

Abstract
53BP1 plays an important role in cellular response to DNA damage. It is thought to be the mammalian homologue of budding yeast Rad9 and/or fission yeast Crb2. Rad9/Crb2 are bona fide checkpoint proteins whose activation requires their corresponding C-terminal tandem BRCT (BRCA1 C-terminal) motifs, which mediate their oligomerization and phosphorylation at multiple sites following DNA damage. Here we show that the function of human 53BP1 similarly depends on its oligomerization and phosphorylation at multiple sites but in a BRCT domain-independent manner. Moreover, unlike its proposed yeast counterparts, human 53BP1 only has limited checkpoint functions but rather acts as an adaptor in the repair of DNA double strand breaks. This difference in function may reflect the higher complexity of the DNA damage response network in metazoa including the evolution of other BRCT domain-containing proteins that may have functions redundant or overlapping with those of 53BP1.
To protect the integrity of their DNA against the potentially deleterious assaults from various endogenous and environmental sources, cells have evolved a genome surveillance network that carefully coordinates DNA repair with cell cycle progression. DNA double strand breaks (DSBs)4 are considered the most toxic type of DNA damage. If left unrepaired or repaired improperly, they cause chromosomal aberrations, which may be lethal or result in oncogenic transformation (1, 2). One of the network components activated early in response to DNA DSBs is 53BP1, a large nuclear protein that was originally identified in a yeast two-hybrid screen as a p53-binding partner (3). Domain analysis revealed the presence of a tandem BRCT motif at the C terminus of 53BP1, a domain frequently found in proteins implicated in DNA damage response pathways (4, 5). Moreover, the N terminus of 53BP1 contains several (S/T)Q motifs, the preferred phosphorylation sites for members of the phosphoinositide 3-kinase-related protein kinase (PIKK) family, which play central roles in DNA damage signal transduction. Subsequent studies showed that 53BP1 is phosphorylated by the PIKK family member ATM and undergoes a rapid relocalization to sites of DNA DSBs upon exposure of cells to DNA-damaging agents (6–9). Mice deficient for 53BP1 show increased radiation sensitivity and an elevated tumor risk (10, 11). Moreover, class switch recombination of immunoglobulin heavy chains is severely impaired in the absence of 53BP1, suggesting a defect in the non-homologous end-joining of DNA DSBs (12, 13).
The repair defect in 53BP1-deficient cells can be quantified by assessing PIKK-dependent phosphorylation of the histone H2A variant, H2AX, a component of the nucleosome core structure (14, 15). In response to DNA damage, H2AX is reversibly phosphorylated at megabase regions flanking the sites of DNA DSBs (16). Once repair has been completed, the histone mark is removed from the chromatin with the help of protein phosphatase 2A (17, 18).
Previous studies showed that relocalization of 53BP1 to the sites of DNA DSBs depends on a region upstream of the BRCT domains (10, 19, 20). This region includes a tandem Tudor domain, which was recently proposed to be required for the initial recruitment of 53BP1 to chromatin by its direct binding to a methylated histone mark (21, 22), as well as an adjacent region required for 53BP1 accumulation (20). However, it is unknown to what extent the 53BP1 BRCT motifs or the multiple N-terminal (S/T)Q sites are needed for the repair function of 53BP1. To address these questions, we stably expressed various 53BP1-deletion mutants in 53BP1-defcient MEFs and analyzed the repair capacity of these cells by determining the numbers of residual P-H2AX foci following treatment with ionizing radiation. Moreover, we fine-mapped the region required for 53BP1 oligomerization and, using the same approach, assessed the role of 53BP1 oligomerization in DNA DSB repair.
EXPERIMENTAL PROCEDURES
Constructs and Transfection
The original plasmid containing HA-tagged human 53BP1 cDNA was obtained from Dr. K. Iwabuchi (pCMH6K53BP1) (23). Mutations or deletions were inserted using site-directed mutagenesis (QuikChange, Stratagene). The 15AQ plasmid, a pCMH6K53BP1 derivative, was provided by Dr. P. B. Carpenter (10). Stable cell lines were established by co-transfecting 53BP1-deficient, immortalized MEF cells (11) with the purified 53BP1 plasmid and a vector containing the puromycin resistance gene under the transcriptional control of the PGK promoter. FuGENE 6 (Invitrogen) was used as the transfection reagent.
Repair and Checkpoint Assays
For the DNA DSB repair assay, cells were plated onto coverslips and grown to confluency. Once the cells had stopped dividing, they were irradiated with 1 or 2 Gy using a 137Cs source and allowed to recover for up to 27 h prior to immunostaining with anti-phopsho H2AX antibodies (24). The cells were viewed on a Nikon Eclipse 800 microscope, and the number of P-H2AX foci was assessed in 100–200 cells/sample. To analyze the integrity of the intra-S-phase checkpoint, logarithmically growing cells were plated into 96-well plates and grown at 37 °C for 2 days in medium containing 10 nCi of [14C]thymidine (ICN Radiochemicals) per ml. Radioactive medium was replaced overnight with fresh medium to chase 14C-labeled precursors into DNA prior exposure of cells to 0, 5, 10, and 20 Gy of IR. 30 min later, [3H]thymidine was added to the medium (20 μCi/ml), and the cells were incubated for another 30 min at 37 °C. After removal of the radioactive medium and two washes with cold phosphate-buffered saline, the cells were trypsinized, harvested on filter paper, and analyzed on a scintillation counter. For the low dose G2/M checkpoint assay, cells were either untreated or irradiated with 0.5 Gy and then incubated for 1 h at 37 °C prior to fixation and staining with anti-phosphohistone H3 antibodies (Upstate Biotechnology) and propidium iodide. The number of mitotic cells was assessed by immunofluorescence microscopy.
Immunoprecipitation and Immunoblotting
Cells were lysed in lysis buffer (20 mM Tris-HCl, pH 8, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) supplemented with 1 mM phenyl-methanesulfonyl fluoride, 2 μg/ml aprotinin, 50 mM NaF, 40 mM β-glycerolphosphate, and 2 mM Na3VO4, and extracts were incubated with the indicated primary antibodies and protein G-Sepharose beads (Amersham Biosciences) or S-agarose beads (EMD Biosciences) for 1 h at 4 °C. The beads were then gently washed three times with lysis buffer, and the precipitated complexes were resuspended in 2× Laemmli buffer. After boiling, the samples were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to polyvinylidene difluoride membrane, and immunoblotted with various antibodies. Monoclonal antibodies against the FLAG (M2) and HA epitope (HA.11) were purchased from Sigma and Covance, respectively. Anti-p53 antibodies DO1 and 421 were purchased from Oncogene, whereas the polyclonal anti-53BP1 antibody was raised against the N terminus of 53BP1 (7).
RESULTS
Localization of 53BP1 to sites of DNA DSBs (the formation of so-called ionizing radiation-induced foci (IRIF)) requires a region upstream of the two BRCT domains (10, 19, 20). The current hypothesis is that a tandem Tudor fold within this region (here referred to as the IRIF region) is required for the initial targeting of 53BP1 to sites of DSBs by recognizing and binding methylated histones exposed at the areas surrounding the DNA lesions (21, 22). Subsequent interactions of the IRIF region with other proteins or protein modifications are then needed to retain and/or stabilize 53BP1 at the break sites (20, 25).
It is speculated that the proper localization of 53BP1 would be required for its function in DNA damage response, although direct evidence is lacking. To investigate the requirement of various 53BP1 regions in the non-homologous repair of DNA DSBs, we generated 53BP1-null MEF lines expressing HA-tagged full-length or mutated h53BP1. Using residual P-H2AX foci as a readout, we assessed the efficiency of DNA DSB repair 27 h after exposure of superconfluent G1-phase cells to 2 Gy IR. As expected, 53BP1 IRIF mutants (Δ1052–1302 and ΔTudor 1477–1632), which are unable to relocate in response to DNA damage, showed the same repair defect as 53BP1-deficient cells (Fig. 1). Moreover, reconstitution with h53BP1 full-length protein (FL) restored the DSB repair to levels that were similar to the ones observed in the 53BP1 wild-type line (Fig. 1, wt), suggesting that human 53BP1 can fully replace its murine counterpart in DNA DSB repair.
FIGURE 1. Deletions within the region required for 53BP1 foci formation abolish the repair function of 53BP1.
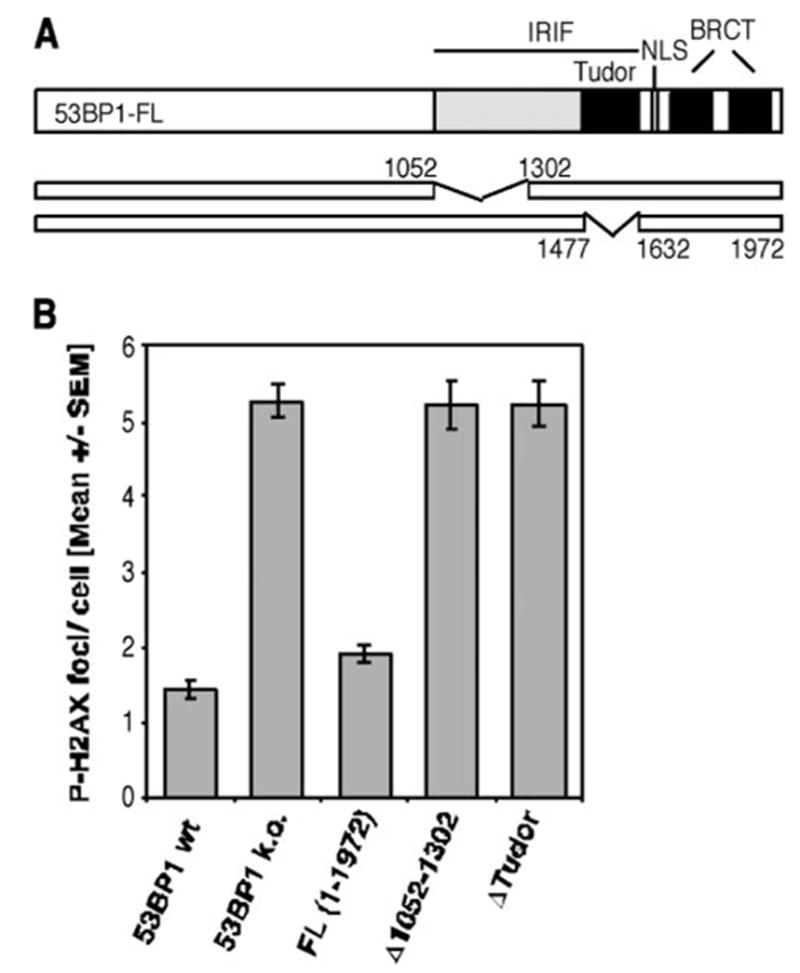
A, schematic diagram of the 53BP1 cDNA constructs Δ1052–1302 and Δ1477–1632 (ΔTudor). Both plasmids have a deletion within the region required for IRIF formation, whereas the nuclear localization sequence (NLS) and tandem BRCT domains remain intact. B, average number of residual P-H2AX foci 27 h after 2 Gy in superconfluent wild-type (wt) or 53BP1-null MEFs (k.o., knockout) and cells transiently reconstituted with wild-type or indicated mutants of human 53BP1. The error bars represent the S.E. of triplicate samples.
The N terminus of 53BP1 contains no functionally defined protein region but has several (S/T)Q sites, which represent potential target sites for ATM or other PIKKs. In fact, we and others had previously shown that 53BP1 becomes hyper-phoshorylated in an ATM-dependent manner in response to DNA damage, and we mapped Ser-6, Ser-25, Ser-29, and Ser-784 as inducible in vivo ATM phosphorylation sites (7, 9, 20). To test whether phosphorylation of these four sites is required for the repair function of 53BP1, we transiently transfected 53BP1-deficient MEFs with a quadruple serine to alanine mutant (S6A,S25A,S29A,S784A). Interestingly, the expression of this particular 53BP1 phospho-mutant almost restored the repair efficiency to wild-type levels, whereas cells expressing the Δ1–1052 mutant showed the repair defect similar to 53BP1-null cells (53BP1−/−, 5.20 ± 0.26; FL, 1.83 ± 0.13; S6A,S25A,S29A,S784A, 2.10 ± 0.18; Δ1–1052, 5.24 ± 0.31, mean ± S.E.).
There are at least two possible explanations for this result. One is that there are additional redundant phosphorylation sites at the N terminus of 53BP1. Phosphorylation of those sites is sufficient for mediating DNA DSB repair. Alternatively, 53BP1 phosphorylation could be independent from the repair function of 53BP1 and might contribute to checkpoint or other function of 53BP1. To distinguish these two possibilities, we analyzed a series of MEF lines stably expressing increasing N-terminal deletions (Δ1–165, Δ1–334, Δ1–500, Δ1–659, Δ1–1052) or a phospho-mutant (10) with all 15 conserved (S/T)Q sites being changed to AQ sites (15AQ) (Fig. 2A). Using the same residual P-H2AX foci assay, we observed a gradual increase in repair deficiency with increasing length of the deletion (Fig. 2B). Consistent with this finding, transient expression of two internal deletion mutants, Δ170–499 and Δ334–659, resulted only in a partial rescue of the repair deficiency (data not shown). Moreover, expression of the 15AQ mutant could not salvage the repair defect in 53BP1-null MEFs, although the average number of residual P-H2AX foci was slightly lower than that observed in the parental line or in cells expressing the Δ1–1052 deletion mutant (Fig. 2B). Based on these observations, we believe that phosphorylation of 53BP1 at multiple sites within the 53BP1 N terminus is required for efficient DNA DSB repair.
FIGURE 2. Deletion of the 53BP1 N terminus or its conserved (S/T)Q sites abolishes the repair function of 53BP1.
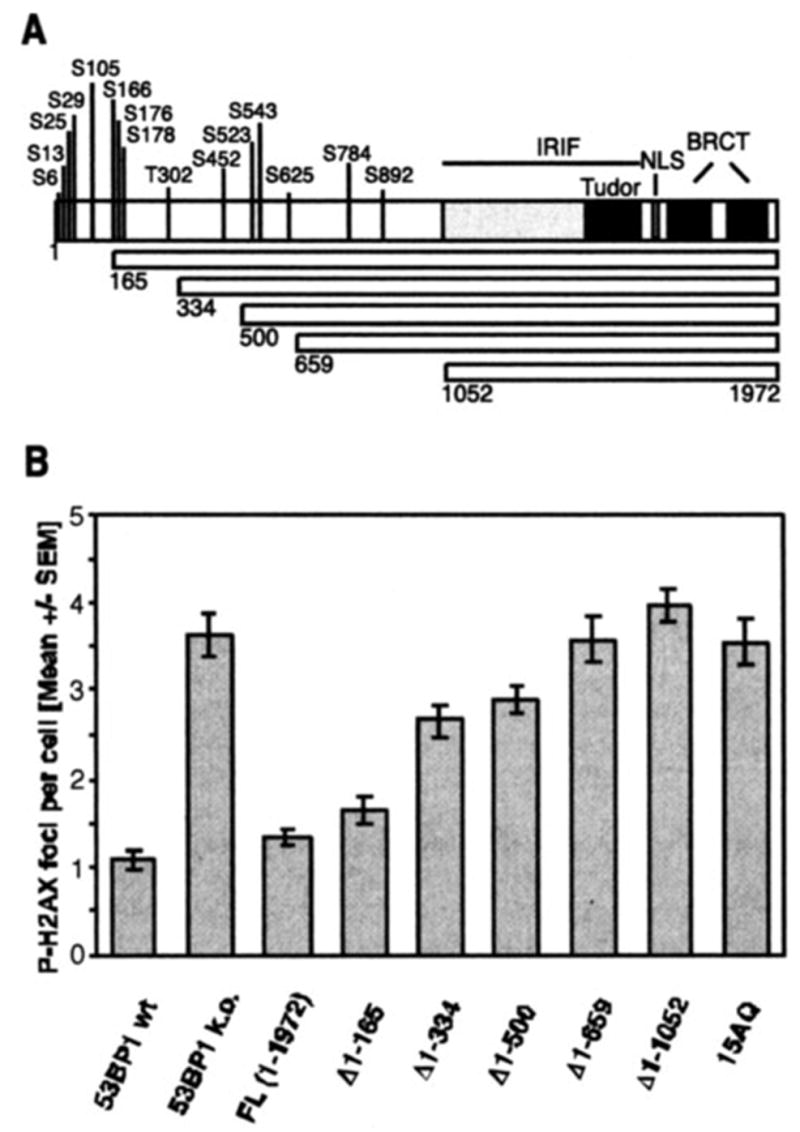
A, schematic representation of the 53BP1 deletion mutants that gradually remove the 15 conserved N-terminal (S/T)Q sites. NLS, nuclear localization sequence. B, average number of residual P-H2AX foci 27 h after 2 Gy in superconfluent wild-type (wt) MEFs, 53BP1-null (k.o.) MEFs, or cells stably reconstituted with wild-type (FL) or various mutants of human 53BP1. The 15AQ mutant has all 15 conserved (S/T)Q sites replaced with AQ sites. The error bars represent the S. E. of triplicate samples.
To test whether 53BP1 phosphorylation would be involved in checkpoint control, we analyzed the ability of cells to repress DNA synthesis in the presence of DNA damage (radio-resistant DNA synthesis (RDS) assay). However, unlike ATM-deficient control cells, neither 53BP1-null nor 53BP1-reconstituted MEFs showed a significant RDS phenotype in response to 5, 10, or 20 Gy IR (Fig. S1 in supplemental materials and data not shown). These findings corroborated our former conclusion that 53BP1 does not play a major role in cell cycle checkpoint control (11).
Next, we investigated to what extent the C-terminal BRCT repeats contribute to the function of 53BP1 in DNA DSB repair. 53BP1 had been identified and named based on the in vitro interaction of its C terminus with the central DNA-binding domain of p53 (3). Although this original observation was made in yeast cells overexpressing these two proteins, subsequent structural analyses confirmed that the first BRCT motif together with the inter-repeat linker bind to the DNA-binding surface of p53 (26). We also observed an interaction of endogenous 53BP1 and p53 in human cells (Fig. 3B). However, the functional significance of this tandem BRCT domain has not been addressed.
FIGURE 3. Deletion of the tandem BRCT motif does not affect the repair function of 53BP1.
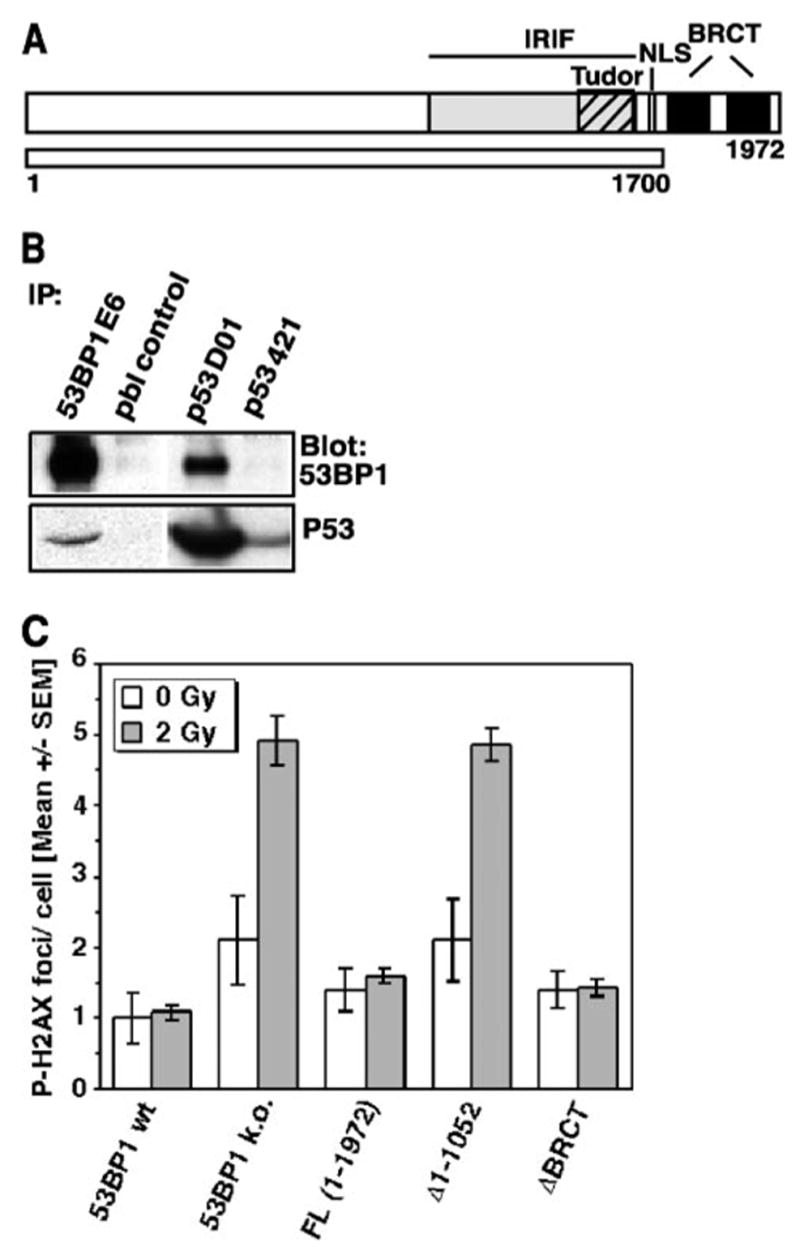
A, schematic diagram of the BRCT deletion construct Δ1700 –1972. NLS, nuclear localization sequence. B, co-immunoprecipitation (IP) of 53BP1 and p53 in A549 cells using 53BP1-specific antiserum E6 or p53-specific antibody DO1. Prebleed serum (pbl) or anti-p53 antibody 421, which recognizes a mutated form of p53, was used as control. C, residual repair foci in superconfluent 53BP1-null MEFs or cells stably reconstituted with the ΔBRCT (Δ1700 –1972) 53BP1 mutant. P-H2AX foci were assessed 27 h after exposure to 0 and 2 Gy of IR. Cells stably expressing full-length or N-terminal deleted 53BP1 (Δ1–1052) are shown as controls. wt, wild type; k.o., knockout. The error bars represent the S. E. of triplicate samples.
To examine the role of the 53BP1 BRCT region in DNA DSB repair, cells stably expressing a truncated form of 53BP1 (ΔBRCT 1700–1972, Fig. 3A) were analyzed for residual P-H2AX foci 27 h after exposure to 2 Gy. Interestingly, expression of the BRCT truncation mutant completely rescued the repair defect in 53BP1-null cells (Fig. 3C). The same observation was made in cells transiently transfected with the ΔBRCT 1700–1972 construct (data not shown). Moreover, the average number of P-H2AX foci in unirradiated ΔBRCT cells was similar to that found in cells expressing full-length 53BP1. In contrast, 53BP1-deficient cells or cells that expressed the N-terminal deletion mutant (Δ1–1052) showed higher baseline levels indicative of a higher number of unrepaired endogenous lesions (Fig. 3C). The rescue effect of the ΔBRCT mutant was not restricted to a late stage of repair but could be seen as early as 1 h following irradiation with 1 Gy (see Fig. S2A in supplemental materials). In addition, a low dose G2/M checkpoint assay showed that the 53BP1 BRCT domains are not involved cell cycle checkpoint control (Fig. S2B in supplemental materials). Together, these findings indicate that the 53BP1 BRCT domains are not required for efficient DSB repair or checkpoint control.
53BP1 has recently been shown to homo-oligomerize in a DNA damage-independent manner. The region required and sufficient for oligomerization has been mapped to residues 1052–1475, an area upstream of the 53BP1 tandem Tudor folds (27). To gain further insight into the function of 53BP1 oligomerization, we attempted to narrow down the region using nuclear magnetic resonance (NMR) spectroscopy. The data gained from this analysis suggested that residues 1231–1270 mediate the oligomerization of 53BP1 (data not shown). To confirm these in vitro findings, we generated an HA-tagged 53BP1 ΔDimer (Δ1231–1270) construct and performed a double-transfection assay. 293T cells were co-transfected with S-FLAG-tagged full-length 53BP1 and the HA-ΔDimer mutant or an HA-tagged control plasmid. 48 h later, aliquots of the cells were collected. Cell lysates were immunoprecipitated with either anti-HA or anti-S antibodies and analyzed by Western blotting using anti-FLAG and anti-HA antibodies, respectively. S-FLAG-53BP1 FL could be readily detected in the HA-FL and HA-Δ1–1052 precipitates but not in the HA-ΔDimer (Δ1231–1270) and HA-Δ1052–1302 precipitates. Correspondingly, HA-FL and HA-Δ1–1052, but not HA-ΔDimer or HA-Δ1052–1302, co-immunoprecipitated with S-FLAG-FL 53BP1 (Fig. 4B). These data confirm that the region comprising residues 1231–1270 is required for 53BP1 oligomerization.
FIGURE 4. 53BP1 oligomerization is required for the efficient repair of DNA DSBs.
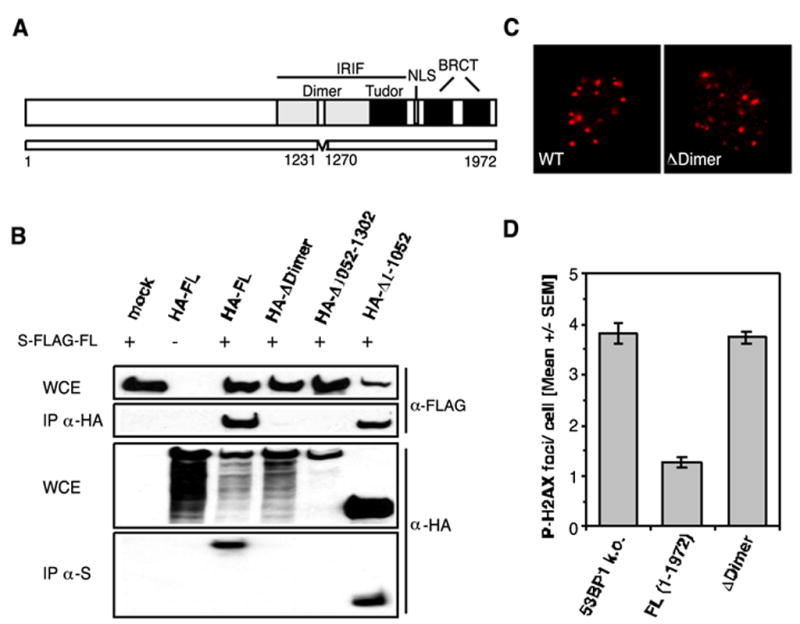
A, schematic representation of the 53BP1 ΔDimer (Δ1231–1270) mutant. NLS, nuclear localization sequence. B, HA-tagged ΔDimer (Δ1231–1270) and Δ1052–1302 mutants fail to co-immunoprecipitate (IP) with S-FLAG-tagged full-length 53BP1 from co-transfected 293T cells. WCE, whole cell extract; mock, mock-transfected. C, IR-induced 53BP1 foci formation (1 Gy/1 h) in wild-type (wt) MEFs and 53BP1-null (k.o.) cells stably expressing the ΔDimer (DDimer) mutant. D, average number of residual P-H2AX foci 27 h after 2 Gy in 53BP1-null MEFs and cells stably reconstituted with either full-length 53BP1 or the ΔDimer (Δ1231–1270) mutant. The error bars represent the S.E. of triplicate samples.
Next, we transfected 53BP1-null MEFs with the ΔDimer (Δ1231–1270) construct and asked whether deletion of the oligomerization region would affect 53BP1 accumulation at the sites of DNA DSBs. Notably, cells expressing the deletion mutant still formed IRIF, although the deleted region resides within the region required for 53BP1 foci formation (Fig. 4, A and C). However, expression of ΔDimer (Δ1231–1270) could not rescue the repair defect in stably transfected 53BP1-deficient MEFs (Fig. 4D). Collectively, these findings suggest that oligomerization of 53BP1 is critical for its function in DNA DSB repair.
DISCUSSION
53BP1 plays a significant role in the repair of DNA DSBs, and here we provide evidence that phosphorylation and homo-oligomerization of 53BP1 are crucial to exert this function. In contrast, the C-terminal tandem BRCT motifs of 53BP1 appear not to be required for DSB repair.
53BP1 has been shown to undergo a dynamic interaction with chromatin following DNA damage. Upon exposure of cells to ionizing radiation, 53BP1 becomes transiently immobilized at the chromosomal regions flanking the DNA DSBs (28). Binding and retention of 53BP1 to chromatin requires an intact Tudor domain, a recently characterized methyl-binding motif (21), as well as a less distinguished upstream region (20). Deletion of the Tudor domain or the preceding region prevent the repair function of 53BP1, indicating that accumulation of 53BP1 at break sites is an absolute requirement for efficient DNA DSB repair. Similarly, mutation of multiple phosphorylation sites at the 53BP1 N terminus impairs DSB repair in an incremental fashion. Although it remains to be determined how many phosphorylation sites have to conspire for full 53BP1 function, it appears that there is no sharp threshold for these phosphorylation events. DNA damage-induced phosphorylation of multiple PIKK sites has also been described in budding yeast Rad9 (29), which is thought to be the homologue of mammalian 53BP1 based on sequence similarities in its tandem BRCT and Tudor motifs. In budding yeast, phosphorylation of Rad9 is required for the survival of genotoxic stress by regulating the activation of the checkpoint kinase Rad53, the functional orthologue of the mammalian signal transducer Chk2 (29). Phosphorylated Rad9 recruits Rad53 to DNA lesions, thus enabling its phosphorylation by the PIKK Mec1. Phosphorylation of Rad53 triggers its activation, autophosphorylation, and subsequent release from Rad9 (30, 31). Similarly, 53BP1 has been shown to interact with Chk2 and promote its phosphorylation (11, 32, 33). However, unlike its yeast counterpart, Chk2 can still be activated in the absence of 53BP1 (11, 34), although the activation or phosphorylation of Chk2 is reduced in 53BP1-deficient cells. These data suggest that 53BP1 is not essential for Chk2 phosphorylation and/or activation following DNA damage, raising the possibility that a more complex and redundant signal adaptor system may be evolved in higher organisms. Indeed, although phosphorylation of Rad9 as well as Rad9-dependent phosphorylation of Rad53 require the C-terminal tandem BRCT domains in budding yeast (35), deletion of the BRCT domains in mammalian 53BP1 did not affect 53BP1 phosphorylation (data not shown) or 53BP1 function in DNA DSB repair. Moreover, homo-oligomerization appears to be the critical function of the Rad9 (budding yeast) or Crb2 (fission yeast) BRCT domains (35, 36), whereas oligomerization of 53BP1 depends on a small region outside of its BRCT domains.
The fact that the BRCT domains of 53BP1 are not required for effective DNA DSB repair raises the question of the specific function of this tandem motif in 53BP1. An obvious possibility would be a role in p53 regulation. The evolution of this tumor suppressor module in higher eukaryotes could have conferred a new role to the C terminus of 53BP1, whereas a novel upstream region assured the homo-oligomerization of the protein. It seems clear that the 53BP1 BRCT domain is able to interact with the DNA-binding domain of p53 and initial studies implied that 53BP1 may stimulate p53-mediated transcriptional activation (23). However, p53 cannot bind simultaneously to 53BP1 and DNA (26, 37). It is therefore unlikely that 53BP1 could directly activate p53-mediated transcription. In addition, IR-induced p21 activation was found to be normal in 53BP1-deficient splenocytes (15). 53BP1 might regulate p53 indirectly by affecting the phosphorylation of p53 following DNA damage. In fact, small interfering RNA-mediated down-regulation of 53BP1 led to impaired p53 accumulation in a human cancer line (32). However, no defect in p53 stabilization was found in primary 53BP1-deficient mouse thymocytes, nor did these cells fail to undergo p53-mediated G1 arrest or apoptosis (11, 15). Thus, it remains a subject of debate as to whether the regulation of p53 is a biological function of 53BP1. Alternatively, the 53BP1 BRCT domains could interact with an as yet unidentified protein, perhaps a phosphoprotein, since tandem BRCT domains bind preferentially to phosphorylated peptides (38, 39). Such a hypothetical complex could function in a subtype of DNA repair that is not measured by our DSB repair assay.
Unlike budding yeast Rad9 or fission yeast Crb2, which are key players in DNA damage checkpoint control (40, 41), 53BP1 appears to have a limited role in DNA damage checkpoints. Although small interfering RNA-mediated down-regulation of 53BP1 in human cancer lines led to defects in the G2/M and intra-S-phase checkpoints (32, 33), minor or no cell cycle checkpoint defects were detected in 53BP1-deficient mouse and chicken cells (11, 34). These differences between 53BP1 and its proposed yeast homologues could have evolved with the increasing number of BRCT-containing proteins in metazoa. For example, checkpoint proteins MDC1/ NFBD1 and MCPH1/BRIT1 are BRCT-containing proteins that do not have any apparent homologues in yeast. These proteins might be evolved to carry out checkpoint functions, whereas 53BP1 gains more importance in DNA DSB repair.
Regardless of their differences, it is still tempting to speculate that the mechanism of action remained similar among 53BP1, budding yeast Rad9, and fission yeast Crb2. All three proteins appear to be oligomeric adaptors that enable/facilitate the activation of effector molecules in response to DNA damage. Further studies will be necessary to elucidate the new role of the 53BP1 BRCT motifs in the complex mammalian damage response network.
Supplementary Material
Acknowledgments
We thank members of the Chen laboratory for helpful discussions and technical support.
Footnotes
The on-line version of this article (available at http://www.jbc.org) contains two supplemental figures.
The abbreviations used are: DSB, double strand breaks; BRCT, BRCA1 C-terminal; PIKK, phosphoinositide 3-kinase-related protein kinase; ATM, ataxia telangiectasia mutated; MEF, mouse embryonic fibroblasts; Gy, grays; HA, hemagglutinin; IR, ionizing radiation; IRIF, IR-induced foci; FL, full length protein.
Supplemental Material can be found at: http://www.jbc.org/cgi/content/full/M607577200/DC1
References
- 1.Difilippantonio MJ, Petersen S, Chen HT, Johnson R, Jasin M, Kanaar R, Ried T, Nussenzweig A. J Exp Med. 2002;196:469–480. doi: 10.1084/jem.20020851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW, Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Cell. 2002;109:811–821. doi: 10.1016/s0092-8674(02)00770-5. [DOI] [PubMed] [Google Scholar]
- 3.Iwabuchi K, Bartel PL, Li B, Marraccino R, Fields S. Proc Natl Acad Sci U S A. 1994;91:6098–6102. doi: 10.1073/pnas.91.13.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bork P, Hofmann K, Bucher P, Neuwald AF, Altschul SF, Koonin EV. FASEB J. 1997;11:68–76. [PubMed] [Google Scholar]
- 5.Callebaut I, Mornon JP. FEBS Lett. 1997;400:25–30. doi: 10.1016/s0014-5793(96)01312-9. [DOI] [PubMed] [Google Scholar]
- 6.Anderson L, Henderson C, Adachi Y. Mol Cell Biol. 2001;21:1719–1729. doi: 10.1128/MCB.21.5.1719-1729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rappold I, Iwabuchi K, Date T, Chen J. J Cell Biol. 2001;153:613–620. doi: 10.1083/jcb.153.3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schultz LB, Chehab NH, Malikzay A, Halazonetis TD. J Cell Biol. 2000;151:1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xia Z, Morales JC, Dunphy WG, Carpenter PB. J Biol Chem. 2001;276:2708–2718. doi: 10.1074/jbc.M007665200. [DOI] [PubMed] [Google Scholar]
- 10.Morales JC, Xia Z, Lu T, Aldrich MB, Wang B, Rosales C, Kellems RE, Hittelman WN, Elledge SJ, Carpenter PB. J Biol Chem. 2003;10:10. doi: 10.1074/jbc.M212484200. [DOI] [PubMed] [Google Scholar]
- 11.Ward IM, Minn K, Van Deursen J, Chen J. Mol Cell Biol. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ward IM, Reina-San-Martin B, Olaru A, Minn K, Tamada K, Lau JS, Cascalho M, Chen L, Nussenzweig A, Livak F, Nussenzweig MC, Chen J. J Cell Biol. 2004;165:459–464. doi: 10.1083/jcb.200403021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manis JP, Morales JC, Xia Z, Kutok JL, Alt FW, Carpenter PB. Nat Immunol. 2004;5:481–487. doi: 10.1038/ni1067. [DOI] [PubMed] [Google Scholar]
- 14.Kuhne M, Riballo E, Rief N, Rothkamm K, Jeggo PA, Lobrich M. Cancer Res. 2004;64:500–508. doi: 10.1158/0008-5472.can-03-2384. [DOI] [PubMed] [Google Scholar]
- 15.Ward IM, Difilippantonio S, Minn K, Mueller MD, Molina JR, Yu X, Frisk CS, Ried T, Nussenzweig A, Chen J. Mol Cell Biol. 2005;25:10079–10086. doi: 10.1128/MCB.25.22.10079-10086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogakou EP, Boon C, Redon C, Bonner WM. J Cell Biol. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J, Kim JA, Downey M, Fillingham J, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, Shen X, Haber JE, Durocher D, Greenblatt JF, Krogan NJ. Mol Cell. 2005;20:801–809. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Keogh MC, Kim JA, Downey M, Fillingham J, Chowdhury D, Harrison JC, Onishi M, Datta N, Galicia S, Emili A, Lieberman J, Shen X, Buratowski S, Haber JE, Durocher D, Greenblatt JF, Krogan NJ. Nature. 2006;439:497–501. doi: 10.1038/nature04384. [DOI] [PubMed] [Google Scholar]
- 19.Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan D, Cao Y, Hamada T, Imamura K, Jeggo PA, Date T, Doherty AJ. J Biol Chem. 2003;278:36487–36495. doi: 10.1074/jbc.M304066200. [DOI] [PubMed] [Google Scholar]
- 20.Ward IM, Minn K, Jorda KG, Chen J. J Biol Chem. 2003;278:19579–19582. doi: 10.1074/jbc.C300117200. [DOI] [PubMed] [Google Scholar]
- 21.Huyen Y, Zgheib O, Ditullio RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- 22.Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 23.Iwabuchi K, Li B, Massa HF, Trask BJ, Date T, Fields S. J Biol Chem. 1998;273:26061–26068. doi: 10.1074/jbc.273.40.26061. [DOI] [PubMed] [Google Scholar]
- 24.Riballo E, Kuhne M, Rief N, Doherty A, Smith GC, Recio MJ, Reis C, Dahm K, Fricke A, Krempler A, Parker AR, Jackson SP, Gennery A, Jeggo PA, Lobrich M. Mol Cell. 2004;16:715–724. doi: 10.1016/j.molcel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- 25.Celeste A, Fernandez-Capetillo O, Kruhlak MJ, Pilch DR, Staudt DW, Lee A, Bonner RF, Bonner WM, Nussenzweig A. Nat Cell Biol. 2003;5:675–679. doi: 10.1038/ncb1004. [DOI] [PubMed] [Google Scholar]
- 26.Joo WS, Jeffrey PD, Cantor SB, Finnin MS, Livingston DM, Pavletich NP. Genes Dev. 2002;16:583–593. doi: 10.1101/gad.959202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams MM, Wang B, Xia Z, Morales JC, Lu X, Donehower LA, Bochar DA, Elledge SJ, Carpenter PB. Cell Cycle. 2005;4:1854–1861. doi: 10.4161/cc.4.12.2282. [DOI] [PubMed] [Google Scholar]
- 28.Bekker-Jensen S, Lukas C, Melander F, Bartek J, Lukas J. J Cell Biol. 2005;170:201–211. doi: 10.1083/jcb.200503043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwartz MF, Duong JK, Sun Z, Morrow JS, Pradhan D, Stern DF. Mol Cell. 2002;9:1055–1065. doi: 10.1016/s1097-2765(02)00532-4. [DOI] [PubMed] [Google Scholar]
- 30.Gilbert CS, Green CM, Lowndes NF. Mol Cell. 2001;8:129–136. doi: 10.1016/s1097-2765(01)00267-2. [DOI] [PubMed] [Google Scholar]
- 31.Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. Curr Biol. 2005;15:1364–1375. doi: 10.1016/j.cub.2005.06.063. [DOI] [PubMed] [Google Scholar]
- 32.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. Science. 2002;298:1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 33.DiTullio RA, Mochan TA, Venere M, Bartkova J, Sehested M, Bartek J, Halazonetis TD. Nat Cell Biol. 2002;4:998–1002. doi: 10.1038/ncb892. [DOI] [PubMed] [Google Scholar]
- 34.Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, Carpenter PB, Bonner WM, Chen J, Nussenzweig A. Nat Cell Biol. 2002;4:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 35.Soulier J, Lowndes NF. Curr Biol. 1999;9:551–554. doi: 10.1016/s0960-9822(99)80242-5. [DOI] [PubMed] [Google Scholar]
- 36.Du LL, Moser BA, Russell P. J Biol Chem. 2004;279:38409–38414. doi: 10.1074/jbc.M403326200. [DOI] [PubMed] [Google Scholar]
- 37.Derbyshire DJ, Basu BP, Serpell LC, Joo WS, Date T, Iwabuchi K, Doherty AJ. EMBO J. 2002;21:3863–3872. doi: 10.1093/emboj/cdf383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Manke IA, Lowery DM, Nguyen A, Yaffe MB, Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Science. 2003;302:636–639. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 39.Yu X, Chini CC, He M, Mer G, Chen J, Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Science. 2003;302:639–642. [Google Scholar]
- 40.Saka Y, Esashi F, Matsusaka T, Mochida S, Yanagida M. Genes Dev. 1997;11:3387–3400. doi: 10.1101/gad.11.24.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinert TA, Hartwell LH, Lou Z, Chini CC, Minter-Dykhouse K, Chen J, Stewart GS, Wang B, Bignell CR, Taylor AM, Elledge SJ, Wu X, Goldberg M, Stucki M, Falck J, D’Amours D, Rahman D, Pappin D, Bartek J, Jackson SP. Science. 1988;241:317–322. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.