

Abstract
Nucleophosmin (NPM) is a nucleolar phosphoprotein that shuttles between the nucleus and cytoplasm during the cell cycle. NPM has several interacting partners and diverse cellular functions, including the processing of ribosomal RNA, centrosome duplication and the control of cellular processes to ensure genomic stability. Subcellular localization of NPM appears to be strongly correlated with NPM functions and cell proliferation. NPM is phosphorylated mainly at its central acidic domain by several upstream kinases, and its phosphorylation appears to be involved in regulating its functions in ribosome biogenesis and centrosome duplication. Recent studies suggest that NPM may act as a licensing factor to maintain proper centrosome duplication and that the Ran/CRM1 nucleocytoplasmic complex regulates local trafficking of NPM to centrosomes by interacting through its nuclear export sequence (NES) motif. Here, we provide a brief overview of NPM functions and its roles in human carcinogenesis, and discuss our recent findings related to the potential mechanisms underlying its regulation of centrosome duplication.
Keywords: Nucleophosmin, Phosphorylation, Centrosome duplication, Ran/CRM1
1. Introduction
Nucleophosmin (NPM), also known as B23 [1], numatrin [2] and NO38 [3], was originally identified as a non-ribosomal nucleolar phosphoprotein found at high levels in the granular regions of the nucleolus. NPM is highly conserved in vertebrates and widely distributed among different species with molecular weight of 35 to 40 kDa and pI of 5.1 to 5. In the human and rat, NPM exists at least as two isoforms, NPM1 and NPM1.2 (B23.1 and B23.2, respectively), which are generated from a single gene via alternative splicing [4]. Both isoforms contain identical sequences in their 5’ regions and major portions of their coding region, but the C-terminal, 35-amino acids of NPM1 are absent from NPM1.2.
To date, several distinct cellular functions for NPM have been described. It is believed to serve as a factor in ribosome biogenesis. NPM binds RNA and DNA [5] and interacts with other nucleolar proteins, including nucleolin [6], protein P120 [7], and human immunodeficiency virus (HIV) Rev and Tat proteins [8, 9]. Its intrinsic endoribonuclease activity, which preferentially cleaves pre-rRNA, suggests that it could participate in the late stages of ribosome biogenesis [10, 11]. In addition, NPM carries out multiple functions outside of the nucleolus. NPM contains a nuclear localization signal (NLS) and shuttles between the nucleus and cytoplasm during the cell cycle [12, 13]. It has been proposed that its shuttling ability is related to its molecular chaperone activities, such as preventing protein aggregation, protecting enzymes during thermal denaturation and facilitating renaturation of chemically-denatured proteins [14]. It was recently demonstrated that NPM is involved in centrosome duplication [15] and acts as a licensing factor to prevent any unscheduled centrosome duplication during the cell cycle [16].
Many studies have suggested that NPM appears to be involved in cancer pathogenesis. In mice, NPM is an essential gene whose inactivation in the germ line leads to a host of developmental defects that cause embryonic lethality at mid-gestation [17]. Haploid-insufficiency of NPM leads to unrestricted centrosome duplication and genomic instability and mice develop myelodysplasia with an acceleration of oncogenesis [17]. Moreover, disruption of the NPM gene by translocation is frequently found in human hematopoietic malignancies [18-20], and NPM appears to contribute to oncogenesis by activating the oncogenic potential of the fused protein partner.
Mutations in its C-terminal region, causing an aberrant cytoplasmic expression, are also frequently detected in cells of primary adult acute myeloid leukemia [21]. NPM is involved in the regulation of the ARF-p53 pathway [22-25], thus the mutation and/or ectopic localization of the protein may be a critical step in malignant transformation. Therefore, understanding the function(s) and the underlying mechanism(s) of NPM regulation may provide novel insights into the molecular pathogenesis of human cancer. A comprehensive review of NPM was recently published [26]. Instead, the primary focus of this review is to describe some of the characteristics and roles of NPM in normal and cancer cells, and to discuss paradoxical activities of NPM being involved in either a tumor suppressor gene or an oncogene with an emphasis of NPM as an early molecular player in human carcinogenesis.
2. Methods
The literature search for this review was performed using the MEDLINE database and reference lists from published papers. Genome assembly and reference sequences for NPM were searched on Genome Browser on the website of UCSC Genome Bioinformatics (http://genome.ucsc.edu).
The search keywords used by combination included B23, NPM, ribosome biogenesis, oligomerization, phosphorylation, centrosome duplication, and human cancer. This review also includes both published and unpublished results from our laboratory.
3. Results
3.1. NPM structure
Human NPM is mapped to chromosome 5q35 and is composed of 12 exons with sizes ranging from 58 to 358 bp. Two isoforms, NPM1 and NPM1.2, have been reported (Fig.1A). The splice variant, NPM1.2, utilizes an alternate 3’-terminal exon, compared with NPM1, resulting in a shorter protein containing 259 amino acids with a distinct C-terminus. NPM1 is mostly nucleolar, while NPM1.2 is present in cells at low levels and is detected both in the cytoplasm and nucleoplasm [27,28], suggesting that the C-terminal ends of NPM are necessary for subcellular localization of the protein. A search of the EST database reveals a new transcript variant (Gene Bank accession #: NM_199185) in the human. This variant lacks an alternate inframe exon 8, resulting in a shorter protein that lacks an internal segment. The functional significance of this variant is unknown.
Fig. 1.

A schematic representation of the human NPM gene. (A) NPM exists as at least three isoforms, variants 1, 2 and 3. The relative sizes and positions of exons are represented by the numbers 1 .12. The line (-) represents multiple alternative splicing events. Blue and yellow areas indicate coding sequences and untranslated regions, respectively. (B) Structure of the wild-type human NPM gene. The regular-spliced NPM gene has 11 exons encoding 294 amino acids. The N-terminal and C-terminal portions of NPM are essential for oligomer formation and nucleic acid binding activity, respectively. The central region of NPM is highly acidic. AD denotes the acidic domain and NAB, the nucleic acid domain. NPM that contains several known and potential phosphorylation sites by cdc2, CKII or N-II kinase, and plk1 are shown in red, green and light blue, respectively. The NES (yellow), NLS (green), and NoLS (red) motifs are highlighted and are conserved among four different species. Prefixes: h, human; m, mouse; r, rat; xl, Xenopus laevis. (C) The C-terminus of NPM is frequently mutated in AML. The positions of the two C-terminal tryptophan (W) residues are represented by red and the mutated residues are shown in pink. The added amino acid sequences common to all the mutated proteins are represented by the light blue box. (D) The NPM N-terminal portions are translocated to ALK, RAR or MLF in lymphoma and leukemia, and their sites are also indicated.
A comparison of the sequences and primary structures of homologous NPM proteins from diverse species from Xenopus laevis to human indicate the presence of an evolutionarily conserved motif that may be relevant to the function and subcellular distribution of the NPM. NPM is a nucleolar protein, which is capable of shuttling in and out of the nucleolus or between the nucleus and cytoplasm. Further sequence analysis indicates that NPM contains physiological hydrophobic leucine-rich nuclear export signal (NES) motif (LxxPxxLxLx), a bipartite nuclear localization signal (NLS) motif, and a nucleolus localization signal (NoLS) (Fig. 1B). We demonstrated that the nucleocytoplasmic shuttling process appears to be directed by these motifs [16]. While wild-type NPM is predominantly localized in the nucleolus, the missense mutant or the deletion mutant of NES is largely distributed in the nucleoplasm, implying that the NES mutant protein lacks the ability to be exported to the cytoplasm and thus, accumulates excessively in the nucleus. Mutations of the NLS motif abolish its localization in the nucleoplasm, as these proteins are mainly found in the cytoplasm and nucleolus. Consistent with a previous study, which proposes that the C-terminal end is important for its nucleolar localization [29], a NoLS mutant that has a substitution in the C-terminal region is no longer localized to the nucleolus [16,29]. These data indicate that the NES, NLS and NoLS motifs are functional and essential for directing NPM subcellular localization.
Several laboratories have observed that NPM exists as monomeric and oligomeric forms in cells. Under native conditions, the molecular weight of the NPM oligomeric form has been estimated to be 230 .250 kDa, which corresponds approximately to a hexamer of NPM [3,30,31]. NPM can be shifted to favor both monomeric and hexameric forms. Low ionic strength and the absence of divalent metal ions promote monomer formation, whereas increased ionic strength or the presence of either monovalent ions or divalent metal ions promotes oligomerization [32]. In HeLa cells, the hexamer of NPM is the major and stable entity, and its oligomerization is not affected by treatments with DNase, RNase, EDTA, reducing agents or lyophilization [33]. However, urea causes a reversible dissociation of the NPM oligomer into a monomer.
Studies using various deletion mutants of NPM isoforms have suggested that the N-terminal portion of the NPM protein is involved in oligomer formation [30,32,34], most likely because NPM1.2 has oligomerization properties virtually identical to those of NPM1 (Fig.1B).
It has been suggested that the oligomerization process of NPM may be a way of modulating its nucleic acid binding and chaperone activities during ribosome biogenesis. Under conditions where significant amounts of the monomer were present, NPM was capable of DNA binding, whereas conditions that strongly favored oligomerization caused a nearly complete abolition of this DNA binding activity [32]. Recently, it was reported that the NPM isoforms, NPM1 and NPM1.2, can form oligomers in vivo and in vitro [28,35,36], but the RNA binding activity of NPM1 is impaired by hetero-oligomerization with NPM1.2 [28]. Another study has shown that the reversible change of oligomerization between the hexameric and monomeric forms of NPM is found to occur most frequently in cells in G1 and G1/S phases [37]. As cells progressed into S, G2 and M phase, the reversible change of the two forms of NPM is decreased. Because rRNA processing occurs mostly at the G1 and G1/S phases, these data taken together indicate that oligomerization may be a necessary step in regulating multiple functions of NPM, especially related to ribosome biogenesis.
3.2. NPM Phosphorylation
Protein phosphorylation and dephosphorylation have been implicated in the regulation of diverse physiological and pathological processes [38]. NPM exists in cells as a phosphoprotein and contains multiple phosphorylation sites. Therefore, phosphorylation of NPM can have major role in the functional regulation of NPM. Early in vitro studies indicate that NPM is phosphorylated by several different kinases, including casein kinase II (CKII), nuclear kinase II (N-II kinase), polo-like kinase (Plk) and cdc2 type kinase [39-42] (Fig.1B). The mid-portion of NPM is highly acidic and is identified as the major phosphorylation region by CKII, cdc2 type kinase or nuclear kinase II. Recent studies show that cdc2 kinase targets a threonine residue at amino acids 185 .240 during the entry into mitosis, while CKII phosphorylates a serine site in the middle acidic portion of the molecule (83 .152) that contains the phosphoserine residue at amino acid 125, identified previously during the G2 phase [43,44]. These changes in NPM phosphorylation during the cell cycle implicates NPM’s major role in controlling the fate of nucleoli and cell growth. Although the regulation of NPM by phosphorylation is not well understood, phosphorylation at the cdc2 sites is believed to be related to the breakdown of the nucleolus during mitosis, whereas CKII phosphorylation has been proposed to play a role in ribosome biogenesis [45]. Recent studies show that NPM phosphorylation by CKII is involved in ribosome biogenesis by increasing its binding affinity to other nucleolar components as well as positively regulating nuclear import of proteins. Phosphorylation by CKII enhances NPM’s affinity to the NLS sequences derived from the SV40 large T antigen and HIV Rev protein [13,46], as well as modulating its molecular chaperoning activity, especially for its interaction with target proteins [47]. In this study, NPM phosphorylated with a cdc2-like protein kinase or mutant forms of NPM in which the nuclear localization signal is either deleted or altered do not stimulate nuclear import of the substrates. Thus, these results suggest that NPM phosphorylation by CKII is necessary step for ribosome biogenesis.
Recently, it has been shown that NPM is a primary target of CDK2/cyclin E in the initiation of centrosome duplication [15]. CDK2/cyclin E phosphorylates NPM at Thr 199, which causes NPM to dissociate from the centrosomes, leading to the initiation of centrosome duplication. NPM associates specifically with unduplicated centrosomes and dissociates from centrosomes upon Thr199 phosphorylation by CDK2/cyclin E at the late G1 phase. Microinjection of an antibody against NPM, which prevents CDK2/cyclin E-mediated phosphorylation and the dissociation of centrosomal NPM, results in the suppression of centrosome duplication. Moreover, the expression of the phosphorylatable deletion mutant NPM results in the inhibition of centrosome duplication. Thus, NPM phosphorylation at T199 may be a necessary step in regulating the function of NPM on centrosome duplication. Interestingly, NPM has been previously shown to be phosphorylated on Thr234 and Thr237 by CDK1/cyclin B. Thus, phosphorylation may play a role in NPM re-association with the centrosome during mitosis.
The role of NPM in regulating centrosome duplication has also been examined by employing NPM-specific siRNA to knock-down the expression of endogeneous NPM [16]. NPM siRNA consistently results in supernumerary centrosomes and amplified centrosomes could be nucleated in mitotic cells expressing NPM siRNA to form spindle poles. In this study, another potential phosphorylation site within the NES motif upstream of the proposed CDK2-cyclin E domain has been identified. In addition, site-directed mutagenesis reveals a potential regulatory role of a NPM phosphorylation site at Thr95 in the NES motif. A potential proline-dependent kinase phosphorylation site mutant (T95A) of NPM is able to associate with the centrosome and suppress premature centrosome duplication induced by NPM siRNA. A mutant mimicking the phosphorylation at T95 (T95D) has a decreased ability to bind to centrosomes and is ineffective in suppressing NPM-siRNA-induced centrosome duplication. Thus, it is possible that the phosphorylation at T95 may regulate NPM’s association with the centrosome. However, the identities of the kinases still remain to be determined.
Another study by Zhang et al [48] revealed that a mitosis-specific phosphorylation event occurs in NPM at a conserved Ser-4 residue by Plk1. Interfering with Ser-4 phosphorylation of NPM in HeLa cells by the overexpression of a NPM mutant carrying point mutations at this phosphorylation site leads to abnormalities in centrosome duplication, centrosome segregation, and cytokinesis. It seems that NPM Ser-4 phosphorylation may play an indirect role in centrosome duplication as such a phosphorylation does not correlate temporally with that of centrosome duplication as well as that it does not exert any effects on Thr199 phosphorylation. It will be interesting to determine how various phosphorylation events cooperate together to regulate NPN functions during cell cycle progression and centrosome duplication.
3.3. Transportation of NPM by Ran/CRM1 Complex in Centrosome Duplication
Centrosomes are the principal microtubule organizing centers of animal cells that establish a microtubule network during interphase and direct bipolar mitotic spindle formation during mitosis. Centrosome duplication in mammalian cells is a tightly controlled process which is coordinated with other cell cycle events. In somatic cells, centrosome duplication begins near the G1/S boundary and is completed during the S phase, coinciding with DNA replication. During cytokinesis, each daughter cell inherits one centrosome, hence centrosomes duplication occurs only once during each cell cycle. To ensure that centrosomes duplicate only once per cell cycle, a complex regulatory network is in place in normal cells, because abnormal centrosome duplication is tightly linked to aneuploidy and is found in virtually every type of human cancer [49]. However, the precise mechanism of how centrosome duplication is controlled is not well defined. Recent studies indicate that the Ran-CRM1 network is involved in regulating centrosome duplication to ensure the formation of a bipolar spindle [50-52]. CRM1 is the export vector for proteins from the nucleus to the cytoplasm and recognizes the NES in these proteins. CRM1 is mainly nuclear, but when soluble proteins are extracted from cells, a small fraction remains attached to the centrosome. A small amount of Ran GTP is tightly associated with the centrosome throughout the cell cycle. The interaction of Ran with the centrosome is mediated by the centrosomal matrix A kinase anchoring protein, AKAP450 [51]. The guanine nucleotide exchange factor, RCC1, facilitates Ran binding to CRM1, while RanBP1, a major regulator of Ran, promotes CRM1 dissociation from Ran. Our recent findings indicate that CRM1 may regulate the fidelity of mitotic centrosome maintenance by acting as a licensing factor to prevent unscheduled duplication [50]. The inactivation of CRM1 function either by a CRM1-specific inhibitor, leptomycin B (LMB), or hepatitis B virus (HBV) HBx protein interaction through its NES motif leads to abnormal centrosome amplication and multipolar spindles. Moreover, the alteration of Ran network components, including Ran and RanBP1, results in multipolar spindles or mitotic centriole splitting, an event leading to the unscheduled initiation of centrosome duplication [52]. Thus, the Ran/CRM1 complex may negatively regulate the initiation of centrosome duplication, possibly through its association with NES-containing proteins [50].
The presence of a functional NES motif in NPM suggests that the Ran/CRM1 complex modulates centrosome duplication by regulating NPM transport and localization. Our studies indicate that NPM may be a substrate for Ran/CRM1 to regulate centrosome duplication and this process is mediated through its NES domain [16]. NPM and CRM1 colocalize on the centrosomes, and NPM coimmunoprecipitates with CRM1. The disruption of CRM1 function by LMB, RanBP1, or HBx leads to NPM dissociation from the centrosomes and a subsequent initiation of premature centrosome duplication. Our study demonstrates that the NPM NES motif is required for CRM1 binding and docking on centrosomes and NPM binding to CRM1 may be regulated by phosphorylation at T95. Together with previous findings, the following scenario for NPM’s role in centrosome duplication can be drawn (Fig. 2). During cell progression from S to G2 phase, a major portion of NPM is located in the nucleolus, mainly functioning as a regulator of ribosome biogenesis and cell proliferation [53]. After the nuclear membrane breaks down at the beginning of mitosis, NPM reassociates with mitotic centrosomes through its interaction with the Ran/CRM1 complex, resulting in the prevention of centrosome reduplication. A newly divided G1 cell contains an NPM-bound centrosome. NPM is then phosphorylated and dissociates from the centrosome to initiate centrosome duplication and DNA replication. Thus, the Ran network together with its substrates may regulate the cell cycle to ensure faithful cell division. Interestingly, a recent study indicates that the leucine residues 42 and 44 of NPM may also serve as an NES to direct Crm1-mediated ribosomal protein L5 nuclear export [54]. It will be interesting to determine the functional connection between these two NESs and how they are contributed to centrosome duplication and other cellular activities.
Fig. 2.
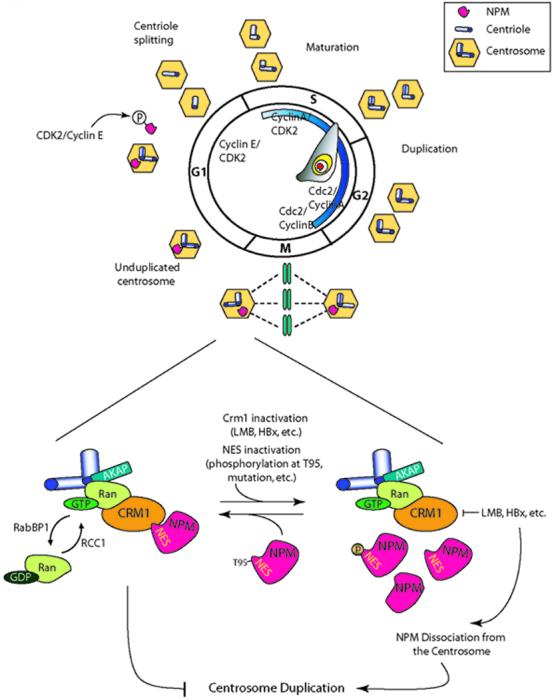
NPM functions as a licensing factor for centrosome duplication and its transportation to the centrosome is regulated by the Ran/CRM1 complex. NPM associates specifically with unduplicated centrosomes, and dissociates from centrosomes following CDK2 kinase phosphorylation during late G1 phase. This event allows centrosome duplication to take place. During cell cycle progression from the S to the G2 phase, NPM is mainly located in the nucleolus where it acts as a regulator of ribosome biogenesis. When the nuclear membrane breaks down at the beginning of mitosis, NPM is relocalized to the centrosome, resulting in the prevention of centrosome reduplication. Newly divided cells contain a NPM-bound centrosome. During mitosis, NPM relocalization to the centrosomes occurs by interacting with the Ran/CRM1 complex through the NES motif. Ran, a small GTPase, can cycle between an active GTP and an inactive GDP-bound form by RCC1 and RanBP1, respectively. GTP-bound Ran localizes at the centrosome with AKAP and CRM1. Inhibition of CRM1 by LMB or HBx, and NES inactivation by phosophorylation at T95, or through mutation of the NPM NES motif of NPM, leads to dissociation from the centrosome and eventually allows centrosome amplication.
3.4. Alteration of NPM in human cancer
NPM is overexpressed in many types of major human solid tumors including tumors of colon, liver, stomach, ovary and prostate [26]. In addition, disruption of the NPM gene by translocation is frequently found in human hematopoietic malignancies [18-20]. Recent studies indicate that about one-third of adult acute myeloid leukemia (AML) contain aberrant cytoplasmic expression of NPM with mutations occurring at the exon-12 of the NPM gene [21]. The recent article by Falini et al [21] and the letter by Nakagawa et al [55] put forward a working model on the mechanism underlying the abnormal cytoplasmic localization of NPM in AML. They suggested that mutated NPM may utilize a Crm1-mediated nuclear export mechanism to lend its cytoplasmic accumulation through a frameshift mutation resulting in the creation of a functional NES motif or by amino acid substitutions at residues 288 and 299 to alter nucleolar localization. Further analyses indicate that the new NES motif created by the mutational event is not in itself sufficient to cause nuclear export of NPM but needs to act in concert with the mutated tryptophan(s) 288 and 290 at the mutant C-terminus [56]. Furthermore, they found that two NESs, i.e., a physiological NES at position 92-104 and a ‘pathological’ NES at the C-terminus created by mutations, are better than one to promote NPM cytoplasmic expression [56]. It remains intriguing whether NPM cytoplasmic accumulation is associated with carcinogenesis, as we and others demonstrated its key role in the maintenance of centrosome duplication contributed by the presence of a physiological NES in NPM.
3.5. NPM as a New Molecular Player for Carcinogenesis
NPM is a multifunctional protein, and its physiological function in tumorigenesis is still controversial as NPM has been ascribed with both tumor suppressive and oncogenic functions. The physiological role of NPM has been demonstrated by the generation of NPM1-deficient mice [17]. NPM1 mutant mice have aberrant oncogenesis and die between embryonic day E11.5 and E16.5 due to severe anemia resulting from defects in primitive haematopoiesis. NPM1 inactivation leads to unrestricted centrosome duplication and genomic instability, implying that NPM is essential for embryonic development and the maintenance of genomic stability. Thus, NPM may serve as a tumor suppressor because abrogating its function leads to tumorigenic phenotypes. Consistent with this view, translocation at the mid-portion and mutation at the C-terminus of the NPM gene occur in a high proportion of hematological disorders, such as promyelocytic leukemia, anaplastic large cell lymphoma (ALCL), AML, chronic myelogenous leukemia and myelodysplastic syndromes (MDS) [18]. Interestingly, translocation only occurs in the NPM N-terminal region where it fuses to different partner genes, such as anaplastic lymphoma kinase (ALK), retinoic acid receptor a (RARa), or myeloid leukemia factor 1 (MLF1) in ALCL, acute promyelocytic leukemia (APL) and MDS, respectively [18-20] (Fig.1D). NPM appears to contribute to oncogenesis by activating the oncogenic potential of the fused protein partner, i.e., ALK, RARa, or MLF1. Furthermore, the deletion of the long arm of chromosome 5 in which NPM is located or all of chromosome 5 is frequently observed in patients with de novo and therapy-related MDS as well as in solid tumors. Another way to block the tumor suppressor function of NPM is to perturb its movement from the nucleus to the cytoplasm that may be critical for malignant transformation. For example, as described above, changes in the subcellular distribution of NPM, such as cytoplasmic dislocation, has been reported in ∼35% of specimens from patients with AML and in a small percentage of pediatric AML patients [21,56]. Analyses of the NPM coding region by RT-PCR and direct sequencing revealed a frame shift mutation in exon 12 of NPM, resulting in the reading frame altered C-terminal portion by replacing seven amino acids with 11 different residues (Fig.1C). In another study, NPM redistribution by UV damage leads to HDM2 interaction, and its binding could affect p53 stabilization by blocking HDM2-p53 complex formation [57]. Thus, NPM may have a role as a tumor suppressor in cells by regulating centrosome duplication or the function of tumor suppressors such as p53.
By contrast, ample evidence has linked NPM with uncontrolled growth, and suggests that NPM may have oncogenic potential when overexpressed. NPM may contribute to oncogenesis by activating the oncogenic potential of the fused protein partner (ALK, RARa, or MLF1)[18-20]. NPM is upregulated when B cells, T cells and Swiss 3T3 cells are stimulated with various mitotic agents [2,58]. NPM is significantly more abundant in many types of tumor and proliferating cells than in normal resting cells [59,60], and cells expressing a large amount of NPM are resistant to apoptosis induced by either UV damage or hypoxia [61,62]. It has been shown that NPM overexpression reduces the percentage of cells in the G1 phase and increases the S-phase population in p53-negative cells, but increases cell cycle arrest in normal cells [53]. Conversely, NPM downregulation is observed in Jurket T-lymphoblasts during apoptosis [63], and delays the cell cycle and M-phase entry [64]. NPM has been shown to be a target of oncogenic c-myc and NPM mRNA can be directly induced by c-Myc through binding to the NPM promoter [65]. Recently, it was demonstrated that NPM targets ARF to nucleoli and blocks ARF-mediated p53 activation and growth suppression in a dose-dependent manner by impairing its ability to interact with nucleoplasmic Mdm2 [24]. Another study using clinical cancer samples also showed a correlation between the level of NPM and the aggressiveness of cell growth as well as cancerous growth [66]. In concordance with these observations, the level of NPM seems to decrease when cells reach contact inhibition. Taken together, these findings suggest that NPM includes a potential role as a positive regulator of cell proliferation. These properties will make NPM a good biomarker for cancer diagnosis. Nevertheless, the apparent paradoxical activities of NPM found in human cancers and in a mouse model with genetic disruption of the NPM gene are intriguing. It remains a challenging task to determine how various functions of NPM are connected to regulate cellular homeostasis.
4. Discussion
NPM is a multifunctional protein involved in an array of biological and pathological processes controlling development, ribosome biogenesis, cell proliferation, transformation and genomic stability. The functions of NPM are strictly regulated by multiple factors (Fig.3). These include its oligomerization status, the expression of various isoforms, phosphorylation or subcellular localization, and these factors can have profound effects on ribosome biogenesis, centrosome duplication and cell cycle progression. NPM functions as an essential regulator for regulating centrosome amplification, genomic stability as well as carcinogenesis. Alterations of NPM function by loss-of-function, mutation or translocation can have differing effects and may contribute to oncogenesis by losing the regulatory potentials of NPM on these processes. Recent studies from our group showed that NES-containing NPM may serve as a crucial licensing factor to ensure the fidelity of centrosome duplication. The Ran/CRM1 complex can interact with NPM through the NES motif, which may allow NPM to localize to the centrosome at the beginning of mitosis. This process may be regulated by phosphorylation of T95.
Fig. 3.

Variable cellular localization of NPMs and its physiological roles. The cellular functions of NPMs are tightly regulated by multiple factors that determine the direction of NPM functions. Oligomerization may modulate the nucleic acid binding activity and chaperone activity of NPM during ribosome biogenesis, and hetero-oligomerization with isoform, NPM1.2, impairs nucleic acid binding activity. NPM phosphorylation by various upstream kinases is necessary to regulate its function in ribosome biogenesis, centrosome duplication and cell cycle progression. NPM can localize in several places and its local trafficking to the specific target sites leads to coordinated ribosome biogenesis, centrosome duplication and cell cycle progression. NPM functions as a pivotal regulator for controlling cell cycle progression, centrosome duplication, genomic instability, as well as carcinogenesis. Alteration of NPM function through mutation or translocation may contribute to oncogenesis.
NPM is strongly linked to the uncontrolled growth of cancer cells. Cancer, which is the leading cause of death in the US according to the American Cancer Society, is a very complex and heterogeneous disease. Its heterogeneity makes cancer resistant to treatments. Theoretically, if all cancers have common traits, then treatments targeting such common traits could be an efficient therapeutic tool capable of killing the majority of cancer cells. The abundance of NPM may serve as a common trait in cancer cells and a high level of NPM may be beneficial to the survival of cancer cells [66]. It is expected that a continued growth of tumor cells requires a continued supply of proteins, requiring continued ribosome biogenesis. Because NPM is an essential factor in this process, it is possible to use an approach targeting NPM for the treatment of different types of cancers. Furthermore, given that NPM has oncogenic potential when overexpressed, NPM may function to translate or amplify multiple oncogenic signaling mechanisms during carcinogenesis. The determination of the in vivo role of NPM during oncogenesis, using model systems such as NPM transgenic mouse models with or without other oncogenes, need to be elucidated in future studies.
Acknowledgements
We thank the editorial assistance of Dorothea Dudek-Creaven and the National Institutes of Health Fellows Editorial Board for editing this manuscript. This research was supported by the Intramural Research Program of Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Footnotes
Source of support: This research was supported by the Intramural Research Program of NCI, NIH.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference
- 1.Yung BY, Busch H, Chan PK. Translocation of nucleolar phosphoprotein B23 (37 kDa/pI 5.1) induced by selective inhibitors of ribosome synthesis. Biochim Biophys Acta. 1985;826:167–173. doi: 10.1016/0167-4781(85)90002-8. [DOI] [PubMed] [Google Scholar]
- 2.Feuerstein N, Mond JJ. “Numatrin,” a nuclear matrix protein associated with induction of proliferation in B lymphocytes. J Biol Chem. 1987;262:11389–11397. [PubMed] [Google Scholar]
- 3.Schmidt-Zachmann MS, Hugle-Dorr B, Franke WW. A constitutive nucleolar protein identified as a member of the nucleoplasmin family. EMBO J. 1987;6:1881–1890. doi: 10.1002/j.1460-2075.1987.tb02447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang JH, Olson MO. Structure of the gene for rat nucleolar protein B23. J Biol Chem. 1990;265:18227–18233. [PubMed] [Google Scholar]
- 5.Wang D, Baumann A, Szebeni A, Olson MO. The nucleic acid binding activity of nucleolar protein B23.1 resides in its carboxyl-terminal end. J Biol Chem. 1994;269:30994–30998. [PubMed] [Google Scholar]
- 6.Li YP, Busch RK, Valdez BC, Busch H. C23 interacts with B23, a putative nucleolar-localization-signal-binding protein. Eur J Biochem. 1996;237:153–158. doi: 10.1111/j.1432-1033.1996.0153n.x. [DOI] [PubMed] [Google Scholar]
- 7.Valdez BC, Perlaky L, Henning D, Saijo Y, Chan PK, Busch H. Identification of the nuclear and nucleolar localization signals of the protein p120. Interaction with translocation protein B23. J Biol Chem. 1994;269:23776–23783. [PubMed] [Google Scholar]
- 8.Fankhauser C, Izaurralde E, Adachi Y, Wingfield P, Laemmli UK. Specific complex of human immunodeficiency virus type 1 rev and nucleolar B23 proteins: dissociation by the Rev response element. Mol Cell Biol. 1991;11:2567–2575. doi: 10.1128/mcb.11.5.2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li YP. Protein B23 is an important human factor for the nucleolar localization of the human immunodeficiency virus protein Tat. J Virol. 1997;71:4098–4102. doi: 10.1128/jvi.71.5.4098-4102.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Herrera JE, Savkur R, Olson MO. The ribonuclease activity of nucleolar protein B23. Nucleic Acids Res. 1995;23:3974–3979. doi: 10.1093/nar/23.19.3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Savkur RS, Olson MO. Preferential cleavage in pre-ribosomal RNA byprotein B23 endoribonuclease. Nucleic Acids Res. 1998;26:4508–4515. doi: 10.1093/nar/26.19.4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borer RA, Lehner CF, Eppenberger HM, Nigg E. Major nucleolar proteins shuttle between nucleus and cytoplasm. Cell. 1989;56:379–390. doi: 10.1016/0092-8674(89)90241-9. [DOI] [PubMed] [Google Scholar]
- 13.Szebeni A, Herrera JE, Olson MO. Interaction of nucleolar protein B23 with peptides related to nuclear localization signals. Biochemistry. 1995;34:8037–8042. doi: 10.1021/bi00025a009. [DOI] [PubMed] [Google Scholar]
- 14.Szebeni A, Olson MO. Nucleolar protein B23 has molecular chaperone activities. Protein Sci. 1999;8:905–912. doi: 10.1110/ps.8.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okuda M, Horn HF, Tarapore P, Tokuyama Y, Smulian AG, Chan PK, et al. Nucleophosmin/B23 is a target of CDK2/cyclin E in centrosome duplication. Cell. 2000;103:127–140. doi: 10.1016/s0092-8674(00)00093-3. [DOI] [PubMed] [Google Scholar]
- 16.Wang W, Budhu A, Forgues M, Wang X. Temporal and spatial control of nucleophosmin by the Ran-Crm1 complex in centrosome duplication. Nat.Cell Biol. 2005;7:823–830. doi: 10.1038/ncb1282. [DOI] [PubMed] [Google Scholar]
- 17.Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K, et al. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005;437:147–153. doi: 10.1038/nature03915. [DOI] [PubMed] [Google Scholar]
- 18.Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 19.Redner RL, Rush EA, Faas S, Rudert WA, Corey SJ. The t(5;17) variant of acute promyelo-cytic leukemia expresses a nucleophosmin-retinoic acid receptor fusion. Blood. 1996;87:882–886. [PubMed] [Google Scholar]
- 20.Yoneda-Kato N, Look AT, Kirstein MN, Valentine MB, Raimondi SC, Cohen KJ, et al. The t(3;5)(q25.1;q34) of myelodysplastic syndrome and acute myeloid leukemia produces a novel fusion gene, NPM-MLF1. Oncogene. 1996;12:265–275. [PubMed] [Google Scholar]
- 21.Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L, et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med. 2005;352:254–266. doi: 10.1056/NEJMoa041974. [DOI] [PubMed] [Google Scholar]
- 22.Bertwistle D, Sugimoto M, Sherr CJ. Physical and functional interactions of the Arf tumor suppressor protein with nucleophosmin/B23. Mol Cell Biol. 2004;24:985–996. doi: 10.1128/MCB.24.3.985-996.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brady SN, Yu Y, Maggi LB, Jr, Weber JD. ARF impedes NPM/B23 shuttling in an Mdm2-sensitive tumor suppressor pathway. Mol Cell Biol. 2004;24:9327–9338. doi: 10.1128/MCB.24.21.9327-9338.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korgaonkar C, Hagen J, Tompkins V, Frazier AA, Allamargot C, Quelle FW, et al. Nucleophosmin (B23) targets ARF to nucleoli and inhibits its function. Mol Cell Biol. 2005;25:1258–1271. doi: 10.1128/MCB.25.4.1258-1271.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colombo E, Marine JC, Danovi D, Falini B, Pelicci PG. Nucleophosmin regulates the stability and transcriptional activity of p53. Nat Cell Biol. 2002;4:529–533. doi: 10.1038/ncb814. [DOI] [PubMed] [Google Scholar]
- 26.Grisendi S, Mecucci C, Falini B, Pandolfi PP. Nucleophosmin and cancer. Nat Rev Cancer. 2006;6:493–505. doi: 10.1038/nrc1885. [DOI] [PubMed] [Google Scholar]
- 27.Wang D, Umekawa H, Olson MO. Expression and subcellular locations of two forms of nucleolar protein B23 in rat tissues and cells. Cell Mol Biol Res. 1993;39:33–42. [PubMed] [Google Scholar]
- 28.Okuwaki M, Tsujimoto M, Nagata K. The RNA binding activity of a ribosome biogenesis factor, nucleophosmin/B23, is modulated by phosphorylation with a cell cycle-dependent kinase and by association with its subtype. Mol Biol Cell. 2002;13:2016–2030. doi: 10.1091/mbc.02-03-0036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nishimura Y, Ohkubo T, Furuichi Y, Umekawa H. Tryptophans 286 and 288 in the C-terminal region of protein B23.1 are important for its nucleolar localization. Biosci Biotechnol Biochem. 2002;66:2239–2242. doi: 10.1271/bbb.66.2239. [DOI] [PubMed] [Google Scholar]
- 30.Yung BY, Chan PK. Identification and characterization of a hexameric form of nucleolar phosphoprotein B23. Biochim Biophys Acta. 1987;925:74–82. doi: 10.1016/0304-4165(87)90149-8. [DOI] [PubMed] [Google Scholar]
- 31.Umekawa H, Chang JH, Correia JJ, Wang D, Wingfield PT, Olson MO. Nucleolar protein B23: bacterial expression, purification, oligomerization and secondary structures of two isoforms. Cell Mol Biol Res. 1993;39:635–645. [PubMed] [Google Scholar]
- 32.Herrera JE, Correia JJ, Jones AE, Olson MO. Sedimentation analyses of the salt- and divalent metal ion-induced oligomerization of nucleolar protein B23. Biochemistry. 1996;35:2668–2673. doi: 10.1021/bi9523320. [DOI] [PubMed] [Google Scholar]
- 33.Chan PK, Chan FY. Nucleophosmin/B23 (NPM) oligomer is a major and stable entity in HeLa cells. Biochim Biophys Acta. 1995;1262:37–42. doi: 10.1016/0167-4781(95)00044-h. [DOI] [PubMed] [Google Scholar]
- 34.Dutta S, Akey IV, Dingwall C, Hartman KL, Laue T, Nolte RT, et al. The crystal structure of nucleoplasmin-core: implications for histone binding and nucleosome assembly. Mol Cell. 2001;8:841–853. doi: 10.1016/s1097-2765(01)00354-9. [DOI] [PubMed] [Google Scholar]
- 35.Hingorani K, Szebeni A, Olson MO. Mapping the functional domains of nucleolar protein B23. J Biol Chem. 2000;275:24451–24457. doi: 10.1074/jbc.M003278200. [DOI] [PubMed] [Google Scholar]
- 36.Zirwes RF, Kouzmenko AP, Peters JM, Franke WW, Schmidt-Zachmann MS, et al. Topogenesis of a nucleolar protein: determination of molecular segments directing nucleolar association. Mol Biol Cell. 1997;8:231–248. doi: 10.1091/mbc.8.2.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chou YH, Yung BY. Cell cycle phase-dependent changes of localization and oligomerization states of nucleophosmin/B23. Biochem Biophys Res Commun. 1995;217:313–325. doi: 10.1006/bbrc.1995.2779. [DOI] [PubMed] [Google Scholar]
- 38.Krebs EG, Beavo JA. Phosphorylation-dephosphorylation of enzymes. Annu Rev Biochem. 1979;48:923–959. doi: 10.1146/annurev.bi.48.070179.004423. [DOI] [PubMed] [Google Scholar]
- 39.Peter M, Nakagawa J, Doree M, Labbe JC, Nigg EA. Identification of major nucleolar proteins as candidate mitotic substrates of cdc2 kinase. Cell. 1990;60:791–801. doi: 10.1016/0092-8674(90)90093-t. [DOI] [PubMed] [Google Scholar]
- 40.Chan PK, Liu QR, Durban E. The major phosphorylation site of nucleophosmin (B23) is phosphorylated by a nuclear kinase II. Biochem J. 1990;270:549–552. doi: 10.1042/bj2700549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones CE, Busch H, Olson MO. Sequence of a phosphorylation site in nucleolar protein B23. Biochim Biophys Acta. 1981;667:209–212. doi: 10.1016/0005-2795(81)90081-7. [DOI] [PubMed] [Google Scholar]
- 42.Feuerstein N, Randazzo PA. In vivo and in vitro phosphorylation studies of numatrin, a cell cycle regulated nuclear protein, in insulin-stimulated NIH 3T3 HIR cells. Exp Cell Res. 1991;194:289–296. doi: 10.1016/0014-4827(91)90367-4. [DOI] [PubMed] [Google Scholar]
- 43.Jiang PS, Chang JH, Yung BY. Different kinases phosphorylate nucleophosmin/B23 at different sites during G(2) and M phases of the cell cycle. Cancer Lett. 2000;153:151–160. doi: 10.1016/s0304-3835(00)00362-1. [DOI] [PubMed] [Google Scholar]
- 44.Chan PK, Aldrich M, Cook RG, Busch H. Amino acid sequence of protein B23 phosphorylation site. J Biol Chem. 1986;261:1868–1872. [PubMed] [Google Scholar]
- 45.Olson MO. The eukaryotic nucleus: molecular biochemistry and macromolecular Assemblies. Telford Press; West Caldwell, NJ: 1990. [Google Scholar]
- 46.Szebeni A, Mehrotra B, Baumann A, Adam SA, Wingfield PT, Olson MO. Nucleolar protein B23 stimulates nuclear import of the HIV-1 Rev protein and NLS-conjugated albumin. Biochemistry. 1997;36:3941–3949. doi: 10.1021/bi9627931. [DOI] [PubMed] [Google Scholar]
- 47.Szebeni A, Hingorani K, Negi S, Olson MO. Role of protein kinase CK2 phosphorylation in the molecular chaperone activity of nucleolar protein b23. J Biol Chem. 2003;278:9107–9115. doi: 10.1074/jbc.M204411200. [DOI] [PubMed] [Google Scholar]
- 48.Zhang H, Shi X, Paddon H, Hampong M, Dai W, Pelech S. B23/nucleophosmin serine 4 phosphorylation mediates mitotic functions of polo-like kinase 1. J Biol Chem. 2004;279:35726–35734. doi: 10.1074/jbc.M403264200. [DOI] [PubMed] [Google Scholar]
- 49.Lingle WL, Salisbury JL. The role of the centrosome in the development of malignant tumors. Curr Top Dev Biol. 2000;49:313–329. doi: 10.1016/s0070-2153(99)49015-5. [DOI] [PubMed] [Google Scholar]
- 50.Forgues M, Difilippantonio MJ, Linke SP, Ried T, Nagashima K, Feden J, et al. Involvement of Crm1 in hepatitis B virus X protein-induced aberrant centriole replication and abnormal mitotic spindles. Mol Cell Biol. 2003;23:5282–5292. doi: 10.1128/MCB.23.15.5282-5292.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keryer G, Di Fiore B, Celati C, Lechtreck KF, Mogensen M, Delouvee A, et al. Part of Ran is associated with AKAP450 at the centrosome: involvement in microtubule-organizing activity. Mol Biol Cell. 2003;14:4260–4271. doi: 10.1091/mbc.E02-11-0773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Fiore B, Ciciarello M, Lavia P. Mitotic Functions of the Ran GTPase Network: The Importance of Being in the Right Place at the Right Time. Cell Cycle. 2004;3:305–313. [PubMed] [Google Scholar]
- 53.Itahana K, Bhat KP, Jin A, Itahana Y, Hawke D, Kobayashi R, et al. Tumor suppressor ARF degrades B23, a nucleolar protein involved in ribosome biogenesis and cell proliferation. Mol Cell. 2003;12:1151–1164. doi: 10.1016/s1097-2765(03)00431-3. [DOI] [PubMed] [Google Scholar]
- 54.Yu Y, Maggi LB, Brady SN, Apicelli AJ, Dai MS, Lu H, Weber JD. Nucleophosmin is essential for ribosomal protein L5 nuclear export. Mol Cell Biol. 2006;26:3798–3809. doi: 10.1128/MCB.26.10.3798-3809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Falini B, Bolli N, Shan J, Martelli MP, Liso A, Pucciarini A, et al. Both carboxy-terminus NES motif and mutated tryptophan(s) are crucial for aberrant nuclear export of nucleophosmin leukemic mutants in NPMc+ AML. Blood. 2006;107:4514–4523. doi: 10.1182/blood-2005-11-4745. [DOI] [PubMed] [Google Scholar]
- 56.Cazzaniga G, Dell’Oro MG, Mecucci C, Giarin E, Masetti R, Rossi V, et al. Nucleophosmin mutations in childhood acute myelogenous leukemia with normal karyotype. Blood. 2005;106:1419–1422. doi: 10.1182/blood-2005-03-0899. [DOI] [PubMed] [Google Scholar]
- 57.Kurki S, Peltonen K, Latonen L, Kiviharju TM, Ojala PM, Meek D, et al. Nucleolar protein NPM interacts with HDM2 and protects tumor suppressor protein p53 from HDM2-mediated degradation. Cancer Cell. 2004;5:465–475. doi: 10.1016/s1535-6108(04)00110-2. [DOI] [PubMed] [Google Scholar]
- 58.Feuerstein N, Chan PK, Mond JJ. Identification of numatrin, the nuclear matrix protein associated with induction of mitogenesis, as the nucleolar protein B23. Implication for the role of the nucleolus in early transduction of mitogenic signals. J Biol Chem. 1988;263:10608–10612. [PubMed] [Google Scholar]
- 59.Feuerstein N, Spiegel S, Mond JJ. The nuclear matrix protein, numatrin (B23), is associated with growth factor-induced mitogenesis in Swiss 3T3 fibroblasts and with T lymphocyte proliferation stimulated by lectins and anti-T cell antigen receptor antibody. J Cell Biol. 1988;107:1629–1642. doi: 10.1083/jcb.107.5.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chan WY, Liu QR, Borjigin J, Busch H, Rennert OM, Tease LA, et al. Characterization of the cDNA encoding human nucleophosmin and studies of its role in normal and abnormal growth. Biochemistry. 1989;28:1033–1039. doi: 10.1021/bi00429a017. [DOI] [PubMed] [Google Scholar]
- 61.Li J, Zhang X, Sejas DP, Pang Q. Hypoxia-induced nucleophosmin protects cell death through inhibition of p53. J Biol Chem. 2004;279:41275–41279. doi: 10.1074/jbc.C400297200. [DOI] [PubMed] [Google Scholar]
- 62.Wu MH, Chang JH, Yung BY. Resistance to UV-induced cell-killing in nucleophosmin/B23 over-expressed NIH 3T3 fibroblasts: enhancement of DNA repair and up-regulation of PCNA in association with nucleophosmin/B23 over-expression. Carcinogenesis. 2002;23:93–100. doi: 10.1093/carcin/23.1.93. [DOI] [PubMed] [Google Scholar]
- 63.Patterson SD, Grossman JS, D’Andrea P, Latter GI. Reduced numatrin/B23/nucleophosmin labeling in apoptotic Jurkat T-lymphoblasts. J Biol Chem. 1995;270:9429–9436. doi: 10.1074/jbc.270.16.9429. [DOI] [PubMed] [Google Scholar]
- 64.Jiang PS, Yung BY. Down-regulation of nucleophosmin/B23 mRNA delays the entry of cells into mitosis. Biochem Biophys Res Commun. 1999;257:865–870. doi: 10.1006/bbrc.1999.0551. [DOI] [PubMed] [Google Scholar]
- 65.Zeller KI, Haggerty TJ, Barrett JF, Guo Q, Wonsey DR, Dang CV. Characterization of nucleophosmin (B23) as a Myc target by scanning chromatin immunoprecipitation. J Biol Chem. 2001;276:48285–48291. doi: 10.1074/jbc.M108506200. [DOI] [PubMed] [Google Scholar]
- 66.Zhang Y. The ARF-B23 connection: implications for growth control and cancer treatment. Cell Cycle. 2004;3:259–262. [PubMed] [Google Scholar]