- phosphate: the sweet side of protein regulation. Current Opinion in Structural Biology. 2003;13:631–636. doi: 10.1016/j.sbi.2003.08.003. [DOI] [PubMed] [Google Scholar]
- [32].Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Exploring the O- GlcNAc proteome: Direct identification of O-GlcNAc-modified proteins from the brain. PNAS. 2004;101:13132–13137. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Whelan SA, Hart GW. Proteomic Approaches to Analyze the Dynamic Relationships Between Nucleocytoplasmic Protein Glycosylation and Phosphorylation. Circ Res. 2003;93:1047–1058. doi: 10.1161/01.RES.0000103190.20260.37. [DOI] [PubMed] [Google Scholar]
- [34].Schroeder MJ, Webb DJ, Shabanowitz J, Horwitz AF, Hunt DF. Methods for the detection of paxillin post-translational modifications and interacting proteins by mass spectrometry. J Proteome Res. 2005;4:1832–41. doi: 10.1021/pr0502020. [DOI] [PubMed] [Google Scholar]
- [35].Morelle W, Canis K, Chirat F, Faid V, Michalski JC. The use of mass spectrometry for the proteomic analysis of glycosylation. Proteomics. 2006;6:3993–4015. doi: 10.1002/pmic.200600129. [DOI] [PubMed] [Google Scholar]
- [36].Ashford D, Dwek RA, Welply JK, Amatayakul S, Homans SW, Lis H, Taylor GN, Sharon N, Rademacher TW. The beta 1-2-D-xylose and alpha 1-3-L- fucose substituted N-linked oligosaccharides from Erythrina cristagalli lectin. Isolation, characterisation and comparison with other legume lectins. Eur J Biochem. 1987;166:311–20. doi: 10.1111/j.1432-1033.1987.tb13516.x. [DOI] [PubMed] [Google Scholar]
- [37].Hogan JM, Pitteri SJ, Chrisman PA, McLuckey SA. Complementary structural information from a tryptic N-linked glycopeptide via electron transfer ion/ion reactions and collision-induced dissociation. J Proteome Res. 2005;4:628–32. doi: 10.1021/pr049770q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Oh H, Breuker K, Sze SK, Ge Y, Carpenter BK, McLafferty FW. Secondary and tertiary structures of gaseous protein ions characterized by electron capture dissociation mass spectrometry and photofragment spectroscopy. PNAS. 2002;99:15863–15868. doi: 10.1073/pnas.212643599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Martinez-Ruiz A, Lamas S. S-nitrosylation: a potential new paradigm in signal transduction. Cardiovascular Research. 2004;62:43–52. doi: 10.1016/j.cardiores.2004.01.013. [DOI] [PubMed] [Google Scholar]
- [40].Loo JA, Edmonds CG, Udseth HR, Smith RD. Effect of Reducing Disulfide-Containing Proteins on Electrospray Ionization Mass-Spectra. Analytical Chemistry. 1990;62:693–698. doi: 10.1021/ac00206a009. [DOI] [PubMed] [Google Scholar]
- [41].Zubarev RA, Kruger NA, Fridriksson EK, Lewis MA, Horn DM, Carpenter BK, McLafferty FW. Electron capture dissociation of gaseous multiply-charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity. Journal of the American Chemical Society. 1999;121:2857–2862. [Google Scholar]
- [42].Chrisman PA, Pitteri SJ, Hogan JM, McLuckey SA. SO2-[middle dot] Electron Transfer Ion/Ion Reactions with Disulfide Linked Polypeptide Ions. Journal of the American Society for Mass Spectrometry. 2005;16:1020–1030. doi: 10.1016/j.jasms.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Breuker K, Oh H, Horn DM, Cerda BA, McLafferty FW. Detailed unfolding and folding of gaseous ubiquitin ions characterized by electron capture dissociation. J Am Chem Soc. 2002;124:6407–20. doi: 10.1021/ja012267j. [DOI] [PubMed] [Google Scholar]