

Abstract
The insulin gene is expressed almost exclusively in pancreatic β-cells. Metabolic regulation of insulin gene expression enables the β-cell to maintain adequate stores of intracellular insulin to sustain the secretory demand. Glucose is the major physiologic regulator of insulin gene expression; it coordinately controls the recruitment of transcription factors [e.g., pancreatic/duodenal homeobox-1 (PDX-1), mammalian homologue of avian MafA/L-Maf (MafA), Beta2/Neuro D (B2), the rate of transcription, and the stability of insulin mRNA. However, chronically elevated levels of glucose (glucotoxicity) and lipids (lipotoxicity) also contribute to the worsening of β-cell function in type 2 diabetes, in part via inhibition of insulin gene expression. The mechanisms of glucotoxicity, which involve decreased binding activities of PDX-1 and MafA and increased activity of C/EBPβ, are mediated by high-glucose–induced generation of oxidative stress. On the other hand, lipotoxicity is mediated by de novo ceramide synthesis and involves inhibition of PDX-1 nuclear translocation and MafA gene expression. Glucotoxicity and lipotoxicity have common targets, which makes their combination particularly harmful to insulin gene expression and β-cell function in type 2 diabetes.
Keywords: B-cell, diabetes, insulin
Insulin is secreted uniquely from the islet β-cells of the pancreas and plays a major role in the maintenance of energy homeostasis. Insulin secretion is tightly regulated to maintain blood glucose levels within a narrow physiological range. Insufficient secretion of insulin from β-cells contributes to the chronic hyperglycemia characteristic of diabetes, a disease that affects > 20 million Americans. Short-term regulation of insulin secretion, such as in response to a meal, occurs mainly at the level of exocytosis. However, the maintenance of adequate intracellular stores of insulin on a long-term basis relies on the transcriptional and translational regulation of insulin biosynthesis. The 2 most important nutrients in mammals, glucose and fatty acids, profoundly affect preproinsulin gene (hereafter referred to as ‘‘insulin gene’’) expression under physiological and pathological circumstances. This review briefly describes the key control elements and their cognate transactivating factors involved in the metabolic regulation of insulin gene transcription. We then describe the manner in which glucose normally regulates insulin gene expression, and how dysregulation occurs upon prolonged exposure to glucose (glucotoxicity) and fatty acids (lipotoxicity).
Structure of the insulin gene and key transcription factors involved in metabolic regulation
In adult mammals, expression of the insulin gene is essentially restricted to the pancreatic β-cell. A highly conserved region lying ~340 bp immediately upstream of the transcription initiation start, hereafter referred to as the insulin promoter, confers both tissue-specific expression and metabolic regulation of the insulin gene. Many transcription factors act upon this region, forming a highly sophisticated transcriptional network that ensures precise regulation. The most critical cis-acting DNA elements involved in transcriptional activation in vitro are referred to the A3, C1, and E1 sites [Fig. 1; reviewed in (1)].
FIGURE 1.
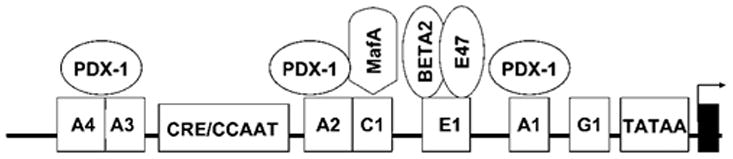
Schematic representation of the proximal region of the rat insulin 2 promoter showing the key elements and trans-activating factors involved in glucose regulation of insulin gene transcription.
Pancreatic/duodenal homeobox-1 (PDX-1)3 is a homeodomain protein that binds to the A3 box of the insulin gene (2). PDX-1 is also essential in the normal development of the pancreas because its deletion results in complete agenesis (3). PDX-1 functionally interacts with proteins of the basic helix-loop-helix (bHLH) family that bind to the E1 box (4). The E1 activator is a heterodimer consisting of ubiquitous class A (E12/E47 and E2/5) and cell-restricted class B (B2) members of the bHLH family (5). The cooperation between PDX-1 and bHLH proteins also involves interactions with other DNA-binding proteins (6) and co-activators (7), thus forming a unique network of protein-protein and protein-DNA interactions. The C1 activator was recently identified as MafA (8). Although a number of additional proteins, such as members of the hepatic nuclear factors and PAX families, contribute to the integrated network of transcription factors that control transcription of the insulin gene, those listed above appear to be the principal regulators of insulin transcription under normal and pathological circumstances.
Glucose regulation of insulin gene expression
Glucose is the major nutrient regulator of pancreatic β-cell function and coordinately regulates insulin gene expression, insulin biosynthesis, and insulin secretion. Glucose controls all steps of insulin gene expression, including transcription, preRNA splicing, and mRNA stability. A3, E1, and C1 are the major glucose-responsive transcription control elements of the insulin gene. In addition, a more distal glucose-responsive element appears to bind a glucose-sensitive complex that is specifically present in primary islets but remains to be identified (9). Glucose promotes the binding of PDX-1 to the A3 site (2) and PDX-1 trans-activating potency (10). In addition, it is now recognized that PDX-1 stimulation of insulin gene transcription involves recruitment of co-activators, such as p300 (11), that affect chromatin structure through posttranslational modifications of histones such as methylation (12) and/or acetylation (13).
The signal transduction mechanisms by which glucose increases PDX-1 binding to the insulin promoter have been studied extensively but remain controversial. Glucose appears to promote translocation and modification of a cytoplasmic, inactive 31-kDa form to the nuclear, active 46-kDa species (14). Although this transformation likely requires phosphorylation, the large increase in apparent molecular mass suggests that PDX-1 undergoes multiple posttranslational modifications, possibly by O-linked N-acetylglucosamine (15) or small ubiquitin-related modifier 1 (16). A number of kinases were proposed to mediate PDX-1 phosphorylation, including p38 mitogen-activated protein kinase (17), phosphatidylinositol-3 kinase (18), and extracellular signal-regulated kinases (19).
MafA and B2/E47 are also stimulated by glucose (20). Although it is unclear how B2/E47 is regulated, MafA expression and binding are activated directly by glucose (21). Importantly, PDX-1, MafA, and B2 do not act in an isolated manner, but interact with each other to induce synergistic activation of insulin transcription (21). Therefore, glucose enhances insulin gene transcription by a number of complementary mechanisms that include recruitment of transcription factors to regulatory sites, histone modifications, and initiation of transcription. Importantly, our understanding of the control of the insulin gene has been gleaned principally from in vitro systems, and their relevance to the endogenous gene in vivo has yet to be established.
In addition to its major effects on the rate of transcription, glucose markedly stabilizes preproinsulin mRNA (22). Two elements located in the 3′-untranslated region of the mRNA molecule were proposed as mediators of this effect, i.e., the conserved UUGAA sequence (23) and a pyrimidine-rich sequence (24). Stabilization appears to involve glucose-regulated binding of a polypyrimidine tract-binding protein to the pyrimidine-rich sequence (24).
Dysregulation of insulin gene expression
As discussed above, glucose is the major physiologic regulator of the insulin gene. In contrast, when β-cells are exposed to elevated levels of glucose for prolonged periods of time, glucose becomes toxic to insulin secretion, gene expression, and β-cell survival. This phenomenon is referred to as glucotoxicity (25). Similarly, chronically elevated levels of fatty acids adversely affect pancreatic β-cell function through a process termed lipotoxicity (26). The relevance of nutrient-induced β-cell dysfunction in humans comes from the fact that chronic hyperglycemia and associated disorders of lipid metabolism are thought to contribute to the deterioration of pancreatic β-cell function observed in patients with type 2 diabetes (27).
Glucotoxicity
Exposure of insulin-secreting cells to elevated glucose levels for several weeks impairs insulin gene expression; this is associated with diminished binding activity of PDX-1 and MafA [reviewed in (28)]. The decrease in PDX-1 binding activity appears to involve post-transcriptional control (29), although the precise mechanism(s) remains to be established. In vivo, PDX-1 expression is also reduced in partially pancreatectomized, hyperglycemic rats (30) and in the diabetic gerbil Psammomys obesus (31), and its binding activity is decreased in islets from Zucker Diabetic Fatty rats (32). The reduction in MafA binding activity in the glucotoxic insulin-secreting HIT-T15 cell was shown recently to be due to a loss of protein expression without changes in mRNA expression, suggesting that glucose reduces MafA activity through a post-translational mode of action (33). Importantly, MafA expression is also reduced in mouse diabetic models (34). In addition, the C/EBPβ transcription factor may directly bind E47 and prevent formation of the B2/E47 activator complex under glucotoxic conditions (35). A recent study further proposed that CEBP/β prevents MafA binding to its cognate sequence under chronic exposure to elevated glucose and, in turn, prevents the cooperative induction of transcription by MafA and B2 (36).
Much progress has been made in recent years in understanding the biochemical mechanisms of glucotoxicity. Ample evidence supports the involvement of oxidative stress in this process, as a result of long-term exposure to elevated glucose [reviewed in (37)]. For example, the decrease in insulin gene transcription (38) and MafA protein expression (33) is prevented by antioxidants in glucotoxic insulin-secreting cells. Moreover, treatment of Zucker Diabetic Fatty rats with anti-oxidants normalizes plasma glucose levels and restores insulin secretion, insulin content, and insulin mRNA levels (38). In addition, activation of the hexosamine pathway decreases insulin gene expression and insulin secretion in isolated islets via generation of oxidative stress but not via O-linked glycosylation (39).
The signaling pathways mediating inhibition of insulin gene expression by oxidative stress appear to involve, at least in part, stress-activated kinases. Overexpression of a dominant-negative form of c-jun N-terminal kinase (JNK) prevents the decrease in PDX-1 binding activity in response to oxidative stress in a c-jun-independent manner (40). On the other hand, c-jun can directly inhibit insulin gene transcription by interfering with bHLH-mediated transcriptional activity (41). It is therefore likely that several interrelated pathways negatively affect insulin gene transcription under conditions of oxidative stress (Fig. 2). Another possibility, not exclusive with the previous one, is that glucotoxicity induces dedifferentiation of the β-cell, as suggested by the observed inhibition of genes associated with β-cell function and derepression of genes not normally expressed in differentiated β-cells (42). For example, the transcription factor c-myc is upregulated in islets from diabetic animals (43), and can inhibit insulin gene transcription by competing for B2 binding at the E-box (44) (Fig. 2).
FIGURE 2.
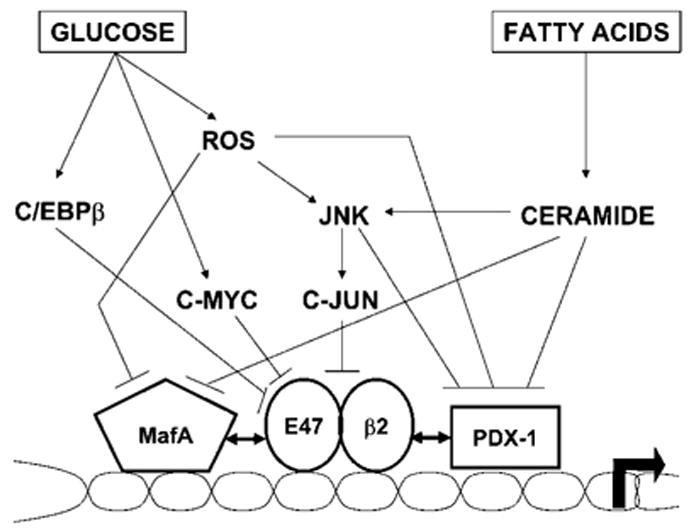
Proposed intracellular mechanisms by which chronically elevated glucose and fatty acids impair insulin gene transcription.
Lipotoxicity
Disorders of lipid metabolism were proposed to contribute to β-cell dysfunction in type 2 diabetes (26). We (45–47) and others (48,49) showed that exposure of isolated islets and insulin-secreting cells to elevated levels of fatty acids in vitro impairs insulin gene expression when glucose concentrations are concomitantly elevated. Deleterious effects of fatty acids on insulin gene expression in isolated islets are associated with an increased accumulation of intracellular triglycerides (TG), suggesting a role for neutral lipid synthesis in this process (45). However, increasing TG synthesis by overexpression of the enzyme diacylglycerol-acyltranferase inhibits glucose-induced insulin secretion but not preproinsulin mRNA levels (50), suggesting that TG accumulation is not directly involved in inhibition of insulin gene expression. Furthermore, because both palmitate and oleate inhibit insulin secretion, but only palmitate impairs insulin gene expression (51), we postulated that distinct mechanisms underlie these 2 functional effects. Because ceramide can be synthesized de novo from palmitate but not from oleate, we hypothesized that palmitate inhibition of insulin gene expression may be mediated by ceramide generation. In fact, exposure of islets to palmitate was shown to result in increased intracellular ceramide content that was largely prevented by inhibitors of de novo ceramide synthesis, which also prevented the decrease in insulin gene expression (47). In contrast, ceramide synthesis does not appear to mediate fatty acid impairment of insulin secretion (51). Because ceramide activates JNK in various cell types, we further hypothesize that ceramide-induced JNK activation might inhibit insulin gene transcription via c-jun-dependent and -independent pathways (Fig. 2). Alternatively, ceramide might inhibit protein kinase B, thereby allowing the transcription factor Fox01 to translocate into the nucleus and repress its target genes. In β cells, PDX-1 and Fox01 exhibit a mutually exclusive pattern of nuclear localization (52), and is it therefore conceivable that an increase in nuclear localization of Fox01 in response to palmitate results in nuclear exclusion of PDX-1. These hypotheses are currently under investigation.
Palmitate impairment of insulin gene expression appears to be mediated by direct inhibition of insulin promoter activity (47). We showed recently in isolated rat islets that palmitate inhibits PDX-1 and MafA binding activities (46). Interestingly, PDX-1 and MafA appear to be affected by palmitate through different mechanisms. Culture of isolated islets in the presence of palmitate prevents the nuclear translocation of PDX-1 that normally occurs upon glucose stimulation, whereas it blocks glucose induction of MafA mRNA expression (46). Importantly, combined, adenovirus-mediated overexpression of PDX-1 and MafA prevents palmitate inhibition of insulin gene expression in isolated rat islets, consistent with an essential role for these 2 transcription factors in the mechanisms of lipotoxicity (46).
It is interesting that MafA and PDX-1 are common glucotoxicity and lipotoxicity targets, although affected by different mechanisms. Hence, glucotoxicity is associated with decreased protein expression of PDX-1 and MafA, whereas under lipotoxic conditions, PDX-1 is expressed but retained in the cytoplasm and MafA mRNA levels are decreased. It is therefore likely that these conditions, which occur concomitantly in most patients with type 2 diabetes, have deleterious effects on insulin gene expression.
Conclusions
Regulation of insulin gene expression under normal circumstances is controlled chiefly by changes in glucose concentrations. Glucose coordinately recruits a highly sophisticated network of transcription factors and co-activators to the insulin promoter, and also prolongs the half-life of insulin mRNA. In vitro and in vivo studies in rodents have provided evidence that under circumstances of chronically elevated levels of glucose and fatty acids, insulin gene expression is greatly reduced. The mechanisms of glucotoxicity and lipotoxicity involve the PDX-1 and MafA transcription factors, with such a convergence having severe adverse consequences on β-cell function (e.g., insulin gene expression). The combined and deleterious effects of glucose and fatty acids on insulin gene expression are likely to contribute to β-cell dysfunction in type 2 diabetes, although this hypothesis has yet to be tested in humans.
Abbreviations used
- bHLH
basic helix-loop-helix
- B2
Beta2/NeuroD
- C/EBPβ
CCAAT/enhancer-binding protein β
- JNK
c-jun N-terminal kinase
- MafA
mammalian homologue of avian MagfA/L-Maf
- PDX-1
pancreatic/duodenal homeobox-1
- TG
triglycerides
Footnotes
Supported by the National Institutes of Health (R01 DK58096 to V.P., R01 DK50203 to R.S., R01 DK38325 to R.P.R.), the American Heart Association to V.P., and the 2003 Thomas R. Lee Career Development Award of the American Diabetes Association to V.P. D.H. is supported by a National Institutes of Health National Research Service Award (F32 DK070406). I.A. is supported by the Juvenile Diabetes Research Foundation (3-2003-579).
LITERATURE CITED
- 1.Poitout V, Stein R, Rhodes CJ. Insulin gene expression and biosynthesis. In: DeFronzo RA, Ferrannini E, Keen H, Zimmet P, editors. International textbook of diabetes mellitus. 3. Chichester: John Wiley & Sons; 2004. pp. 98–123. [Google Scholar]
- 2.Melloul D, Ben-Neriah Y, Cerasi E. Glucose modulates the binding of an islet-specific factor to a conserved sequence within the rat I and the human insulin promoters. Proc Natl Acad Sci U S A. 1993;90:3865–9. doi: 10.1073/pnas.90.9.3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter factor 1 is required for pancreatic development in mice. Nature. 1994;371:606–9. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- 4.Peers B, Leonard J, Sharma S, Teitelman G, Montminy MR. Insulin expression in pancreatic islet cells relies on cooperative interactions between the helix loop helix factor E47 and the homeobox factor STF-1. Mol Endocrinol. 1994;8:1798–806. doi: 10.1210/mend.8.12.7708065. [DOI] [PubMed] [Google Scholar]
- 5.Aronheim A, Ohlsson H, Park CW, Edlund T, Walker MD. Distribution and characterization of helix-loop-helix enhancer-binding proteins from pancreatic β cells and lymphocytes. Nucleic Acids Res. 1991;19:3893–9. doi: 10.1093/nar/19.14.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ohneda K, Mirmira RG, Wang J, Johnson JD, German MS. The homeodomain of PDX-1 mediates multiple protein-protein interactions in the formation of a transcriptional activation complex on the insulin promoter. Mol Cell Biol. 2000;20:900–11. doi: 10.1128/mcb.20.3.900-911.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qiu Y, Guo M, Huang S, Stein R. Insulin gene transcription is mediated by interactions between the p300 coactivator and PDX-1, B2, and E47. Mol Cell Biol. 2002;22:412–20. doi: 10.1128/MCB.22.2.412-420.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olbrot M, Rud J, Moss LG, Sharma A. Identification of β-cell-specific insulin gene transcription factor RIPE3b1 as mammalian MafA. Proc Natl Acad Sci U S A. 2002;99:6737–42. doi: 10.1073/pnas.102168499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sander M, Griffen SC, Huang J, German MS. A novel glucose-responsive element in the human insulin gene functions uniquely in primary cultured islets. Proc Natl Acad Sci U S A. 1998;95:11572–7. doi: 10.1073/pnas.95.20.11572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Petersen HV, Serup P, Leonard J, Michelsen BK, Madsen OD. Transcriptional regulation of the human insulin gene is dependent on the homeodomain protein STF1/IPF1 acting through the CT boxes. Proc Natl Acad Sci U S A. 1994;91:10465–9. doi: 10.1073/pnas.91.22.10465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iype T, Francis J, Garmey JC, Schisler JC, Nesher R, Weir GC, Becker TC, Newgard CB, Griffen SC, Mirmira RG. Mechanism of insulin gene regulation by the pancreatic transcription factor Pdx-1: application of pre-mRNA analysis and chromatin immunoprecipitation to assess formation of functional transcriptional complexes. J Biol Chem. 2005;280:16798–807. doi: 10.1074/jbc.M414381200. [DOI] [PubMed] [Google Scholar]
- 12.Francis J, Chakrabarti SK, Garmey JC, Mirmira RG. Pdx-1 links histone H3-Lys-4 methylation to RNA polymerase II elongation during activation of insulin transcription. J Biol Chem. 2005;280:36244–53. doi: 10.1074/jbc.M505741200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mosley AL, Ozcan S. The pancreatic duodenal homeobox-1 protein (Pdx-1) interacts with histone deacetylases Hdac-1 and Hdac-2 on low levels of glucose. J Biol Chem. 2004;279:54241–7. doi: 10.1074/jbc.M410379200. [DOI] [PubMed] [Google Scholar]
- 14.Rafiq I, Kennedy HJ, Rutter GA. Glucose-dependent translocation of insulin promoter factor-1 (IPF-1) between the nuclear periphery and the nucleoplasm of single MIN6 β-cells. J Biol Chem. 1998;273:23241–7. doi: 10.1074/jbc.273.36.23241. [DOI] [PubMed] [Google Scholar]
- 15.Gao Y, Miyazaki J, Hart GW. The transcription factor PDX-1 is post-translationally modified by O-linked N-acetylglucosamine and this modification is correlated with its DNA binding activity and insulin secretion in min6 β-cells. Arch Biochem Biophys. 2003;415:155–63. doi: 10.1016/s0003-9861(03)00234-0. [DOI] [PubMed] [Google Scholar]
- 16.Kishi A, Nakamura T, Nishio Y, Maegawa H, Kashiwagi A. Sumoylation of Pdx1 is associated with its nuclear localization and insulin gene activation. Am J Physiol Endocrinol Metab. 2003;284:E830–40. doi: 10.1152/ajpendo.00390.2002. [DOI] [PubMed] [Google Scholar]
- 17.Macfarlane WM, McKinnon CM, Felton-Edkins ZA, Cragg H, James RF, Docherty K. Glucose stimulates translocation of the homeodomain transcription factor PDX1 from the cytoplasm to the nucleus in pancreatic β-cells. J Biol Chem. 1999;274:1011–6. doi: 10.1074/jbc.274.2.1011. [DOI] [PubMed] [Google Scholar]
- 18.da Silva Xavier G, Varadi A, Ainscow EK, Rutter GA. Regulation of gene expression by glucose in pancreatic β-cells (MIN6) via insulin secretion and activation of phosphatidylinositol 3′-kinase. J Biol Chem. 2000;275:36269–77. doi: 10.1074/jbc.M006597200. [DOI] [PubMed] [Google Scholar]
- 19.Khoo S, Griffen SC, Xia Y, Baer RJ, German MS, Cobb MH. Regulation of insulin gene transcription by ERK1 and ERK2 in pancreatic β cells. J Biol Chem. 2003;278:32969–77. doi: 10.1074/jbc.M301198200. [DOI] [PubMed] [Google Scholar]
- 20.Sharma A, Stein R. Glucose-induced transcription of the insulin gene is mediated by factors required for β-cell-type-specific expression. Mol Cell Biol. 1994;14:871–9. doi: 10.1128/mcb.14.2.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao L, Guo M, Matsuoka TA, Hagman DK, Parazzoli SD, Poitout V, Stein R. The islet β cell-enriched MafA activator is a key regulator of insulin gene transcription. J Biol Chem. 2005;280:11887–94. doi: 10.1074/jbc.M409475200. [DOI] [PubMed] [Google Scholar]
- 22.Welsh M, Nielsen DA, MacKrell AJ, Steiner DF. Control of insulin gene expression in pancreatic β-cells and in an insulin producing cell line, RIN-5F cells. 2. Regulation of insulin mRNA stability. J Biol Chem. 1985;260:13590–4. [PubMed] [Google Scholar]
- 23.Wicksteed B, Herbert TP, Alarcon C, Lingohr MK, Moss LG, Rhodes CJ. Cooperativity between the preproinsulin mRNA untranslated regions is necessary for glucose-stimulated translation. J Biol Chem. 2001;276:22553–8. doi: 10.1074/jbc.M011214200. [DOI] [PubMed] [Google Scholar]
- 24.Tillmar L, Carlsson C, Welsh N. Control of insulin mRNA stability in rat pancreatic islets. Regulatory role of a 3′-untranslated region pyrimidine-rich sequence. J Biol Chem. 2002;277:1099–106. doi: 10.1074/jbc.M108340200. [DOI] [PubMed] [Google Scholar]
- 25.Robertson RP, Zhang HJ, Pyzdrowski KL, Walseth TF. Preservation of insulin mRNA levels and insulin secretion in HIT cells by avoidance of chronic exposure to high glucose concentrations. J Clin Invest. 1992;90:320–5. doi: 10.1172/JCI115865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Unger RH. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes. 1995;44:863–70. doi: 10.2337/diab.44.8.863. [DOI] [PubMed] [Google Scholar]
- 27.Poitout V, Robertson RP. Minireview: secondary β-cell failure in type 2 diabetes—a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–42. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- 28.Robertson RP, Harmon JS, Tanaka Y, Sacchi G, Tran POT, Gleason CE, Poitout V. Glucose toxicity of the β-cell: cellular and molecular mechanisms. Diabetes mellitus. In: Le Roith D, Taylor SI, Olefsky JM, editors. A fundamental and clinical text. 2. Philadelphia: Lippincott Williams & Wilkins; 2000. pp. 125–132. [Google Scholar]
- 29.Olson LK, Sharma A, Peshavaria M, Wright CVE, Towle HC, Robertson RP, Stein R. Reduction of insulin gene transcription in HIT-T15 cells chronically exposed to a supraphysiologic glucose concentration is associated with loss of STF-1 transcription factor expression. Proc Natl Acad Sci U S A. 1995;92:9127–31. doi: 10.1073/pnas.92.20.9127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zangen DH, Bonner-Weir S, Lee CH, Latimer JB, Miller CP, Habener JF. Reduced insulin, GLUT2, and IDX-1 in β-cells after partial pancreatectomy. Diabetes. 1997;46:258–64. doi: 10.2337/diab.46.2.258. [DOI] [PubMed] [Google Scholar]
- 31.Leibowitz G, Ferber S, Apelqvist A, Edlund H, Gross DJ, Cerasi E, Melloul D, Kaiser N. IPF1/PDX1 deficiency and β-cell dysfunction in Psammomys obesus, an animal with type 2 diabetes. Diabetes. 2001;50:1799–806. doi: 10.2337/diabetes.50.8.1799. [DOI] [PubMed] [Google Scholar]
- 32.Harmon JS, Gleason CE, Tanaka Y, Oseid EA, Hunter-Berger KK, Robertson RP. In vivo prevention of hyperglycemia also prevents glucotoxic effects on PDX-1 and insulin gene expression. Diabetes. 1999;48:1995–2000. doi: 10.2337/diabetes.48.10.1995. [DOI] [PubMed] [Google Scholar]
- 33.Harmon JS, Stein R, Robertson RP. Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic β cells. J Biol Chem. 2005;280:11107–13. doi: 10.1074/jbc.M410345200. [DOI] [PubMed] [Google Scholar]
- 34.Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D. FoxO1 protects against pancreatic β cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–63. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 35.Lu M, Seufert J, Habener JF. Pancreatic β-cell-specific repression of insulin gene transcription by CCAAT/Enhancer-binding protein β. Inhibitory interactions with basic helix-loop-helix transcription factor E47. J Biol Chem. 1997;272:28349–59. doi: 10.1074/jbc.272.45.28349. [DOI] [PubMed] [Google Scholar]
- 36.Lawrence MC, McGlynn K, Park BH, Cobb MH. ERK1/2-dependent activation of transcription factors required for acute and chronic effects of glucose on the insulin gene promoter. J Biol Chem. 2005;280:26751–9. doi: 10.1074/jbc.M503158200. [DOI] [PubMed] [Google Scholar]
- 37.Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet B cells in diabetes. J Biol Chem. 2004;279:42351–4. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- 38.Tanaka Y, Gleason CE, Tran POT, Harmon JS, Robertson RP. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc Natl Acad Sci U S A. 1999;96:10857–62. doi: 10.1073/pnas.96.19.10857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaneto H, Xu G, Song KH, Suzuma K, Bonner-Weir S, Sharma A, Weir GC. Activation of the hexosamine pathway leads to deterioration of pancreatic β-cell function through the induction of oxidative stress. J Biol Chem. 2001;276:31099–104. doi: 10.1074/jbc.M104115200. [DOI] [PubMed] [Google Scholar]
- 40.Kaneto H, Xu G, Fujii N, Kim S, Bonner-Weir S, Weir GC. Involvement of c-Jun N-terminal kinase in oxidative stress-mediated suppression of insulin gene expression. J Biol Chem. 2002;277:30010–8. doi: 10.1074/jbc.M202066200. [DOI] [PubMed] [Google Scholar]
- 41.Henderson E, Stein R. c-jun inhibits transcriptional activation by the insulin enhancer, and the insulin control element is the target of control. Mol Cell Biol. 1994;14:655–62. doi: 10.1128/mcb.14.1.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laybutt DR, Sharma A, Sgroi DC, Gaudet J, Bonner-Weir S, Weir GC. Genetic regulation of metabolic pathways in β-cells disrupted by hyperglycemia. J Biol Chem. 2002;277:10912–21. doi: 10.1074/jbc.M111751200. [DOI] [PubMed] [Google Scholar]
- 43.Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patane G, Laybutt R, Bonner-Weir S, Weir GC. Chronic hyperglycemia triggers loss of pancreatic β cell differentiation in an animal model of diabetes. J Biol Chem. 1999;274:14112–21. doi: 10.1074/jbc.274.20.14112. [DOI] [PubMed] [Google Scholar]
- 44.Kaneto H, Sharma A, Suzuma K, Laybutt DR, Xu G, Bonner-Weir S, Weir GC. Induction of c-Myc expression suppresses insulin gene transcription by inhibiting NeuroD/B2-mediated transcriptional activation. J Biol Chem. 2002;277:12998–3006. doi: 10.1074/jbc.M111148200. [DOI] [PubMed] [Google Scholar]
- 45.Briaud I, Harmon JS, Kelpe CL, Segu VB, Poitout V. Lipotoxicity of the pancreatic β-cell is associated with glucose-dependent esterification of fatty acids into neutral lipids. Diabetes. 2001;50:315–21. doi: 10.2337/diabetes.50.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hagman DK, Hays LB, Parazzoli SD, Poitout V. Palmitate inhibits insulin gene expression by altering PDX-1 nuclear localization and reducing MafA expression in isolated rat islets of Langerhans. J Biol Chem. 2005;280:32413–8. doi: 10.1074/jbc.M506000200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kelpe CL, Moore PC, Parazzoli SD, Wicksteed B, Rhodes CJ, Poitout V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J Biol Chem. 2003;278:30015–21. doi: 10.1074/jbc.M302548200. [DOI] [PubMed] [Google Scholar]
- 48.Gremlich S, Bonny C, Waeber G, Thorens B. Fatty acids decrease IDX-1 expression in rat pancreatic islets and reduce GLUT2, glucokinase, insulin, and somatostatin levels. J Biol Chem. 1997;272:30261–9. doi: 10.1074/jbc.272.48.30261. [DOI] [PubMed] [Google Scholar]
- 49.Ritz-Laser B, Meda P, Constant I, Klages N, Charollais A, Morales A, Magnan C, Ktorza A, Philippe J. Glucose-induced preproinsulin gene expression is inhibited by the free-fatty acid palmitate. Endocrinology. 1999;140:4005–14. doi: 10.1210/endo.140.9.6953. [DOI] [PubMed] [Google Scholar]
- 50.Kelpe CL, Johnson LM, Poitout V. Increasing triglyceride synthesis inhibits glucose-induced insulin secretion in isolated rat islets of Langerhans. A study using adenoviral expression of diacylglycerol acyltransferase. Endocrinology. 2002;143:3326–32. doi: 10.1210/en.2002-220402. [DOI] [PubMed] [Google Scholar]
- 51.Moore PC, Ugas MA, Hagman DK, Parazzoli SD, Poitout V. Evidence against the involvement of oxidative stress in fatty acid inhibition of insulin secretion. Diabetes. 2004;53:2610–6. doi: 10.2337/diabetes.53.10.2610. [DOI] [PubMed] [Google Scholar]
- 52.Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH, 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic β cell growth. J Clin Invest. 2002;110:1839–47. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]