

Abstract
Conventional treatments are not adequate for the majority of lung cancer patients. Conditionally replicating adenoviruses (CRAds) represent a promising new modality for the treatment of neoplastic diseases, including non-small cell lung cancer. Specifically, following cellular infection, the virus replicates selectively in the infected tumor cells and kills the cells by cytolysis. Next, the progeny virions infect a new population of surrounding target cells, replicate again and eradicate the infected tumor cells while leaving normal cells unaffected. However, to date there have been two main limitations to successful clinical application of these CRAd agents; i.e. poor infectivity and poor tumor specificity. Here we report the construction of a CRAd agent, CRAd-CXCR4.RGD, in which the adenovirus E1 gene is driven by a tumor-specific CXCR4 promoter and the viral infectivity is enhanced by a capsid modification, RGD4C. This agent CRAd-CXCR4.RGD, as expected, improved both of the viral infectivity and tumor specificity as evaluated in an established lung tumor cell line and in primary tumor tissue from multiple patients. As an added benefit, the activity of the CXCR4 promoter was low in human liver as compared to three other promoters regularly used for targeting tumors. In addition, this agent has the potential of targeting multiple other tumor cell types. From theses data, the CRAd-CXCR4.RGD appears to be a promising novel CRAd agent for lung cancer targeting with low host toxicity.
Keywords: CXCR4 gene, tumor-specific promoter, transcriptional targeting, adenoviral vector
Introduction
Conventional treatments are not adequate for a majority of lung cancer patients. The failure of conventional therapy occurs because these tumors are remarkably resistant to chemotherapy and radiation, both of which work by damaging the DNA of rapidly dividing tumor cells.1 The use of replicative viral agents (virotherapy) represents a novel approach to such neoplastic diseases. In this strategy, target tumor cell killing by the viral agent is achieved as a direct consequence of viral replication.2 Most specifically, adenoviral vectors have been used extensively for virotherapy, including the Ad5 24 vector, containing a 24 base pair deletion in the E1A gene that targets replication to retinoblastoma gene (Rb) defective tumors.3 Suzuki et al. have extended this work, describing that an enhanced infectivity yield was observed by using a capsid modified Ad5 24.RGD vector in a human lung cell line, A549, and a human prostate cancer cell line, LNCap.4 An ideal viral agent would, thus, possess two characteristics: 1) such viral vectors would have the capacity to selectively infect tumor versus non-tumor cells, which we denote as “infectivity”; 2) the viral vectors would possess a relative preference for replication in tumor versus non-tumor cells, which we denote as “specificity”. However, both viral infectivity and specificity are poor in currently available conditionally replicative viral vectors. To develop infectivity enhanced conditionally replicative viral vectors with high specificity for lung carcinoma is thus the goal.
The limitation of poor infectivity with current non-replicative and replicative Ad systems has been found to result from a relative paucity on tumor cell surfaces of the primary receptor for adenovirus type 5, the Coxsackie-Adenovirus Receptor (CAR), on primary tumor cells relative to their cell line counterparts.5, 6 On this basis, it has been proposed that gene delivery via “CAR-independent” pathways may be required to circumvent this key aspect of tumor biology.7, 8 Native Ad5 tropism is mediated by two capsid proteins: the fiber and the penton base. These proteins bind to the primary high affinity cellular receptor CAR and to the integrins ανβ3 and ανβ5, respectively. Based on these molecular interactions, a concerted effort has been made to modify Ad5 tropism, resulting in enhanced tumor cell transduction by retargeting cellular entry through heterologous pathways. Consideration of the studies designed to retarget Ad5 tropism has established the concept that cell-specific transduction can be accomplished with genetic modifications of the viral domains that dictate cellular attachment. In this regard, a trimeric fiber protein protrudes from each of the 12 vertices of the icosahedral Ad particle, where it is attached non-covalently to the penton base.9 It has been shown that the globular carboxy-terminal (C-terminal) knob domain of the Ad fiber protein is the ligand for attachment to the Ad5 cellular receptor CAR, the first step in infection.10, 11, 12, 13 Based on the demonstration that infection can be blocked both by a recombinant Ad5 trimeric knob expressed in E. coli, and by an anti-knob antibody, 14 the knob is both necessary and sufficient for Ad5 binding to host cells. The amino-terminal tail domain of the fiber is separated from the knob domain by a long rod-like shaft comprising a 15-amino acid residue motif repeated twenty-two times in Ad5.15 Following attachment, the next step in Ad5 infection is internalization of the virion by receptor-mediated endocytosis. This process is mediated by the interaction of Arg-Gly-Asp (RGD) sequences in the penton base with secondary host cell receptors, the integrins ανβ3 and ανβ5.16 Thus, understanding of Ad structure-function and the cellular entry pathway has facilitated attempts to modify the tropism of Ad5 vectors and permitted the enhanced transduction of tumor cells.
The improvement of poor tumor specificity with current replicative Ad systems has recently been exploited by using a tumor specific promoter (TSP) to drive the E1 expression resulting in the viral replication being restricted in normal host cells, but not in tumor cells. In this method, a CRAd replicates only in tumor cells, killing cells by oncolysis, but not in normal host cells, thereby avoiding the toxicity of the CRAd agent. An ideal tumor specific promoter (TSP) for transcriptional targeting exhibits selective high activity in tumor cells [termed a “tumor on” phenotype]. To mitigate hepatoxicity upon systemic delivery, candidate promoters additionally need to exhibit low activity in the endogenous sink, i.e. the liver [termed a “liver off” phonotype]. Many TSPs have been explored for specific cancers, such as prostate-specific antigen for prostate cancer, and α-fetoprotein promoter for hepatocarcinoma.17, 18 Müller et al. have demonstrated that CXCR4 expression was undetectable in normal epithelial cells but markedly up regulated in cancer cells.19 Others have described the chemokine, SDF-1, and its receptor, CXCR4, to have critical roles in determining the metastastic destination of tumor cells. 20, 21, 22, 23 Recent data further showed that CXCR4 gene expression is linked to lung cancer.24 Specifically, Phillps 25 and Burger 26 et al. demonstrated that CXCR4 gene expression was detected in multiple non-small cell lung cancer cell lines, including A459, by using real-time PCR for CXCR4 gene transcription, immunohistochemistry for localization of CXCR4, FACS for analysis of CXCR4 cell surface expression and Western blotting for expression of CXCR4 protein. Other authors have discovered that transcriptional targeting in renal cancer cell lines is possible via the human CXCR4 promoter with low background activity in normal keratinocytes and human mesothelial cells 27 and that the CXCR4 gene is highly expressed in breast cancer cell lines and primary cells but not in normal mammary cells. 19 In our previous work,28 CXCR4 promoter activities were analyzed in pancreatic, breast, and ovarian cancer cell lines along with melanoma and breast cancer cell lines and in primary cells derived from patient material. The CXCR4 promoter had a “tumor on” and “liver off” phenotype in both in vitro and in vivo experiments when a recombinant adenoviral vector (reAd5-CXCR4.Luc), in which a luciferase reporter gene is driven by the CXCR4 promoter, was employed. However, we have not previously tested the CXCR4 promoter activity in lung cancer cells.
In this study, we constructed a conditionally replicative adenoviral vector, in which the Ad E1 gene was regulated by using the CXCR4 promoter as a TSP and viral infectivity was enhanced with the capsid modification, RGD4C, by which the adenoviral vector targeted to tumor cells via a CAR -independent pathway. We verified that infectivity was enhanced and that the selected vector replicated in both the lung cancer cell line, A549, and in three primary non-small cell lung tumor samples obtained from patients. In addition, the activity of the CXCR4 promoter was seen to be low in human liver as compared to three other tumor promoters which previously had been used for targeting to other cancers. The data indicated that the CRAd-CXCR4-RGD construct is an excellent candidate for translation into a clinical trial for treatment of non-small cell lung carcinoma and other cancers by virotherapy.
Materials and Methods
Cells and Tissues
Human tumor cell lines A549 (lung cancer, ATCC), OVSK3ip1 [ovarian cancer, American Type Culture Collection (ATCC)], D65 (glioma, ATCC), and Hs-766T (pancreatic cancer, a kind gift from Dr. Masato Yamamoto) were each cultured in medium recommended by the ATCC. The D65MG cell line was a kind gift from Dr. G. Y. Gillespie (Department of Neurosurgery, University of Alabama at Birmingham). 911 cells (a kind gift from Dr. Van Der Eb, Leiden University, The Netherlands) were maintained in Dulbecco’s modified Eagle’s medium. Each medium was also supplemented with 10% fetal calf serum, penicillin (100 IU/ml), and streptomycin (100 μg/ml). Cells were incubated at 37° C in a 5% CO2 environment under humidified conditions. A human mammary epithelial cell line (HMEC) was used as a control and purchased from Cambrex BioScience (Walkersville, MD) and cultured in the medium specially purchased for that purpose from the same company.
Human liver specimens were obtained from donor remnants after liver transplantation. Specimens of lung, similarly not needed for diagnostic purposes, were collected by Dr. Robert Cerfolio, Department of Surgery at UAB, both following IRB approval. To generate tissue slices, tissue was cut in consecutive 0.5 mm-thick slices using the Krumdieck tissue slicer (Alabama Research Development, Munford, AL). Sequential slices were then cultured in 24 well-plates in RPMI medium supplemented with 10% bovine fetal serum, 100U/ml penicillin, 100μg/ml streptomycin and 5μg/ml insulin. Cultures were maintained at 37° C in a humidified atmosphere of 95% air and 5% CO2. Three tissue slices were examined per group.
Recombinant Adenoviruses
All recombinant adenoviruses including Ad5-CXCR4.Luc,28 Ad5-Cox-2.Luc,29 Ad5-SLPI.Luc,30 Ad5-MK.Luc,31 and Ad5-CMV.Luc were generated in this laboratory at UAB. The CRAd genomes were constructed via homologous recombination in Escherichia coli (Figure 1). The CRAd-CXCR4.RGD vector has the following characteristics: 1) The CRAd agent contains the human CXCR4 promoter nucleotide -191/+8826 to replace Ad5 nt 346–521 (NCBI nucleotide database accession # BK000408), which is the region of the Ad5 native E1 promoter. The CXCR4-controlled E1 expression cassette was placed in the original E1 region of the Ad5 gene; 2) A RGD-4C capsid modification was inserted into the Ad fiber knob region for enhancement of Ad infectivity;32 3) The E3 gene was retained in the Ad genome for elevating the oncolytic effect of the CRAd agents;33 and 4) A poly-A signal was inserted between the inverted terminal repeat (ITR) and the CXCR4 promoter to stop the non-specific transcriptional activity of the ITR, and to retain the tumor specificity of the CXCR4 promoter.
Figure 1.

Characterization of CRAd-CXCR4.RGD.
(A) Construction of the CRAd agents. A 279 bp segment of the CXCR4 promoter was amplified from the clone pBSKCAT/CXCR4 3B/4-1[5′Δ3], and inserted into a pScE1 vector. A poly-A signal sequence was inserted between the ITR and CXCR4 promoter to terminate the transcription signal from the ITR. The constructed clone was recombinated with pVK503C, which contains the E3 gene and a RGD4C fiber motif, to generate the CRAd-CXCR4.RGD. The a, b, and c, arrowheads are specific oligos of poly-A and the CXCR4 promoter sequences. The arrow bars indicate the sizes of the PCR products amplified by using specific oligo pairs. The PCR products were run on an agarose gel (1%). The PCR products were amplified with the oligo pairs a/b and a/c from the template DNAs of CRAd-CXCR4.RGD and a negative control Ad5-CMV.Luc, respectively. The sizes of PCR products are shown at the right bottom.
Briefly, DNA fragments containing nucleotides −191/+88 were cut with Bam HI and Hind III restriction endonucleases from the clone pBSKCAT/CXCR4 3B/4-1[5′Δ3],34 and subcloned into the plasmid pBSSK (Stratagene, La Jolla, CA) by use of the same restriction sites. A SV40 poly-A (PA) fragment was then cut with Xba I/Bam HI from a pGL3B vector (Invitrogen, Carlsbad, CA) and inserted into the pBSSK by use of the same restriction sites. A generated clone named pBSSK/PA/CXCR4 was used to create shuttle vectors. DNA fragments containing both a SV40 PA and the CXCR4 promoter were cut with Not I/ Xho I, and subcloned into the same sites of the pScsE1 plasmid (from Dr. D.M. Nettelbeck, Erlangen, Germany) which contained a DNA fragment from nucleotide number 522 to 3924 (GenBank sequence AD5001). This fragment covered both the Ad5 E1A and E1B coding region and was amplified from the genomic vector pXC-1.35 Thus the plasmid, pScsE1/PA/CXCR4 was generated.
The Ad vector, pVK503c,4 was a kind gift from Dr. V. Krasnykh (M.D. Anderson, Houston, TX), and contained both the E3 gene and a capsid modified RGD4C. After cleavage with Pme I, the shuttle vector, pScsE1/PA/CXCR4, was recombined with Cla I linerized pVK503c to generate a CRAd genome with a RGD4C-modified fiber. The resultant plasmids encoding the CXCR4 promoter in the CRAd was linearized with Pac I and transfected into 911 cells using Lipofectamine (Qiagen, Valencia, CA). Generated viruses were propagated in A549 cells, a lung tumor cell line, in which the CXCR4 gene is positively expressed 25 and the promoter is active, and purified by double CsCl density gradient centrifugation, followed by dialysis against phosphate-buffered saline (PBS) containing 10% glycerol. The viruses were titrated by plaque assay in 293 cells, and vp number was determined spectrophotometrically based on absorbance at a wavelength of 260nm. The viruses were stored at −80° C until use.
Wild-type Ad5 (Adwt) and Ad5-CXCR4.Luc were used as replication positive and negative controls, respectively, in the CRAd agent analysis. The characteristics of all adenovirus vectors used in this study are shown in Table 1.
Table 1.
The characteristics of adenoviral vectors used in this study
| Virus name | Promoter | Reporter | E1 | E3 | Modification | Replication | Reference |
|---|---|---|---|---|---|---|---|
| AdCMVLuc | CMV | Luciferase | No | No | No | No | 29 |
| AdCox-2Luc | Cox-2 | Luciferase | No | No | No | No | 29 |
| AdCXCR4Luc | CXCR4 | Luciferase | No | No | No | No | 28 |
| AdMkLuc | Midkine | Luciferase | No | No | No | No | 31 |
| AdSLPILuc | SLPI | Luciferase | No | No | No | No | 30 |
| AdCMVLuc.RGD | CMV | Luciferase | No | No | RGD4C | No | 30 |
| Adwt | Native | No | Yes | Yes | No | Yes | 29 |
| CRAd-CXCR4.RGD | CXCR4 | No | Yes | Yes | RGD4C | Yes | Present study |
Analysis of the CRAd Construct by PCR and Sequencing
The structure of the CRAds were verified by PCR and sequence data. The virus (5.0 x 108 viral particles [vp]) was processed with a blood DNA kit (Qiagen), and 1/50 of the DNA solution was analyzed by PCR for various regions with Taq polymerase (Qiagen). Thermal cycling conditions were as follows: initial denaturing, 5 minutes at 95° C; 30 cycles of 30 seconds each at 95° C, 30 seconds at 55° C, and 1 minute at 72° C. The primers used for each region were as follows: a) PA-F, 5′GATGAGTTTGGACAAACCAC; b) CXCR4-F, 5′TACCGACCACCCGCAAACA; c) CXCR4-R, 5′AACCGCTGGTTCTCCAGATGC. The PCR products were 279 bp amplified with an oligo pair b/c and 541 bp with a/c as verified on a 1% agarose gel (Fig 1). The PCR products were also analyzed by automatic sequence analysis performed by using the BigDye Terminator v3.1 cycle sequencing ready reaction kit and an Applied Biosystems 3100 Genetic Analyzer (Foster City, CA) at the Genomics Core of the Howell and Elizabeth Heflin Center for Human Genetics of the University of Alabama at Birmingham. A negative control, Ad5-CMV.Luc, was performed in parallel.
Detection of the E1 Gene from the CRAd Agents by Using Quantitative Real-Time PCR
The CRAd-CXCR4.RGD was diluted to 103, 104 and 105 viral particles (vp)/μl with distilled water, and then added to real-time PCR reactions as described in the Materials and Methods. Real-time PCR was performed as described below, and the E1 gene copy number was determined by using an oligo pair (forward primer-5′AACCAGTTGCCGTGAGAGTTG, reverse primer-5′CTDGTTAAGCAAG TCCTCG ATACA) and a probe (5′ 6-FAM-CACAGCTGGCGACGCCCATAMRA-3′). The oligos were designed using Primer Express 1.0 (Perkin-Elmer, Foster City, CA) and synthesized by Applied Biosystems. The real-time PCR reaction specifics have been described by us elsewhere.28, 36 A negative control, Ad5-CXCR4.Luc, was used in parallel, in which the Ad5 E1 gene was deleted.
Analysis of Replication of CRAd Agents in Tumor Cell Lines
A549 cells (105)/ well were cultured as described above, infected with 100 vp/cell of CRAd-CXCR4.RGD, Adwt, or Ad5-CXCR4.Luc in infection medium containing 2 % FBS, and incubated at 37° C in a 5 % CO2 environment. After a 3 hour incubation, infection medium was taken off, the cells were washed three times to remove uninternalized viruses, and the cells then placed in fresh culture medium with 10 % FBS. Media from triplicate wells were collected 1 (24 hours after viral infection), 3, and 9 days later, DNA was extracted from 200 μl of media with the DNeasy Tissue Kit (Qiagen, Valencia, CA). The Ad5 E4 gene was detected in DNA samples by using an oligo pair (forward primer-5′GGAGTGCGCCGAGACAAC, and reverse primer-5′ACTACGTCCGGCGTTCCAT) and a probe (5′ 6-FAM- TGGCATGACACTACGACCAACACG ATCT-TAMRA-3′). Real-time PCR was performed as described above. Ad E4 gene copy numbers were detected and normalized with human β-actin.
Analysis of Promoter Activities in Human Liver
Excess human liver not needed for diagnostic purposes was obtained from hepatectomy specimens following liver transplantation. To generate liver tissue slices, tissue was cut in consecutive 0.5mm-thick slices using the Krumdieck tissue slicer. Sequential slices were then cultured in 24 well-plates in RPMI medium supplemented with 10% bovine fetal serum, 100 U/ml penicillin, 100 μg/ml streptomycin and 5 μg/ml insulin. Each slice contains approximately one million cells as determined by Kirby et al.37 The tissue slices were infected with A) 500 vp/cell of Ad5-CXCR4.Luc, Ad5-MK.Luc, Ad5-SLPI.Luc, Ad5-Cox-2.Luc, or Ad5-CMV.Luc and B) 500 vp/cell of Ad5-CMV.Luc or Ad5-CMV.Luc.RGD. After two days of incubation at 37° C in a humidified atmosphere of 95% air and 5% CO2, luciferase activity was determined by using the Luciferase Assay System (Promega) on the Luminocount (Packard) instrument. Three tissue slices were included per group. Protein determination was performed by using the Bio-Rad Protein Assay Kit (Bio-Rad Laboratories, Inc. Hercules, CA). The luciferase activities were presented as RLU (relative light units).
Replication of CRAd-CXCR4 in human lung tumor slices
Excess tissue from three human lung tumor specimens not needed for diagnostic purposes were obtained from Dr. Robert Cerfolio (Department of Surgery, UAB) after operation. To generate lung tissue slices, tissue was cut using the Krumdieck tissue slicer under the tissue culture conditions as described above. The tissue slices were infected with 500 vp/cell of CRAd-CXCR4.RGD, Adwt or Ad5-CXCR4.Luc in fresh infection medium as described above. Three tissue slices were included per group. After 24 hour and 72 hour incubation times, respectively, total DNA was extracted from the human lung slices via the DNeasy Tissue Kit (Qiagen, Valencia, CA). DNA samples were treated with DNase free RNase to remove possible RNA contamination and stored at −80° C until use. Ad E4 gene copy numbers were quantified as described and normalized with human beta-actin. We reported viral copy number as E4 copies/ng DNA. The amount of DNA in each sample was determined by using a human β-actin probe, oligo pair and a series of diluted genomic DNA samples (50ng, 5ng, 0.5ng and 0.05ng DNA).
In vitro Analysis of Cytocidal Effects
The in vitro cytocidal effect of the CRAd-CXCR4.RGD was analyzed by determining the viability of the test cells with crystal violet staining after infection. Briefly, 25,000 cells (including Hs-766T pancreatic adenocarcinoma cells, A549 lung cancer cells, D65 glioma cells, OVSK3ip1 ovarian carcinomacells and a normal control cell line HMEC) / well were plated on a 12-well plate, cells were infected at 100, 10, 1, or 0 vp/cell with CRAd-CXCR4.RGD, or Ad5-CXCR4.Luc in infection medium. Three hours later, the infection medium was replaced with the appropriate complete medium. After 10 days of cultivation, the cells were fixed with 10% buffered formalin for 10 minutes and stained with 1% crystal violet in 70% ethanol for 20 minutes, followed by washing 3 times with tap water and air drying. Trypan blue exclusion experiments were also performed as described elsewhere.38
Statistic analysis
The Student’s test was employed for statistical analysis where P<0.05 was considered to be statistically significant.
Results
Attributes of the CXCR4 CRAds
The vector shown in Figure 1 was constructed, amplified in 911 cells and scaled up in A549 cells. The yields were 2.3 x 1011 vp/ml with a vp/plaque-forming unit ratio of 49. The vector structure was confirmed by PCR of the viral DNA (Figure 1, right bottom) and sequence data (not shown) from PCR products. PCR products of 541 bp were amplified from the DNA template of CRAd-CXCR4.RGD by using the oligo pair a/c (as described in Methods and Materials) which contained a poly-A signal sequence (262 bp) and a short version of the CXCR4 promoter (279 bp or −191/+88). PCR products of 279 bp were amplified from the DNA template of CRAd-CXCR4.RGD by using the oligo pair b/c contained which the CXCR4 promoter only. As expected, both PCR amplifications with the same oligo pairs were negative when the non-replicative vector, Ad5-CMV.Luc, was used as the template. The CRAd agent, CRAd-CXCR4.RGD, possessed the E1 gene (Figure 2). The E1 gene was detected by real-time PCR at all tested Ad dilutions (103, 104, and 105 vp) in a dose responsive manner, but not in the non-replicating viral vector Ad5-CXCR4.Luc, in which the E1 gene was deleted as shown in Table 1. In the CRAd-CXCR4.RGD, the native promoter of the E1 gene (nt 346-521, NCBI nucleotide database accession # BK000408), was replaced by the CXCR4 promoter. Thus, the virus E1 gene was driven by the CXCR4 promoter instead of the native promoter, which was evidenced by the CRAds propagated in the lung tumor cell line, A549, in which the CXCR4 gene was over-expressed. The Ad type 5 backbone, pVK503C, was a kind gift from Dr. V. Krasnyhk (MD Anderson Cancer Center, Houston, TX) and used for generating the CRAd agents in this study. This vector contains the E3 gene and a RGD4C motif, peptide CDCRGDCFC, which was inserted into the HI loop of the Ad knob.29 The E3 11.6 kDa adenovirus death protein has a key role in cell-killing and promotes the release of progeny virions from the cell.30 RGD4C is a capsid modification, which allows one to retarget adenoviral vector to integrins via a CAR-independent pathway, for Ad infectivity enhancement, and was verified by sequencing data (not shown).
Figure 2.
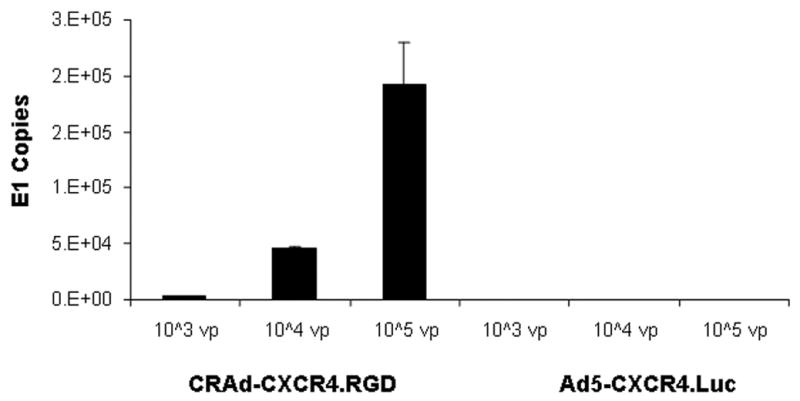
Adenovirus E1 copy detection.
The CRAd-CXCR4.RGD and a non-replicating Ad vector, Ad5-CXCR4.Luc were diluted with distilled water to either 105, 104, or 103vp/μl, and the E1 gene was sought by real-time PCR as described in the Materials and Methods. E1 levels were detected at all dilutions with the CRAd agent but not in the non-replicative Ad vector. All experiments in were performed in triplicate.
The Evidence for CRAd Agent Replication in Tumor Cell Lines
The CRAd-CXCR4.RGD agent was rescued in A549 cells as described previously. To further assay CRAd replication, A549 lung cancer cells (105) were plated in a 24 well plate and cells were infected with 100 vp/cell of CRAd-CXCR4.RGD, Adwt, or Ad5-CXCR4.Luc. After a 3 hr infection, the cells were washed three times with PBS to remove free non-internalized viruses and provided with fresh medium. The presence of the E4 gene was determined by using real-time PCR from DNA samples extracted from the medium after 1, 3, and 9 days post-infection. The Ad5 E4 copy numbers in the medium should relate to the levels of released virions from the tumor cells and also to the replication rates of the CRAd agents. The data shown in Figure 3 demonstrated that the Ad5 E4 copies were detected after both 3 and 9 days post-infection but were undetectable at day 1 for CRAd-CXCR4.RGD or Adwt. As predicted, the Ad5 E4 copy was undetectable at 1, 3, and 9 days post-infection for Ad5-CXCR4.Luc, a non replicating Ad vector. The replication levels of Adwt were 17 million copies at day 3 and 61 million copies at day 9, respectively, with a 3.6 fold incremental rate. Compared to CRAd-CXCR4.RGD, replication levels were 17 million at day 3 post-infection and 147 million at day 9, respectively, with an 8.6 fold incremental rate. These data indicated that both the CRAd agent and Adwt replicated in A549 lung cancer cells but that the replication rate in CRAd-CXCR4.RGD was higher than that in Adwt.
Figure 3.

Replication of CRAd-CXCR4.RGD in the A549 cell line.
Lung tumor cells (5 x 104) were plated in 24 well plates. Cells were infected by the CRAd-CXCR4.RGD, a replicating Ad control (Adwt), or a non-replicating Ad control, Ad5-CXCR4.Luc, at 100 v.p./cell. 200 μl of medium were taken from each well at days one (24 hours after infection), three, and nine. DNA was isolated with a Qiagen column, and the E4 gene copy number was determined by real-time PCR as described. All experiments were performed in triplicate.
A “Liver Off” Profile is seen with the CXCR4 Promoter in Human Liver Slices
Because Ad replication is species-specific and because the major Ad toxicity in gene therapy trials is hepatic, we tested the CXCR4 promoter activity in human liver specimens in organ culture. For that, we measured luciferase expression by using a conventional assay in human liver slices after a 2 day infection period with Ad5-CXCR4.Luc, Ad5-MK.Luc, Ad5-SLPI.Luc, Ad5Cox-2.Luc, or Ad5-CMV.Luc, respectively (Figure 4A). The first four vectors are regularly used in our center for tumor targeting, and the last one, Ad5-CMV.Luc, was used as a positive control. All these vectors have a luciferase reporter gene under the control of a different promoter. The luciferase expression level is tied to the activity of the promoter. Compared to the CMV promoter, the CXCR4 promoter activity (4.1% RLU of CMV) is the lowest with a P value of < 0.01 among the four tumor specific promoters in human liver slices. We also compared Ad5-CMV.Luc with Ad5-CMV.Luc.RGD in human liver slices (Figure 4B). The luciferase activities of both Ad vectors are of similar levels without a statistically significant difference. From these experiments, it is evident that the CXCR4 promoter has a “liver off” phenotype and that should result in low toxicity to the human host liver.
Figure 4.

Transcriptional activity of the CXCR4 promoter in human liver slices.
Human liver slices were infected with A) 500 vp/cell of Ad vector, AdCMVLuc, AdCXCR4Luc, AdSLPILuc, AdMkLuc, or AdCox-2Luc; B) 500VP/cell of Ad-CMV.Luc and Ad-CMV.Luc.RGD. Two days following infection, luciferase activities were detected by a conventional assay and shown as RLU (relative light units). *, P<0.05; **, P<0.01.
The Evidence for CRAd Agent Replication in Lung Tumor Tissue Slices
To verify the replication of the test CRAd agent under near-clinical conditions, we determined the replication of the CRAd-CXCR4.RGD in three lung tumor specimens from patients. All three lung tumor samples were conventional squamous cell carcinomas diagnosed by Pathologists in the Department of Pathology at UAB. The histopathological appearance of tumor from two of the patients is shown in Figure 5. The samples were sliced as described in Materials and Methods, and infected with 500vp/cell of CRAd-CXCR4.RGD, Adwt or the non-replicative control, Ad5-CXCR4.Luc. The slices were collected after 24 hours and 72 hours, respectively, and DNAs were isolated from tissue as described in the Materials and Methods. The E4 copy numbers were determined by real-time PCR which served as a surrogate for the levels of adenoviruses released from infected tumor cells. The data showed (Figure 6) that E4 DNA copy number per ng DNA of CRAd-CXCR4.RGD increased between day 1 and 3, to 140%, 140%, and 360% in the three patient samples, p87, p90, and p92, respectively. The E4 DNA copy number per ng DNA of Adwt increased between day 1 and 3, to 100 %, 670 % and 870 %, respectively. The DNA copy numbers of CRAd-CXCR4.RGD at day 1 are 1217, 3371 and 3805, respectively compared to that of Adwt, 685, 267 and 588, respectively, in all the three lung tumor samples. As predicted, Ad5-CXCR4.Luc had low viral copy levels in the three lung tumor samples. A statistically significant difference was noted between the CRAd agent and both the Adwt and the non-replicative Ads in all three patient samples. There were no significant differences between Adwt and non-replicative Ads, although the values of the former were higher than that of the later in the patient tumor samples.
Figure 5.

Pathologic diagnosis of lung cancer from two patients.
Representative sections of squamous cell carcinoma from two patients, p87 and p90, and one non-neoplastic normal lung, n4, are demonstrated. All sections were stained with hematoxylin and eosin (H & E). The magnifications are x 100~200.
Figure 6.

Replication activity of the CXCR4 promoter in patient lung tissue slices.
Patient lung slices were infected with 500 vp/cell of Ad vector (Adwt, Ad5-CXCR4.Luc, or CRAd-CXC4.RGD). 24 hours and 72 hours after infection, DNAs were isolated from each lung section and the E4 levels were determined by real-time PCR after normalization against beta-actin and shown as E4 copy number per ng DNA.
*, P<0.05; **, P<0.01.
CXCR4 CRAds induce cytotoxicity in multiple tumor cell lines
Next, we used the CRAd-CXCR4.RGD as an oncolytic anti-tumor agent. CRAd-CXCR4.RGD was evaluated for its cell-killing effect in a variety of tumor cell lines. Cytotoxicity was evaluated after 10 days of incubation via crystal violet staining (Fig. 7). While the replication-incompetent AdCXCR4Luc vector had no cytotixic effect even at 100 vp/cell, the CXCR4-based CRAds induced strong cytotoxicity in the lung cancer cell line, A549 as well as in Hs-766T cells (pancreatic cancer cell line). Almost 100% of cells were killed even at the minimal dose, 1vp/cell. Intermediate cytoxicities were observed in the ovarian cancer SKOV3ip1 cell line and in glioma D65 cells in which 100% of cells died at 10 vp/cell. As expected, however, no cytoxicity was observed in the non-transformedl mammary epithelial cell line, HMEC, even at the highest dose, 100 vp/cell tested.
Figure 7A.


Cytotoxic efficiency of CRAd-CXCR4.RGD on multiple tumor cell lines.
5 x 104 cells from each cell line, including the lung cell line, A549, pancreatice cancer cell line, Hs-766T, the glioma cell line, D65, the ovarian cancer cell line, SKOV3ip1, and a non-tranformed mammary epithelial cell line (HMEC) were plated onto 24-well plates, and infected with CRAd-CXCR4.RGD or Ad5-CXCR4.Luc at the indicated MOIs or mock-infected. Cells were stained with crystal violet after a 10 day incubation as described in the Materials and Methods.
7B. Transductional levels of Ad agents in HMEC and A549 cell lines.
5 x 104 cells of HMEC and A549 were plated onto 24-well plates, and infected with CRAd-CXCR4.RGD and Adwt at MOI=100. After a 3 hour incubation, DNA was isolated from the cells and E4 and Actin were detected by real-time PCR.
The transductional level of the CXCR4-based CRAd was only 57% in HMEC cells as compared to A549 cells (100%) and showed a similar transductional level as Adwt in both HMEC and A549 cells (Figure 7B). The results indicate that the CXCR4 based CRAd transduced into HMEC cells at a lower level as compared to that in the lung cancer cell line, A549, and that both integrins and CAR are present on their cell surfaces.
Discussion
Recent advances in our understanding of growth factors, molecular oncology, tumor immunology and gene therapy have provided the rationale for cancer gene therapy.39 Some of these approaches in the gene therapy field, including adenovirus-mediated p53 gene transfer and adenovirus-mediated herpes simplex virus thymidine kinase/ganciclovir gene therapy, are being tested in clinical trials in patients with lung cancer and malignant mesothelioma. 16, 40, 41 The major challenges of gene therapy include inefficient gene delivery,5,6 toxicity32 and immune responses,42 although we did not focus on the immune response of adenovirus type5 CRAd agents in this study.
Recently, replication competent adenoviruses have come into focus as promising novel anti-tumor agents for viral oncolysis and enhanced transfer of therapeutic genes.4, 43 Tumor specificity is the key to the realization of replicating viruses as cancer therapeutics. This is especially relevant in the context of systemic therapy, as anticipated for treatment of most malignant neoplasms. Despite the theoretical utility of conditionally replicative adenoviruses (CRAds), the overall tumor response in practice has been limited. This is based on the lack of native adenovirus receptor, CAR, on most tumor cell surfaces and lack of TSPs to selectively drive Ad E1 expression. Ad E1 protein corresponds to viral replication and deletion of the E1A coding region from the Ad genome makes the virus non-replication competent; allowing replication only in the E1A transcomplementing cell lines, such as 293 and 911. In this study, we constructed a novel CRAd agent, CRAd-CXCR4.RGD, in which the Ad E1 gene is under the control of a tumor specific promoter, the CXCR4 promoter, while viral infectivity is enhanced via a CAR-independent pathway by an RGD4C capsid modification. We evaluated the replication of this agent in tumor cell lines and in lung tumor samples from patients. We also evaluated the activity of the promoter in human liver.
We used CXCR4 as a tumor specific promoter to regulate the Ad E1 gene expression of CRAd-CXCR4.RGD in this study based on our previously published data. We had shown that the CXCR4 promoter has a phenotype of “tumor on and liver off” both in vitro and in vivo, and that CXCR4 should prove to be a good candidate TSP for lung cancer gene therapy approaches.28 Here we successfully constructed a CRAd agent in which the E1 gene expression was under the control of the CXCR4 promoter, a TSP for regulatory tumor targeting. As a tumor killing effect (oncolysis) is known to parallel viral replication in tumor cells, replication remains the key to evaluating the tumor killing effect of the CRAd-CXCR4.RGD. We infected the CRAd-CXCR4.RGD and controls into A549 cells, a lung cancer cell line, and the DNAs were purified from the medium after 1, 3, and 9 days of incubation. The Ad DNAs were detected in day 3 and day 9 medium samples by using real-time PCR and we showed that the Ad E4 copy number of CRAd-CXCR4.RGD increased 8.6 fold while that of Adwt increased only 3.6 fold. From these data, we conclude that 1) the replication of the CRAd agent is more efficient than that of Adwt in the tumor cell line, A549; 2) because the transductional level of CRAd-CXCR4.RGD was almost double amount as compared to that of Adwt in A549 cells in Figure 7B, and the oncolytic activity of CRAd-CXCR4.RGD was more than 100 fold higher in A549 cells as compared to that in HMECs in Figure 7A. As described above, the tumor killing effect is parallel to viral replication which is correlated to Ad E1 protein level driven by the CXCR4 promoter in this study. Thus, the CRAd higher replication rate in A549 cells as observed that most probably is regulated at both transductional and transcriptional levels, but mainly at the transcriptional level. Both of them could enhance viral spread in tumor cells. This point would require addition support with further studies as it is at odds with the data of Hitt et al. 44 that very low levels of E1 protein are sufficient for virus production in cultured cells and the finding that wide variation in E1A expression levels has little effect on virus replication; 3) the Ad E4 gene was not detected in the media in day 1 samples for both the CRAd agent and Adwt. This suggests that the whole life cycle from adenovirus internalization to viral release from infected cells takes longer than 24 hours; 4) the viruses detected in the media are viable because they were capable being released from tumor cells with intact biological function.
Infection with adenovirus causes profound changes in host-cell macromolecular synthesis that ultimately leads to cell death. Virion fiber protein inhibits macromolecular synthesis when applied directly to cells bearing the adenovirus receptor.45 Cell-specific DNA synthesis, export of cellular mRNAs from the nucleus to the cytoplasm, and cell-specific translation are all inhibited after infection, but the precise mechanisms are not completely understood. Also, high expression of the CXCR4 gene in some blood elements, such as NK cells and lymphocytes, have been reported. However, the main damage to the host is that the majority (>95%) of viruses released into the blood stream home to the liver 32, 46 which leads to lethal host toxicity. We have reported28 that the activity of the CXCR4 promoter is “off” in mouse liver. To mimic the in vivo condition of the human host, we used human liver slices instead of murine liver in this study to rigorously evaluate the activity of the CXCR4 promoter. Compared to three other promoters regularly used for tumor targeting in this Center, the activity of the CXCR4 promoter is the lowest among them in this organ. In other words, the CXCR4 promoter has the lowest toxicity to human liver among the available tested promoters.
Xenograft animal models are the conventional system to evaluate cancer therapeutics in the gene therapy field. However, this is not a perfect model system for several reasons. Multiples studies have been performed in athymic nude mice and severely combined immonodeficient mice (scid).47, 48 Although nude mice lack functional T lymphocytes, they retain natural killer (NK) cell activity that can impede the normal patterns of growth and metastasis from xenograft implants.49 Thus, this model system, lacking a competent immue response, does not provide a reliable model for direct extrapolation to the clinical environment.45 For this reason, we used human lung tumor tissue to evaluate the viral replication of the CRAd agent in this study from three patients with primary squamous cell carcinoma of lung. All three patients were diagnosed by UAB Department of Pathology Surgical Pathology and confirmed by one of us (G.P.S.). The patterns are similar in that the adenovirus E4 copy number of the CRAd-CXCR4.RGD was higher than that of Adwt in all three samples. Of note, the replication rates as measured by DNA copy at day 3/DNA copy at day 1 are lower than that of Adwt. That argues that this higher DNA copy number in the CRAd agent might only reflect the infectivity potential in primary tumor cells, but not replication.
In the CRAd-CXCR4.RGD vector, the Ad E1 is regulated by a TSP, the CXCR4 promoter. The E1A –induced activation of the apoptosis pathway(s) must be modulated by E1B protein to ensure efficient virus replication prior to cell death.50 Activation of the interferon-indicible RNase L pathway by an adenovirus-associated type I (VAI) RNA51 may also contribute to the stimulation of apoptotic pathways in adenovirus-infected cells.52 The E3 11.6-kDa adenovirus death protein, which exists in CRAd-CXCR4.RGD, also has a role in cell killing and promotes the release of progeny virions from cells. Taken together, cell killing is related to virus replication which, in turn, is related to E1 expression level. As noted, the E1 expression of the CRAd-CXCR4.RGD is under the control of the CXCR4 promoter. We detected tumor cell killing with an oncolytic assay by staining with crystal violet. The data showed the CRAd-CXCR4 agent has strong cytoxicity to the lung cancer cell line A549 and the pancreatic cancer cell line, Hs-766T, as well as intermediate cytoxicity to the D65 glioma cell line and SKOV3ip1 ovarian cancer cell line, but not to the non-transformed mammary epithelial cell line HMEC. The transductional level of CRAd-CXCR4.RGD was 57% in HMEC cells as compared to that in A549 cells (100%) (Figure 7B); however, the cell killing activity (oncolusis) of CRAd-CXCR4.RGD, which is achieved by direct consequence of the viral replication, was more than 100 fold higher in A549 cells than that in HMEC cells (Figure 7A). From these data, the generated CXCR4-CRAd appears to be able to target multiple tumors in addition to squamous cell carcinoma of the lung. Because many papers have described the relationship of the CXCR4 gene as related to tumor metastasis, we anticipate evaluating whether metastatic lung lesions will be modulated by this agent in a future study. In this study, we didn’t show any data of tumor killing by CRAd agents in primary cells because it is technically not feasible. From our unpublished data we know that untreated primary tumor cells are alive for less than four days in vitro, thus, viability assays of virally infected primary cells would be nearly impossible to perform.
In conclusion, we identified the human CXCR4 promoter as a tumor-specific regulatory element for lung cancer and others solid neoplasms. Viral replication is active in an established lung cancer cell line and primary lung tumor tissues. Thus, the CXCR4 promoter emerges as an especially promising transcriptional targeting gene approach for lung cancer. It is apparent that the CXCR4 promoter in a CRAd context should be potentially useful for future experimental clinical applications for squamous cell carcinoma of the lung and other cancer types.
Acknowledgments
We are especially indebted to Drs. N. L. Michael and M. Yamamoto for the gifts of the plasmids, pLuc-1430c, pLuc-cyc1.2 and AdCox-2Luc, used in this study. Also we thank Drs. S.D. Barker, and Y. Adachi for AdSLPILuc and AdMkLuc used in this study as controls.
The abbreviations used are
- bp
base pairs
- CMV
cytomegalovirus
- CRAd
conditionally replicating adenovirus
- MOI
multiplicity of infection
- pfu
plaque-forming units
- vp
viral particles
- TSP
tumor-specific promoter
Conflict of Interest Statement
None Declared
Supported by the National Institute of Health Grants: R01 CA83821, R01 CA 94084, K12 HD01261-02, R01 HL67962, and R01 CA93796 and The Mesothelioma Applied Research Foundation, Inc.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Moon C, Oh Y, Roth JA. Current status of gene therapy for lung cancer and head and neck cancer. Clin Cancer Res. 2003;9:5055–67. [PubMed] [Google Scholar]
- 2.Webb HE, Smith CE. Viruses in the treatment of cancer. Lancet. 1970;1(1):206–8. doi: 10.1016/s0140-6736(70)91790-3. [DOI] [PubMed] [Google Scholar]
- 3.Fueyo J, Gomez-Manzano C, Alemany R, Lee PS, McDonnell TJ, Mitlianga P, Shi YX, Levin VA, Yung WK, Kyritsis AP. A mutant oncolytic adenovirus targeting the Rb pathway produces anti-glioma effect in vivo. Oncogene. 2000;19:2–12. doi: 10.1038/sj.onc.1203251. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki K, Fueyo J, Krasnykh V, Reynolds PN, Curiel DT, Alemany R. A conditionally replicative adenovirus with enhanced infectivity shows improved oncolytic potency. Clin Cancer Res. 2001;7:120–6. [PubMed] [Google Scholar]
- 5.Hemmi S, Geertsen R, Mezzacasa A, Peter I, Dummer R. The presence of human coxsackievirus and adenovirus receptor is associated with efficient adenovirus-mediated transgene expression in human melanoma cell cultures. Hum Gene Ther. 1998;9:2363–73. doi: 10.1089/hum.1998.9.16-2363. [DOI] [PubMed] [Google Scholar]
- 6.Miller CR, Buchsbaum DJ, Reynolds PN, Douglas JT, Gillespie GY, Mayo MS, Raben D, Curiel DT. Differential susceptibility of primary and established human glioma cells to adenovirus infection: targeting via the epidermal growth factor receptor achieves fiber receptor-independent gene transfer. Cancer Res. 1998;58:5738–48. [PubMed] [Google Scholar]
- 7.Reynolds PN. Targeting gene delivery for pulmonary disease. In: Albelda SM, Marcel Dekker, editors. Gene therapy for diseases of the lung. Vol. 169. 2002. pp. 119–144. [Google Scholar]
- 8.Krasnykh V, Dmitriev I, Mikheeva G, Miller CR, Belousova N, Curiel DT. Characterization of an adenovirus vector containing a heterologous peptide epitope in the HI loop of the fiber knob. J Virol. 1998;72:1844–52. doi: 10.1128/jvi.72.3.1844-1852.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devaux C, Adrian M, Berthet-Colominas C, Cusack S, Jacrot B. Structure of adenovirus fiber. I. Analysis of crystals of fiber from adenovirus serotypes 2 and 5 by electron microscopy and X-ray crystallography. J Mol Biol. 1990;215:567–88. doi: 10.1016/S0022-2836(05)80169-X. [DOI] [PubMed] [Google Scholar]
- 10.Bergelson JM, Cunningham JA, Droguett G, Kurt-Jones EA, Krithivas A, Hong JS, Horwitz MS, Crowell RL, Finberg RW. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–3. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 11.Frey BM, Hackett NR, Bergelson JM, Finberg R, Crystal RG, Moore MA, Rafii S. High-efficiency gene transfer into ex vivo expanded human hematopoietic progenitors and precursor cells by adenovirus vectors. Blood. 1998;91:2781–92. [PubMed] [Google Scholar]
- 12.Hong SS, Karayan L, Tournier J, Curiel DT, Boulanger PA. Adenovirus type 5 fiber knob binds to MHC class I alpha2 domain at the surface of human epithelial and B lymphoblastoid cells. Embo J. 1997;16:2294–306. doi: 10.1093/emboj/16.9.2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomko RP, Johansson CB, Totrov M, Abagyan R, Frisen J, Philipson L. Expression of the adenovirus receptor and its interaction with the fiber knob. Exp Cell Res. 2000;255:47–55. doi: 10.1006/excr.1999.4761. [DOI] [PubMed] [Google Scholar]
- 14.Henry LJ, Xia D, Wilke ME, Deisenhofer J, Gerard RD. Characterization of the knob domain of the adenovirus type 5 fiber protein expressed in Escherichia coli. J Virol. 1994;68:5239–46. doi: 10.1128/jvi.68.8.5239-5246.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruigrok RW, Barge A, Albiges-Rizo C, Dayan S. Structure of adenovirus fiber. II. Morphology of single fibres. J Mol Biol. 1990;215:589–96. doi: 10.1016/S0022-2836(05)80170-6. [DOI] [PubMed] [Google Scholar]
- 16.Wickham TJ, Mathias P, Cheresh DA, Nemerow GR. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 1993;73:309–19. doi: 10.1016/0092-8674(93)90231-e. [DOI] [PubMed] [Google Scholar]
- 17.Lee SE, Jin RJSG, Lee SJ, Yoon MS, Park DS, Heo, Choi H. Development of a new plasmid vector with PSA-promoter and enhancer expressing tissue-specificity in prostate carcinoma cell lines. Anticancer Res. 2000;20:417–22. [PubMed] [Google Scholar]
- 18.Huber BE, Richards CA, Krenitsky TA. Retroviral-mediated gene therapy for the treatment of hepatocellular carcinoma: an innovative approach for cancer therapy. Proc Natl Acad Sci U S A. 1991;88:8039–43. doi: 10.1073/pnas.88.18.8039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, McClanahan T, Murphy E, Yuan W, Wagner SN, Barrera JL, Mohar A, Verastegui E, Zlotnik A. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410:50–6. doi: 10.1038/35065016. [DOI] [PubMed] [Google Scholar]
- 20.Moore MA. The role of chemoattraction in cancer metastases. Bioessays. 2001;23:674–6. doi: 10.1002/bies.1095. [DOI] [PubMed] [Google Scholar]
- 21.Payne AS, Cornelius LA. The role of chemokines in melanoma tumor growth and metastasis. J Invest Dermatol. 2002;118:915–22. doi: 10.1046/j.1523-1747.2002.01725.x. [DOI] [PubMed] [Google Scholar]
- 22.Murakami T, Maki W, Cardones AR, Fang H, Tun Kyi A, Nestle FO, Hwang ST. Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res. 2002;62:7328–34. [PubMed] [Google Scholar]
- 23.Taichman RS, Cooper C, Keller ET, Pienta KJ, Taichman NS, McCauley LK. Use of the stromal cell-derived factor-1/CXCR4 pathway in prostate cancer metastasis to bone. Cancer Res. 2002;62:1832–7. [PubMed] [Google Scholar]
- 24.Spano JP, Andre F, Morat L, Sabatier L, Besse B, Combadiere C, Deterre P, Martin A, Azorin J, Valeyre D, Khayat D, Le Chevalier T, Soria JC. Chemokine receptor CXCR4 and early-stage non-small cell lung cancer: pattern of expression and correlation with outcome. Ann Oncol. 2004;15:613–7. doi: 10.1093/annonc/mdh136. [DOI] [PubMed] [Google Scholar]
- 25.Phillips RJ, Burdick MD, Lutz M, Belperio JA, Keane MP, Strieter RM. The stromal derived factor-1/CXCL12-CXC chemokine receptor 4 biological axis in non-small cell lung cancer metastases. Am J Respir Crit Care Med. 2003;167:1676–86. doi: 10.1164/rccm.200301-071OC. [DOI] [PubMed] [Google Scholar]
- 26.Burger M, Glodek A, Hartmann T, Schmitt-Graff A, Silberstein LE, Fujii N, Kipps TJ, Burger JA. Functional expression of CXCR4 (CD184) on small-cell lung cancer cells mediates migration, integrin activation, and adhesion to stromal cells. Oncogene. 2003;22:8093–101. doi: 10.1038/sj.onc.1207097. [DOI] [PubMed] [Google Scholar]
- 27.Haviv YS, van Houdt WJ, Lu B, Curiel DT, Zhu ZB. Transcriptional targeting in renal cancer cell lines via the human CXCR4 promoter. Mol Cancer Ther. 2004;3:687–91. [PubMed] [Google Scholar]
- 28.Zhu ZB, Makhija SK, Lu B, Wang M, Kaliberova L, Liu B, Rivera AA, Nettelbeck DM, Mahasreshti PJ, Leath CA, 3rd, Yamaoto M, Alvarez RD, Curiel DT. Transcriptional targeting of adenoviral vector through the CXCR4 tumor-specific promoter. Gene Ther. 2004;11:645–8. doi: 10.1038/sj.gt.3302089. [DOI] [PubMed] [Google Scholar]
- 29.Yamamoto M, Davydova J, Wang M, Siegal GP, Krasnykh V, Vickers SM, Curiel DT. Infectivity enhanced, cyclooxygenase-2 promoter-based conditionally replicative adenovirus for pancreatic cancer. Gastroenterol. 2003;125:1203–18. doi: 10.1016/s0016-5085(03)01196-x. [DOI] [PubMed] [Google Scholar]
- 30.Barker SD, Coolidge CJ, Kanerva A, Hakkarainen T, Yamamoto M, Liu B, Rivera AA, Bhoola SM, Barnes MN, Alvarez RD, Curiel DT, Hemminki A. The secretory leukoprotease inhibitor (SLPI) promoter for ovarian cancer gene therapy. J Gene Med. 2003;5:300–10. doi: 10.1002/jgm.341. [DOI] [PubMed] [Google Scholar]
- 31.Adachi Y, Reynolds PN, Yamamoto M, Grizzle WE, Overturf K, Matsubara S, Muramatsu T, Curiel DT. Midkine promoter-based adenoviral vector gene delivery for pediatric solid tumors. Cancer Res. 2000;60:4305–10. [PubMed] [Google Scholar]
- 32.Dmitriev I, Krasnykh V, Miller CR, Wang M, Kashentseva E, Mikheeva G, Belousova N, Curiel DT. An adenovirus vector with genetically modified fibers demonstrates expanded tropism via utilization of a coxsackievirus and adenovirus receptor-independent cell entry mechanism. J Virol. 1998;72:9706–13. doi: 10.1128/jvi.72.12.9706-9713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu DC, Chen Y, Seng M, Dilley J, Henderson DR. The addition of adenovirus type 5 region E3 enables calydon virus 787 to eliminate distant prostate tumor xenografts. Cancer Res. 1999;59:4200–3. [PubMed] [Google Scholar]
- 34.Wegner SA, Ehrenberg PK, Chang G, Dayhoff DE, Sleeker AL, Michael NL. Genomic organization and functional characterization of the chemokine receptor CXCR4, a major entry co-receptor for human immunodeficiency virus type 1. J Biol Chem. 1998;273:4754–60. doi: 10.1074/jbc.273.8.4754. [DOI] [PubMed] [Google Scholar]
- 35.Nettelbeck DM, Rivera AA, Balague C, Alemany R, Curiel DT. Novel oncolytic adenoviruses targeted to melanoma: specific viral replication and cytolysis by expression of E1A mutants from the tyrosinase enhancer/promoter. Cancer Res. 2002;62:4663–70. [PubMed] [Google Scholar]
- 36.Zhu ZB, Makhija SK, Lu B, Wang M, Kaliberova L, Liu B, Rivera AA, Nettelbeck DM, Mahasreshti PJ, Leath CA, Barker S, Yamamoto M, Li F, Alvarez RD, Curiel DT. Transcriptional targeting of tumors with a novel tumor-specific survivin promoter. Cancer Gene Ther. 2004;11:256–62. doi: 10.1038/sj.cgt.7700679. [DOI] [PubMed] [Google Scholar]
- 37.Kirby TO, Rivera AA, Rein D, Wang M, Ulasov I, Breidenbach M, Kataram M, Contreras JL, Krumdieck C, Yamamoto M, Rots MG, Haisma HJ, Alvarez RD, Mahasreshti PJ, Curiel DT. A novel ex vivo model system for evaluation of conditionally replicative adenoviruses therapeutic efficacy and toxicity. Clin Cancer Res. 2004;10:8697–703. doi: 10.1158/1078-0432.CCR-04-1166. [DOI] [PubMed] [Google Scholar]
- 38.Fueyo J, Gomez-Manzano C, Yung WK, Liu TJ, Alemany R, McDonnell TJ, Shi X, Rao JS, Levin VA, Kyritsis AP. Overexpression of E2F-1 in glioma triggers apoptosis and suppresses tumor growth in vitro and in vivo. Nat Med. 1998;4:685–90. doi: 10.1038/nm0698-685. [DOI] [PubMed] [Google Scholar]
- 39.Buchsbaum DJ, Curiel DT. Gene therapy for the treatment of cancer. Cancer Biother Radiopharm. 2001;16:275–88. doi: 10.1089/108497801753131354. [DOI] [PubMed] [Google Scholar]
- 40.Carbone DP, Minna JD. The molecular genetics of lung cancer. Adv Intern Med. 1992;37(1):53–71. [PubMed] [Google Scholar]
- 41.Swisher SG, Roth JA, Nemunaitis J, Lawrence DD, Kemp BL, Carrasco CH, Connors DG, El-Naggar AK, Fossella F, Glisson BS, Hong WK, Khuri FR, Kurie JM, Lee JJ, Lee JS, Mack M, Merritt JA, Nguyen DM, Nesbitt JC, Perez-Soler R, Pisters KM, Putnam JB, Jr, Richli WR, Savin M, Schrump DS, Shin DM, Shulkin A, Walsh GL, Wait J, Weill D, Waugh MK. Adenovirus-mediated p53 gene transfer in advanced non-small-cell lung cancer. J Natl Cancer Inst. 1999;91(7):63–71. doi: 10.1093/jnci/91.9.763. [DOI] [PubMed] [Google Scholar]
- 42.Sumida SM, Truitt DM, Lesniak MS. Neutralizing antibodies to adenovirus serotype 5 vaccine vectors are directed primarily against the adenovirus hexon protein. J Immunol. 2005;174(11):7179–85. doi: 10.4049/jimmunol.174.11.7179. [DOI] [PubMed] [Google Scholar]
- 43.Sonabend AM, I, Ulasov V, Lesniak MS. Conditionally replicative adenoviral vectors for malignant glioma. Rev Med Virol. 2006;16:99–115. doi: 10.1002/rmv.490. [DOI] [PubMed] [Google Scholar]
- 44.Hitt MM, Graham FL. Adenovirus E1A under the control of heterologous promoters: wide variation in E1A expression levels has little effect on virus replication. Virology. 1990;179:667–78. doi: 10.1016/0042-6822(90)90134-d. [DOI] [PubMed] [Google Scholar]
- 45.Levine AJ, Ginsberg HS. Mechanism by which fiber antigen inhibits multiplication of type 5 adenovirus. J Virol. 1967;1:747–57. doi: 10.1128/jvi.1.4.747-757.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reynolds P, Dmitriev I, Curiel DT. Insertion of an RGD motif into the HI loop of adenovirus fiber protein alters the distribution of transgene expression of the systemically administered vector. Gene Ther. 1999;6:1336–9. doi: 10.1038/sj.gt.3300941. [DOI] [PubMed] [Google Scholar]
- 47.Croy BA, Linder KE, Yager JA. Primer for non-immunologists on immune-deficient mice and their applications in research. Comp Med. 2001;51:300–13. [PubMed] [Google Scholar]
- 48.Williams SS, Alosco TR, Croy BA, Bankert R. The study of human neoplastic disease in severe combined immunodeficient mice. Lab Anim Sci. 1993;43:139–46. [PubMed] [Google Scholar]
- 49.Stakleff KD, V, Von Gruenigen E. Rodent models for ovarian cancer research. Int J Gynecol Cancer. 2003;13:405–12. doi: 10.1046/j.1525-1438.2003.13317.x. [DOI] [PubMed] [Google Scholar]
- 50.Han J, Sabbatini P, Perez D, Rao L, Modha D, White E. The E1B 19K protein blocks apoptosis by interacting with and inhibiting the p53-inducible and death-promoting Bax protein. Genes Dev. 1996;10:461–77. doi: 10.1101/gad.10.4.461. [DOI] [PubMed] [Google Scholar]
- 51.Desai SY, Patel RC, Sen GC, Malhotra P, Ghadge GD, Thimmapaya B. Activation of interferon-inducible 2′-5′ oligoadenylate synthetase by adenoviral VAI RNA. J Biol Chem. 1995;270:3454–61. doi: 10.1074/jbc.270.7.3454. [DOI] [PubMed] [Google Scholar]
- 52.Diaz-Guerra M, Rivas C, Esteban M. Activation of the IFN-inducible enzyme RNase L causes apoptosis of animal cells. Virology. 1997;236:354–63. doi: 10.1006/viro.1997.8719. [DOI] [PubMed] [Google Scholar]