

Abstract
Quantitation of wild-type and deleted mitochondrial DNA (mtDNA) co-existing within the same cell, (a.k.a. heteroplasmy), is important in mitochondrial disease and aging. We report the development of a multiplex three-primer PCR assay that is capable of absolute quantitation of wild-type and deleted mtDNA simultaneously. Molecular beacons were designed to hybridize with either mtDNA molecule, which allows real-time detection during PCR amplification. The assay is specific and can detect down to 6 copies of mtDNA, making it suitable for single-cell analyses. The relative standard deviation in the threshold cycle number is ~0.6%. Heteroplasmy was quantitated in individual cytoplasmic hybrid cells (cybrids), containing a large mtDNA deleteion, and bulk cell samples. Individual cybrid cells contained 100–2600 copies of wild-type mtDNA, 950–4,700 copies of deleted mtDNA and the percent of heteroplasmy ranged from 43±16% to 95±16%. The average amount of total mtDNA was 3800±1600 copies/cybrid cell and the average percent of heteroplasmy correlated well with the bulk cell sample. The single-cell analysis also revealed that heteroplasmy in individual cells is highly heterogeneous. This assay will be useful to monitor clonal expansions of mtDNA deletions and investigate the role of heteroplasmy in cell-to-cell heterogeneity in cellular models of mitochondrial disease and aging.
Keywords: mitochondrial DNA quantitation, heteroplasmy, real-time PCR, molecular beacons
Introduction
Mitochondria contain their own genome, which codes for 13 polypeptides necessary for oxidative phosphorylation [1]. Mitochondrial DNA (mtDNA) mutations are linked with many myopathies and encephalopathies [2, 3] and are implicated in aging and age related diseases [4–7]. In particular, large mtDNA deletions result in mitochondrial diseases such as Pearson’s syndrome, Kearns-Sayre syndrome, progressive external ophthalmoplegia and the “common deletion” (i.e. mtDNA4977) has been shown to accumulate with age [2, 4, 8].
A single mitochondrion may contain 0-21 mtDNA molecules as reported by PCR [9] and fluorescence microscopy [10]. Single cells may contain hundreds of mitochondria and therefore hundreds to thousands of mtDNA molecules. In healthy cells all the mtDNA molecules are identical, which is termed homoplasmy. However, due to the polyploid nature of the mitochondrial genome, wild-type and mutated mtDNA may coexist in a single cell; this condition is known as heteroplasmy. The degree of heteroplasmy, i.e. the percent of mutated mtDNA, has been shown to be fundamental to the expression of the mutation phenotype [11–14]. However, bulk assays that use millions of cells to quantitate deleted mtDNA have proven unsuccessful in relating cell function with heteroplasmy levels in non-dividing tissues; single-cell studies were required [15]. Likewise, investigation of clonal expansion of mtDNA deletions can only be performed on individual cells. Therefore, quantitation of mtDNA heteroplasmy in single cells remains important in the study of mitochondrial disease and aging.
Several methods have been developed to identify and quantitate mtDNA mutations and have been reviewed previously [16]. The most common method for quantitation of deletion mutations is Southern blotting, but it lacks the sensitivity to detect deletions when the ratio of deleted to wild-type mtDNA is small or when the total amount of mtDNA is small, as in the case of single cells. Therefore, PCR-based assays must be applied in these cases to amplify and quantitate deleted and wild-type mtDNA. A typical PCR assay produces two mtDNA fragments; one fragment is used to measure the total amount of mtDNA and the other measures the amount of deleted mtDNA. This is routinely accomplished using two primer sets, but can be simplified to a three-primer PCR strategy [17]. Following PCR amplification the products are routinely separated by agarose gel electrophoresis or capillary electrophoresis and quantitated.
Real-time detection of PCR products has several advantages over conventional methods. Namely, real-time detection (i) is less labor intensive, (ii) has higher throughput, (iii) does not require post-amplification sample handling, which reduces carry-over contamination, and (iv) has a large dynamic range, typically over 6 orders of magnitude. In addition, it has shown excellent accuracy in quantitating mtDNA deletions and correlates well with traditional Southern blot and competitive three-primer PCR analyses for bulk samples [18].
We report the development of a multiplex PCR assay that is used to quantitate wild-type and deleted mtDNA from a cytoplasmic hybrid (cybrid) cell line in a single reaction. PCR is performed using a three-primer PCR method [17], which allows amplification of wild-type and deleted mtDNA simultaneously. Detection is accomplished using two molecular beacons [19] that hybridize specifically to the wild-type and deleted mtDNA PCR products. Absolute quantitation of heteroplasmy was performed from bulk and single cybrid cells. This is the first report of mtDNA deletion quantitation employing a three-primer PCR assay in conjunction with multiplex real-time detection using molecular beacons.
Materials and Methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum and agarose were purchased from Invitrogen (Carlsbad, CA, USA). Uridine was purchased from MP Biomedicals (Solon, OH, USA). Trypsin solution (10×, 5.0 g/L Trypsin, 2.0 g/L EDTA·4Na, 8.5 g/L NaCl), N-[2-hydroxyethyl]-[piperazine]-N-[ethanesulfonic acid] (HEPES), phosphate buffered saline (10x PBS, containing 100 mM KH2PO4 / Na2HPO4 solution, pH 7.4, 27 mM KCl, 1370 mM NaCl), gentamycin, ethylenediaminetetraacetic acid (EDTA), ethyleneglycol-bis(β-aminoethyl)-N,N,N′,N′-tetraacetic acid (EGTA), mannitol, poly(vinyl alcohol) (PVA) average MW 31,000–50,000 and 98–99% hydrolyzed, Triton X-100 and proteinase K were purchased from Sigma (St. Louis, MO, USA). Sucrose was purchased from Roche Diagnostics (Indianapolis, IN, USA). Ethidium bromide was purchased from Bio-Rad Laboratories (Hercules, CA, USA). The 100 bp DNA ladder was purchased from New England Biolabs (Ipswich, MA, USA).
Cell Lines and Culturing
A human cytoplasmic hybrid (cybrid) cell line, containing a 7522 bp deletion (ΔH2-1) spanning positions 7982–15,504, was a generous gift from Dr. Carlos Moraes (University of Miami). The construction of the ΔH2-1 cell line has been described previously [20]. Briefly, enucleated fibroblasts from a patient with an infantile metabolic disorder were fused with mtDNA-less 143B/206 ρ0 cells. 143B human osteosarcoma cells were purchased from American Type Culture Collection (Manassas, VA, USA, CRL-8303). ΔH2-1 cells were cultured to confluence in 0.22-μm-filtered DMEM medium containing 10% (v/v) fetal bovine serum, 50 μg/mL uridine and 10 μg/mL gentamycin stored at 4 °C. The high-glucose DMEM medium was used to compensate for the dysfunctional oxidative phosphorylation system due to the mtDNA deletion. All cells were cultured in 75-cm2 vented culture flasks at 37 °C and 5% CO2 and split every 3–4 days. For splitting, the cells were rinsed with PBS, lifted with 0.25 g/L trypsin for 5 min and diluted in fresh growth medium.
Sample Preparation
Prior to PCR analysis cells were washed three times in ice cold 220 mM mannitol, 70 mM sucrose, 0.5 mM EGTA, 2 mM HEPES, pH 7.4 buffer (MSHE buffer). For single cell PCR, cells were diluted in 250 mM sucrose, 10 mM HEPES at pH 7.4 (S/H buffer) and deposited on a PVA-coated microscope slide. For bulk PCR analysis, cells were counted with a Fuchs-Rosenthal hemacytometer (Hausser Scientific, Horsham, PA, USA) and diluted in S/H buffer. To isolate mitochondria cells were disrupted using N2 cavitation (Parr Instrument, Moline, IL, USA). Whole cells, nuclei and membrane debris were removed by centrifugation at 600×g for 10 min at 4 °C in an Eppendorf 5415D centrifuge (Eppendorf, Westbury, NY, USA). The supernatant was removed and mitochondria were pelleted by centrifugation at 16,000×g for 10 min at 4 °C. Mitochondria were resuspended in ice cold S/H buffer.
MtDNA Standard Construction
The wild-type and deleted mtDNA standards were constructed by performing PCR directly from isolated 143B and ΔH2-1 mitochondria, respectively, based on a modification of the methods described by Melov et al. [21] and Khrapko et al. [22]. Briefly, 1 μL of 10 mM EDTA, 0.5% Triton X-100, 2 mg/mL proteinase K at pH 7.4 (lysis solution) was added to 5 μL of suspended mitochondria and incubated at 37 °C for 60 min. Following incubation, proteinase K was heat inactivated by incubation at 95 °C for 2 min. Brilliant® SYBR® Green QPCR master mix (Stratagene, La Jolla, CA, USA), DNase-free water and primers (300 nM final concentration) were added to the reaction mixture to a final volume of 50 μL. Primers were designed using FastPCR (http://www.biocenter.helsinki.fi/bi/Programs/fastpcr.htm) and synthesized by the MicroChemical Facility (University of Minnesota). Table 1 displays the sequences and positions at which the forward primer (S1) and the reverse primers (S2 and S3) hybridize to the mtDNA molecules. Primers S1 and S2 are used to amplify a fragment of the wild-type mtDNA molecule. Primers S1 and S3 encompass the deletion found in the ΔH2-1 cybrid cells and amplify a fragment of the deleted mtDNA molecule. The forward primer hybridizes to the heavy strand and the reverse primers hybridize to the light strand. In order to produce standards, amplification was performed using primers S1 and S2 for wild-type mtDNA (143B mitochondria) and primers S1 and S3 for deleted mtDNA (ΔH2-1 mitochondria) in separate reactions. Thermal cycling was performed using the Mx3000P™ QPCR system (Stratagene) as follows; an initial denaturation cycle for 2 min at 95 °C, followed by 40 cycles of denaturation for 30 seconds at 95 °C, annealing for 60 seconds at 55 °C and elongation for 90 seconds at 72 °C.
Table 1.
Primers and molecular beacons
| Primers | Position | Sequence |
|---|---|---|
| S1 | 7743–7763 | 5′-CTAACATCTCAGACGCTCAGG |
| S2 | 8290–8310 | 5’-AGTTAGCTTTACAGTGGGCTC |
| S3 | 15,909–15,928 | 5′-CCGGTTTACAAGACTGGTGT |
| F1 | 7798–7820 | 5′-CATCCTAGTCCTCATCGCCCTCC |
| R1 | 8186–8209 | 5′-GGGCATGAAACTGTGGTTTGCTCC |
| R2 | 15,727–15,749 | 5′-GAATGAGGAGGTCTGCGGCTAGG |
|
| ||
| Molecular Beacons | Position | Sequence |
|
| ||
| MB1a | 8072–8092 | FAM-5′-CCAGCGGCCTAATGTGGGGACAGCTCACGCTGG-BHQ1 |
| MB2a | 15,534–15,553 | HEX-5′-CCAGCGCTTGATGTGGGGAGGGGTGTCGCTGG-BHQ1 |
The stem sequence is shown in bold
Following PCR amplification of deleted and wild-type mtDNA standards, the PCR products were separated at 100V on 2% agarose gels for 1.5 h. PCR products were imaged with 0.75 μg/mL ethidium bromide and a Bio-Rad Molecular Imager FX (Bio-Rad Laboratories, Hercules, CA). The appropriate bands were excised and purified from the gel using the High Pure PCR Product Purification Kit (Roche Diagnostics, Indianapolis, IN, USA). Standards were quantitated with the Quant-iT™ PicoGreen® dsDNA assay kit (Invitrogen) and a spectrofluorometer (FP 6200, Jasco, Easton MD, USA). Quantitation was performed according to the supplier’s directions using λ-DNA (Invitrogen) to construct the calibration curve.
Three-Primer Multiplex Real-Time PCR Assay
To quantitate heteroplasmy, deleted and wild-type mtDNA were amplified simultaneously using primers F1, R1 and R2, shown in Table 1. Primer F1 hybridizes to the heavy strand and the reverse primers R1 and R2 hybridize to the light strand. Primers F1 and R2 lay outside the deletion and primer R1 lays inside the deletion. The wild-type and deleted PCR products were detected using molecular beacons MB1 and MB2 as displayed in Table 1. The stem sequence, shown in bold, was identical for both molecular beacons and has been described previously [23]. The FAM and HEX fluorescence was quenched by Black Hole Quencher-1 (BHQ1) when the molecular beacon was not hybridized to DNA. Molecular beacons were synthesized by Integrated DNA Technologies (Coralville, IA, USA).
Unless stated otherwise, PCR assays were performed using the FastStart TaqMan Probe Master Mix with ROX (Roche Diagnostics) and contained 300 nM of each molecular beacon and primer with DNase-free water added to make 25 μL reactions. Thermal cycling was performed by the Mx3000P™ QPCR system (Stratagene) as follows: 1 cycle of 95 °C for 10 min and 50 cycles of 95 °C for 30 seconds, 60 °C for 1 minute, 72 °C for 30 seconds. The fluorescence intensity was measured and recorded in real-time after the annealing stage in each PCR cycle.
Capture of Single Cells
Microscope slides were coated with PVA to reduce cell adhesion by spreading 10 μL of a 5% (w/v) PVA solution over the slide and allowing the slides to dry at 140 °C for 1 hour. ΔH2-1 cells were deposited on the microscope slide and a single cell was positioned beneath the lumen of a poly(acryloylamino propanol) modified capillary [24] (50 μm i.d., Polymicro Technologies, Phoenix, AZ, USA). Negative pressure was applied with a syringe pump to the outlet of the capillary to capture a single cell. An Olympus IX-81 inverted fluorescence microscope (Melville, NY, USA) and a C9100–01 EM CCD camera (Hamamatsu Corp., Bridgewater, NJ, USA) were used to visualize the capture of single cells in the bright-field. Image sequences were acquired with SimplePCI 5.3 software (Compix Imaging Systems, Cranberry Township, PA, USA) at 10 frames per second. Following capture, single cells were deposited in the PCR wells by applying positive pressure at the capillary outlet with the syringe pump. The PCR wells contained 5 μL of S/H buffer and 1 μL of lysis solution. Between capture of each cell the capillary was flushed with S/H buffer for ~1 min with the syringe pump.
Direct PCR Amplification from Cells
To release mtDNA from ΔH2-1 cells, cells were incubated in the lysis solution for 60 min at 37 °C, followed by heat inactivation of proteinase K at 95 °C for 2 min as described previously [21, 22]. Following incubation, the PCR reagents were added and PCR was performed as described above. For PCR from bulk ΔH2-1 cells the cell density was adjusted to 5×106 cells/mL in S/H buffer prior to incubation in the lysis solution and diluted prior to PCR.
Data Analysis
Data was analyzed using Mx3000P version 2.0 software (Stratagene). The threshold cycle was determined by measuring the cycle number at which the fluorescence reached a defined threshold. The optimal fluorescence threshold was determined by the Mx3000P software using the amplification-based algorithm. Once the threshold was optimized it remained constant for all samples within the experiment.
Results
Multiplex Three-Primer PCR
We report the development of a multiplex three-primer PCR assay that allows simultaneous quantitation of wild-type and deleted mtDNA at the single cell level. A cytoplasmic hybrid (cybrid) cell line (ΔH2-1), which contains a 7.5 kb deletion, was used as the cell model. Cybrid cell lines are a useful tool for studying mtDNA distribution, inheritance and the effects of heteroplasmy on cellular functions, because a specific deletion can be studied with a stable nuclear background and the degree of heteroplasmy can be manipulated [25]. The cybrid cells used in this study were constructed to contain ~75% deleted mtDNA. The large deletion of the ΔH2-1 cybrid cells was from a patient with an infantile metabolic disorder without oxidative phosphorylation dysfunction [20]. This deletion has been detected in patients with Kearn’s Sayre syndrome (KSS) [26–28]. KSS is a multisystem disorder characterized by paralysis of extraocular muscles, pigmentary retinal degeneration and early onset (before age 20).
Amplification was performed with a three-primer PCR strategy. This procedure is simpler than a conventional amplification in which two primer sets are used. The three-primer PCR amplification system is represented schematically in Figure 1. DNA is amplified between primers F1 and R1 when the wild-type mtDNA molecule is present. Similarly, DNA is amplified from primers F1 and R2 for the deleted mtDNA molecule. DNA cannot amplify between primers R1 and R2, since they hybridize to the same strand of mtDNA. Primers F1 and R2 are far enough apart so that no PCR product is formed from wild-type mtDNA using the described thermal cycling conditions.
Figure 1.
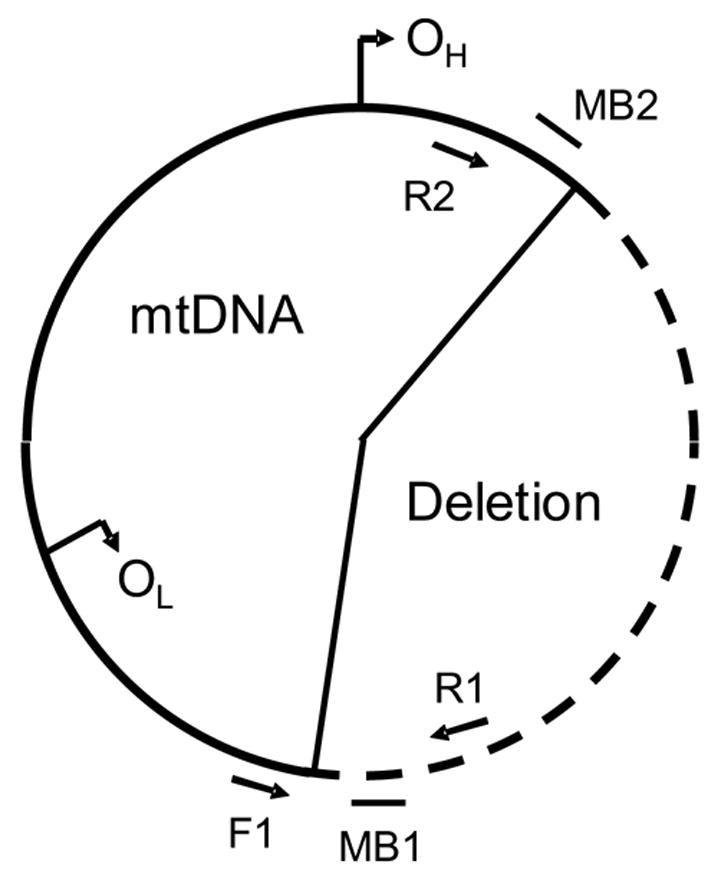
A schematic representation of the multiplex three-primer PCR assay that illustrates the approximate locations of the forward primer (F1), reverse primers (R1 and R2) and molecular beacons MB1 and MB2. PCR amplification results in amplification between primers F1 and R1 for wild-type mtDNA and between F1 and R2 for deleted mtDNA. MB1 is labeled with FAM and hybridizes with the PCR product from the wild-type mtDNA. Similarly, MB2 is labeled with HEX and is specific to deleted mtDNA.
A unique aspect of this assay is that quantitation is performed using molecular beacons with real-time detection. The molecular beacons were designed with loop sequences that were specific to wild-type and deleted mtDNA. As illustrated, MB1 hybridizes to the wild-type mtDNA PCR product and is fluorescently labeled with FAM. Similarly, MB2 hybridizes to the deleted mtDNA PCR product and is fluorescently labeled with HEX. Both molecular beacons have identical stem sequences, which were previously reported for the real-time PCR quantitation of a point mutation in mtDNA [23].
mtDNA Standard Construction
To enable absolute quantitation of wild-type and deleted mtDNA, standards were constructed using PCR. A fragment of the wild-type mtDNA molecule from 143B osteosarcoma cells was amplified with primers S1 and S2 (Table 1). Similarly, primers S1 and S3 amplified a fragment of the deleted mtDNA molecule from ΔH2-1 cybrid cells. The PCR products from each reaction were separated by agarose gel electrophoresis and the appropriate bands were excised, purified and quantitated to produce mtDNA standards that could be used to form calibration curves. The purity of both standards was confirmed by amplifying each standard with primers F1, R1 and R2 (Figure 1) using real-time detection with MB1 and MB2. The PCR products were also separated on an agarose gel. No contamination was observed in either mtDNA standard.
PCR Specificity
Since wild-type and deleted mtDNA coexists in single cells, it was necessary to prove that the primers used in the three-primer amplification system are specific to their intended mtDNA targets. This specificity was demonstrated by amplifying wild-type and deleted mtDNA standards using various combinations of the primers. Figure 2 displays an image of the resulting PCR products following agarose gel electrophoresis. As shown, PCR products are formed when primers F1 and R1 are used to amplify wild-type mtDNA and when primers F1 and R2 are used to amplify deleted mtDNA. Since the wild-type and deleted mtDNA PCR products are very similar in size, 411 and 429 bp, respectively, they could not be resolved from each other by agarose gel electrophoresis after PCR amplification from a mixture containing both mtDNA standards. No other PCR products are observed for any other combinations of primers and template, which indicates that each primer set is specific to its intended target. As expected, primers R1 and R2 do not form any PCR products and no products are detected without template. These results conclusively show that the three-primer PCR assay specifically amplifies its intended targets.
Figure 2.
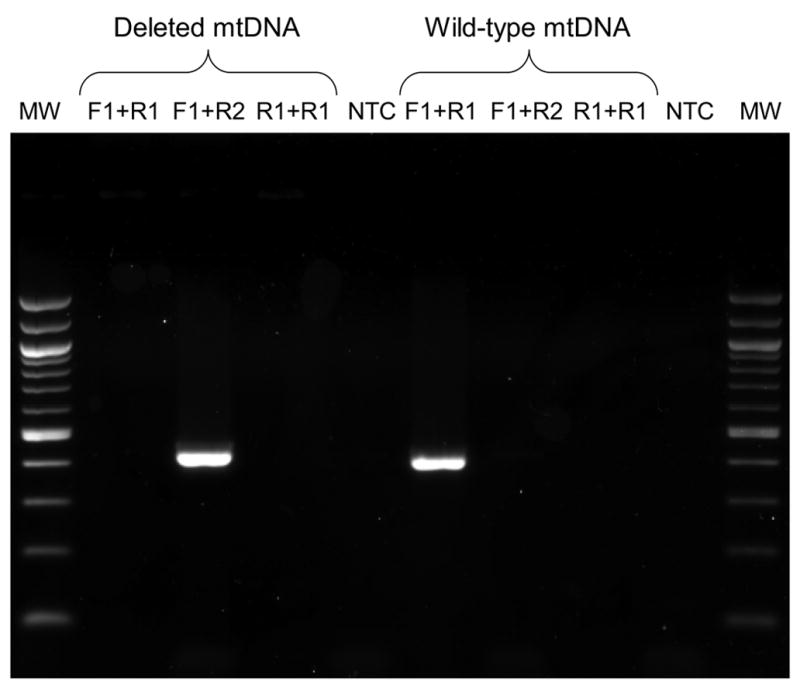
An image of PCR products following separation on a 2% agarose gel and imaged with ethidium bromide. Lane MW is a 100 base pair DNA ladder in which the 1,000 bp and 500 bp bands appear twice as intense. The wild-type and deleted mtDNA products are 411 and 429 bp, respectively. Lanes 1–4 contained deleted mtDNA and lanes 5–8 contained wild-type mtDNA. Lanes 1 and 5 contained primers F1 and R1, lanes 2 and 6 contained primers F1 and R2, lanes 3 and 7 contained primers R1 and R2 and lanes 4 and 8 contained all three primers but neither template.
Real-Time Fluorescence Detection
To accurately quantitate DNA the PCR assay must also amplify the DNA template efficiently. Theoretically, the amount of DNA in each PCR cycle should double. Amplification efficiencies that are significantly lower than theoretically predicted indicate the presence of a PCR inhibitor or the necessity to redesign the primers and molecular beacons. To measure the amplification efficiencies for wild-type and deleted mtDNA, serial dilutions of both mtDNA standards were prepared from ~60,000 to 6 copies. Figures 3A and 3B display the amplification curves for 5 serial 10× dilutions of each mtDNA standard. The amplification curves plot the fluorescence intensity, measured after the annealing step in each PCR cycle, versus the cycle number. The fluorescence threshold that was used to determine the threshold cycle (Ct) is shown as a dashed line. Figure 3C is a semi-logarithmic plot of the corresponding Ct versus the copy number. A PCR assay that has 100% efficiency will have a slope of −3.322 (−log210). Based on the slopes of the calibration curves in Figure 3C, we calculated that both mtDNA standards are amplified with 97% efficiency down to 6 copies. These calibration curves highlight the sensitivity and large dynamic range of the PCR assay and indicate that absolute quantitation of heteroplasmy in single cells is possible.
Figure 3.
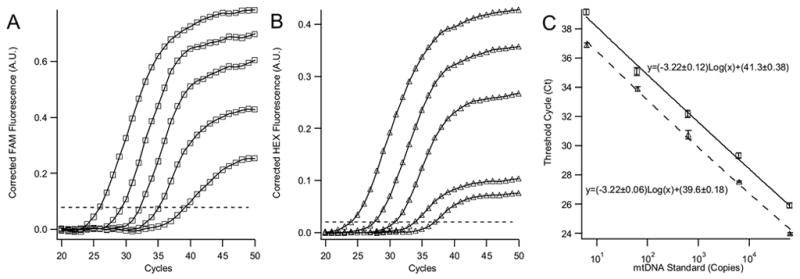
Panel A and B present the amplification curves for wild-type (□) and deleted (▵) mtDNA standards, respectively. The dotted line in panels A and B is the fluorescence threshold that was used to determine the threshold cycles. Panel C displays the corresponding amplification efficiency curves in a semi-log plot. Amplification efficiencies were 104% and 105% for the wild-type and deleted mtDNA, respectively. Wild-type mtDNA standard copy numbers ranged from 62,300 to 6 copies and deleted from 64,400 to 6 copies. The equations for the least-squares fit line are shown in panel C. Correlation coefficients for both fitted lines were >0.99. In all panels the experiments were performed in triplicate and error bars are shown on the graphs, although some are obscured by data point markers.
It is noted that in Figure 3C there is a small difference in the y-intercepts for the wild-type and deleted mtDNA calibration curves. This is an artifact that is caused by different optimal fluorescence thresholds in the FAM and HEX channels. The small difference in thresholds manifests itself in a small shift of the calibration curve, but does not reflect a higher sensitivity or efficiency for the deleted mtDNA and does not influence mtDNA quantitation.
Single-Cell PCR
Investigation of clonal expansion of mtDNA deletions and correlation of heteroplasmy with cell function excludes bulk analysis of hundreds of cells. Single-cell PCR assays are required to measure heteroplasmy in these cases. To demonstrate the usefulness of the newly developed PCR assay, we used it to quantitate heteroplasmy in bulk and single ΔH2-1 cybrid cells. To allow single-cell analysis, the PCR assay was combined with reported procedures to release mtDNA directly from cells without nucleic acid isolation [21, 22].
Single cells were sampled from a PVA coated microscope slide using a capillary in conjunction with a micromanipulator and visually aided with a microscope. A movie displaying the selection and capture of a single cell using the capillary is provided in the supplementary information (Supplementary Movie 1). The cell was positioned directly beneath the capillary lumen and introduced into the capillary using slight negative pressure with a syringe pump. Following capture the cell was expelled into a PCR well containing the lysis solution and S/H buffer. The capillary surface was covalently modified with poly(acryloylamino propanol) [24] to reduce cell adsorption once it had been introduced into the capillary, which results in better reproducibility in isolating single cells.
To allow absolute quantitation of wild-type and deleted mtDNA, a calibration curve was performed simultaneously with the bulk and single-cell samples. Table 2 tabulates the amount of wild-type and deleted mtDNA for 10 individual ΔH2-1 cells and approximately 46 ΔH2-1 cells that were lysed in bulk. Single cells were heterogeneous in both the amount of wild-type and deleted mtDNA and percent heteroplasmy. Despite the small number of single cells that were sampled, the average percent of heteroplasmy, average wild-type and deleted mtDNA copy numbers and the average number of total mtDNA copies for single cells are statistically similar to the bulk cell sample using Student’s t-test (p>0.05). The large error in the averages for single-cells is due to the heterogeneity amongst the cells and is not primarily due to the PCR assay. The mtDNA copy number per cell is ~50% less for the ΔH2-1 cybrid cell line than was reported for 143B cells [29].
Table 2.
Quantitation of heteroplasmy
| ΔH2-1 Cells | Wild-type mtDNA Copiesa | Deleted mtDNA Copiesa | Total mtDNA Copiesb | Percent Heteroplasmy |
|---|---|---|---|---|
| Cell 1 | 260 | 4600 | 4800 | 95 |
| Cell 2 | 600 | 3600 | 4200 | 85 |
| Cell 3 | 1000 | 3900 | 4900 | 80 |
| Cell 4 | 1500 | 2600 | 4100 | 60 |
| Cell 5 | 600 | 950 | 1600 | 60 |
| Cell 6 | 80 | 1300 | 1400 | 95 |
| Cell 7 | 850 | 1900 | 2800 | 70 |
| Cell 8 | 450 | 2600 | 3000 | 85 |
| Cell 9 | 2600 | 1900 | 4500 | 45 |
| Cell 10 | 2000 | 4700 | 6700 | 70 |
| Average of Single Cellsc | 1000±800 | 2800±1300 | 3800±1600 | 75±16 |
| Bulk Cellsc,d | 900±200 | 3200±300 | 4100±400 | 80±13 |
Based on propagation of errors associated with the calibration curve, errors are 15% and 11% RSD for wild-type and deleted mtDNA quantitation.
Sum of wild-type and deleted mtDNA copies
mtDNA copies/cell
Quantitation perfomed in quadruplicate for ~46 ΔH2-1 cells lysed in bulk
It has been reported that heteroplasmy levels from 50–90% can be tolerated by the cell before significant bioenergetic functions are inhibited [11, 12]. Therefore, it is expected that the majority of cells used in this study are bioenergetically dysfunctional. Indeed, the cybrid cells must be cultured in high glucose medium supplemented with uridine to compensate for reduced oxidative phosphorylation. Further investigation will be needed to determine if the percent of heteroplasmy in the single cells is manifested in different bioenergetic functions, e.g. membrane potential, between the cells.
Discussion
The role of mitochondrial DNA (mtDNA) deletions and heteroplasmy in disease is well documented [2, 3]. However, their role in aging is more controversial. In aging an important correlation that must be elucidated is the link between mtDNA heteroplasmy and cell function. This precludes the use of bulk cell samples; single-cell analyses are required due to the cell-to-cell heterogeneity in function and heteroplasmy [17, 21, 22]. In that regard, we report a multiplex three-primer PCR assay that was shown to be specific, reproducible and sensitive, which makes it suitable for single-cell analysis. It is also significantly less labor intensive than conventional methods that require post-amplification analysis, e.g. gel electrophoresis; therefore, the reported assay has higher throughput. The assay also allows absolute quantitation of both wild-type and deleted mtDNA molecules, as opposed to merely measuring the ratio of wild-type to deleted mtDNA.
However, a limitation of the assay is that the PCR primers must be designed for a specific deletion and cannot be used to screen for unknown deletions. Yet, since the throughput is high and there is very little sample handling and no post-amplification analysis, several deletions could be screened with additional primers and molecular beacons simultaneously. As such, the assay allows correlation between a specific mtDNA deletion and cellular function. The reported PCR assay shows great potential due to its specificity and sensitivity. Combination of PCR assays with other techniques, i.e. histochemistry and laser capture microdissection, have provided insight into the effects of heteroplasmy in single muscle fibers [30]. However, other techniques could be combined with the reported PCR assay. For example, we have previously reported on sampling and separation of mitochondria from muscle tissue cross section by CE-LIF [31–33]; this method could be combined with the PCR assay to yield valuable information on the heteroplasmy level in individual muscle fibers. In fact, the sensitivity of the PCR assay may allow quantitation of heteroplasmy in individual mitochondria collected post CE-LIF analysis, which would shed light on mtDNA distribution, inheritance and clonal expansion.
Supplementary Material
Figure 4.

Bright field images of a singleΔH2-1 cybrid cell being captured in a 50 μm i.d. poly(acryloylamino propanol) modified capillary. The capillary lumen is positioned directly above the cell. Panels A–D show the cell beneath the capillary lumen; in panel E the cell is detaching from the microscope slide and moving out of focus into the capillary. Lastly, the cell is no longer visible as it has been captured by the capillary in panel F. The total time for collection is 1.5 seconds with each image corresponding to 0.25 second intervals.
Acknowledgments
This work was supported by the National Institute of Health (R01-AG20866). B.G.P. acknowledges support from NIH-Chemistry/Biology Interface Training Grant (GM08700). E.A.A. is supported by an NIH Career Award (1K02-AG21453). The authors thank Dr. Carlos Moraes (University of Miami) for providing the ΔH2-1 cybrid cell line.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 2.DiMauro S. Mitochondrial diseases. Biochim Biophys Acta. 2004;1658:80–88. doi: 10.1016/j.bbabio.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 3.Taylor RW, Turnbull DM. Mitochondrial DNA mutations in human disease. Nat Rev Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chomyn A, Attardi G. MtDNA mutations in aging and apoptosis. Biochem Biophys Res Commun. 2003;304:519–529. doi: 10.1016/s0006-291x(03)00625-9. [DOI] [PubMed] [Google Scholar]
- 5.Howell N, Elson JL, Chinnery PF, Turnbull DM. mtDNA mutations and common neurodegenerative disorders. Trends Genet. 2005;21:583–586. doi: 10.1016/j.tig.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 6.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 7.Szibor M, Holtz J. Mitochondrial ageing. Basic Res Cardiol. 2003;98:210–218. doi: 10.1007/s00395-003-0421-z. [DOI] [PubMed] [Google Scholar]
- 8.McKenzie D, Bua E, McKiernan S, Cao Z, Aiken JM. Mitochondrial DNA deletion mutations: a causal role in sarcopenia. Eur J Biochem. 2002;269:2010–2015. doi: 10.1046/j.1432-1033.2002.02867.x. [DOI] [PubMed] [Google Scholar]
- 9.Cavelier L, Johannisson A, Gyllensten U. Analysis of mtDNA copy number and composition of single mitochondrial particles using flow cytometry and PCR. Exp Cell Res. 2000;259:79–85. doi: 10.1006/excr.2000.4949. [DOI] [PubMed] [Google Scholar]
- 10.Satoh M, Kuroiwa T. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp Cell Res. 1991;196:137–140. doi: 10.1016/0014-4827(91)90467-9. [DOI] [PubMed] [Google Scholar]
- 11.Bentlage HA, Attardi G. Relationship of genotype to phenotype in fibroblast-derived transmitochondrial cell lines carrying the 3243 mutation associated with the MELAS encephalomyopathy: shift towards mutant genotype and role of mtDNA copy number. Hum Mol Genet. 1996;5:197–205. doi: 10.1093/hmg/5.2.197. [DOI] [PubMed] [Google Scholar]
- 12.Porteous WK, James AM, Sheard PW, Porteous CM, Packer MA, Hyslop SJ, Melton JV, Pang CY, Wei YH, Murphy MP. Bioenergetic consequences of accumulating the common 4977-bp mitochondrial DNA deletion. Eur J Biochem. 1998;257:192–201. doi: 10.1046/j.1432-1327.1998.2570192.x. [DOI] [PubMed] [Google Scholar]
- 13.Wallace DC. Mitochondrial diseases in man and mouse. Science. 1999;283:1482–1488. doi: 10.1126/science.283.5407.1482. [DOI] [PubMed] [Google Scholar]
- 14.Wallace DC. Mitochondrial defects in cardiomyopathy and neuromuscular disease. Am Heart J. 2000;139:S70–85. doi: 10.1067/mhj.2000.103934. [DOI] [PubMed] [Google Scholar]
- 15.Johnston W, Karpati G, Carpenter S, Arnold D, Shoubridge EA. Late-onset mitochondrial myopathy. Ann Neurol. 1995;37:16–23. doi: 10.1002/ana.410370106. [DOI] [PubMed] [Google Scholar]
- 16.Moraes CT, Atencio DP, Oca-Cossio J, Diaz F. Techniques and pitfalls in the detection of pathogenic mitochondrial DNA mutations. J Mol Diagn. 2003;5:197–208. doi: 10.1016/S1525-1578(10)60474-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sciacco M, Bonilla E, Schon EA, DiMauro S, Moraes CT. Distribution of wild-type and common deletion forms of mtDNA in normal and respiration-deficient muscle fibers from patients with mitochondrial myopathy. Hum Mol Genet. 1994;3:13–19. doi: 10.1093/hmg/3.1.13. [DOI] [PubMed] [Google Scholar]
- 18.He L, Chinnery PF, Durham SE, Blakely EL, Wardell TM, Borthwick GM, Taylor RW, Turnbull DM. Detection and quantification of mitochondrial DNA deletions in individual cells by real-time PCR. Nucleic Acids Res. 2002;30:e68. doi: 10.1093/nar/gnf067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyagi S, Kramer FR. Molecular beacons: probes that fluoresce upon hybridization. Nat Biotechnol. 1996;14:303–308. doi: 10.1038/nbt0396-303. [DOI] [PubMed] [Google Scholar]
- 20.Moraes CT, Kenyon L, Hao H. Mechanisms of human mitochondrial DNA maintenance: the determining role of primary sequence and length over function. Mol Biol Cell. 1999;10:3345–3356. doi: 10.1091/mbc.10.10.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melov S, Schneider JA, Coskun PE, Bennett DA, Wallace DC. Mitochondrial DNA rearrangements in aging human brain and in situ PCR of mtDNA. Neurobiol Aging. 1999;20:565–571. doi: 10.1016/s0197-4580(99)00092-5. [DOI] [PubMed] [Google Scholar]
- 22.Khrapko K, Bodyak N, Thilly WG, van Orsouw NJ, Zhang X, Coller HA, Perls TT, Upton M, Vijg J, Wei JY. Cell–by-cell scanning of whole mitochondrial genomes in aged human heart reveals a significant fraction of myocytes with clonally expanded deletions. Nucleic Acids Res. 1999;27:2434–2441. doi: 10.1093/nar/27.11.2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Szuhai K, Ouweland J, Dirks R, Lemaitre M, Truffert J, Janssen G, Tanke H, Holme E, Maassen J, Raap A. Simultaneous A8344G heteroplasmy and mitochondrial DNA copy number quantification in myoclonus epilepsy and ragged–red fibers (MERRF) syndrome by a multiplex molecular beacon based real-time fluorescence PCR. Nucleic Acids Res. 2001;29:E13. doi: 10.1093/nar/29.3.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gelfi C, Curcio M, Righetti PG, Sebastiano R, Citterio A, Ahmadzadeh H, Dovichi NJ. Surface modification based on Si-O and Si-C sublayers and a series of N-substituted acrylamide top-layers for capillary electrophoresis. Electrophoresis. 1998;19:1677–1682. doi: 10.1002/elps.1150191026. [DOI] [PubMed] [Google Scholar]
- 25.King MP. Use of ethidium bromide to manipulate ratio of mutated and wild-type mitochondrial DNA in cultured cells. Methods Enzymol. 1996;264:339–344. doi: 10.1016/s0076-6879(96)64032-4. [DOI] [PubMed] [Google Scholar]
- 26.Mita S, Rizzuto R, Moraes CT, Shanske S, Arnaudo E, Fabrizi GM, Koga Y, DiMauro S, Schon EA. Recombination via flanking direct repeats is a major cause of large-scale deletions of human mitochondrial DNA. Nucleic Acids Res. 1990;18:561–567. doi: 10.1093/nar/18.3.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakase H, Moraes CT, Rizzuto R, Lombes A, DiMauro S, Schon EA. Transcription and translation of deleted mitochondrial genomes in Kearns-Sayre syndrome: implications for pathogenesis. Am J Hum Genet. 1990;46:418–427. [PMC free article] [PubMed] [Google Scholar]
- 28.Zupanc ML, Moraes CT, Shanske S, Langman CB, Ciafaloni E, DiMauro S. Deletion of mitochondrial DNA in patients with combined features of Kearns-Sayre and MELAS syndromes. Ann Neurol. 1991;29:680–683. doi: 10.1002/ana.410290619. [DOI] [PubMed] [Google Scholar]
- 29.King MP, Attardi G. Human cells lacking mtDNA: repopulation with exogenous mitochondria by complementation. Science. 1989;246:500–503. doi: 10.1126/science.2814477. [DOI] [PubMed] [Google Scholar]
- 30.Bua E, Johnson J, Herbst A, Delong B, McKenzie D, Salamat S, Aiken JM. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am J Hum Genet. 2006;79:469–480. doi: 10.1086/507132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahmadzadeh H, Johnson RD, Thompson L, Arriaga EA. Direct sampling from muscle cross sections for electrophoretic analysis of individual mitochondria. Anal Chem. 2004;76:315–321. doi: 10.1021/ac034809g. [DOI] [PubMed] [Google Scholar]
- 32.Ahmadzadeh H, Thompson LV, Arriaga EA. On-column labeling for capillary electrophoretic analysis of individual mitochondria directly sampled from tissue cross sections. Anal Bioanal Chem. 2006;384:169–174. doi: 10.1007/s00216-005-0171-x. [DOI] [PubMed] [Google Scholar]
- 33.Johnson RD, Navratil M, Poe BG, Xiong G, Olson KJ, Ahmadzadeh H, Andreyev D, Duffy CF, Arriaga EA. Analysis of mitochondria isolated from single cells. Anal Bioanal Chem. 2006 doi: 10.1007/s00216-006-0689-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.