

Abstract
Excessive glutamate receptor stimulation can produce rapid disruption of dendritic morphology, including dendritic beading. We recently showed that transient NMDA exposure resulted in irreversible loss of synaptic function and loss of MAP2 from apical dendrites. The present study examined the initiation and progression of dendritic injury in slice following this excitotoxic stimulus. NMDA exposure (30μM, 10 min) produced irregularly shaped dendritic swellings, evident first in distal apical dendrite branches, and later (20–90min) involving most proximal dendrites. Over the same timecourse, immunoreactivity for the microtubule associated protein MAP2 was progressively lost from apical dendrites, and increased in CA1 somata. This damage and MAP2 loss was Ca2+-dependent, and was not reversible within the timecourse of these experiments (90 min post-NMDA washout). Formation of regularly-spaced, spherical dendritic varicosities (dendritic beading) was rarely observed, except when NMDA was applied in Ca2+-free ACSF. Under these conditions, beading appeared predominant in interneurons, as assessed from experiments with GAD67-GFP (Δneo) mice. Ca2+-removal was associated with significantly better preservation of dendritic structure (MAP2) following NMDA exposure, and other ionic fluxes (sensitive to Gd3+ and spermine) may contribute to residual damage occurring in Ca2+-free conditions. These results suggest that irregularly shaped dendritic swelling is a Ca2+-dependent degenerative event that may be quite different from Ca2+-independent dendritic beading, and can be a predominant type of injury in CA1 pyramidal neurons in slice.
Keywords: Beading, excitotoxicity, hippocampus, CA1, dendrite, interneuron
INTRODUCTION
Excitotoxic injury of neuronal dendrites (“dendrotoxicity” (Olney et al., 1979)) may be an important part of the initiation of neuronal injury following a range of neurological insults. The formation of focal dendritic swellings, appearing as a regularly-alternating pattern of rounded varicosities (dendritic beading) has been observed following various types of injury, including in vivo ischemia (Hori and Carpenter, 1994; Matesic and Lin, 1994) and excitatory amino acid agonist exposure in vivo (Olney et al., 1979; Sloviter and Dempster, 1985) and in vitro (Park et al., 1996; Hasbani et al., 1998; Al-Noori and Swann, 2000; Ikegaya et al., 2001; Oliva et al., 2002). While many detailed studies of mechanisms of dendritic beading have used neuronal cultures or slice culture preparations, less is known about the progression of dendritic damage in mature slice preparations. Regular, spherical dendritic beading occurs in mature neurons exposed to cold, and is rapidly reversible (within ~5 min) with this non-lethal stimulus (Kirov et al., 2004). In contrast, other studies report irreversible dendritic beading in mature CA1 pyramidal neurons following oxygen-glucose deprivation (OGD) or glutamate agonist exposure, and it was concluded that this beading underlies irrecoverable changes in slice light transmittance (Andrew et al., 1999; Jarvis et al., 1999; Obeidat et al., 2000). However, in those studies reporting irreversible beading, at least some of the individual varicosities shown appeared irregularly shaped, with more heterogeneous distribution of sizes and dendrite constrictions than typically has been described for dendritic beading in neuronal cultures.
We recently examined responses to NMDA in acute hippocampal slices, to investigate mechanisms underlying loss of MAP2 from dendrites (Hoskison and Shuttleworth, 2006). In that study, most preparations were examined at late timepoints after a brief NMDA exposure, and it was difficult to evaluate dendritic structure due to extensive MAP2 loss. However, in one experiment, immediately following NMDA exposure, significant irregular swelling was observed, with little evidence of regularly-spaced dendritic beads. The aim of the present work was to examine the characteristics of dendritic structural changes in CA1 dendrites following this excitotoxic stimulation, with particular emphasis on the progression of effects, and the dependence on extracellular Ca2+ concentration. Using a stimulus (30μM NMDA, 10 min) matched to that producing reversible dendritic beading in isolated cultures (Park et al., 1996; Faddis et al., 1997; Hasbani et al., 1998) and hippocampal slice cultures (Ikegaya et al., 2001), we have tested the hypothesis that a predominant type of dendritic injury in mature CA1 neurons is Ca2+-dependent and persistent, with structural characteristics quite dissimilar from organized dendritic beading. The enduring nature of pervasive irregular swellings that we describe here suggests that this type of dendritic injury may contribute to persistent neuronal damage and dysfunction associated with excitotoxic injury in these slices. In contrast, challenges under Ca2+-free conditions produced some beading, but this distinct injury was restricted to a subpopulation of dendrites in CA1 that appeared to include processes of interneurons.
EXPERIMENTAL PROCEDURES
Hippocampal slice preparation and stimulation
Experiments were carried out in accordance with the National Institute of Health guidelines for the humane treatment of laboratory animals, and the protocol for these procedures was reviewed annually by the Institutional Animal Care and Use Committee at the University of New Mexico School of Medicine. All efforts were made to minimize animal suffering and the number of animals used. Acute hippocampal slices (250μm) were prepared from adult mice (4–6 weeks of age) and handled in a similar manner as used for electrophysiological studies, as described previously (Hoskison and Shuttleworth, 2006). Briefly, mice were deeply anesthetized with a mixture of ketamine and xylazine (85mg/ml and 15mg/ml, respectively; 200μl s.c.) and decapitated. Brains were removed and placed in ice-cold cutting solution and coronal sections were cut using a Vibratome (Technical Products International, St Louis MO). Slices from each animal were divided between control and treatment groups, and initially transferred into room-temperature ACSF. After warming to 37°C and holding for 1 hour, ACSF was changed again, and slices were maintained at room temperature for 1.5–3 hours before beginning experiments. FVB/N mice (Harlan) were used for most experiments. Because this strain is demonstrated to show vulnerability to hippocampal neuron cell death following in vivo kainate administration (Schauwecker and Steward, 1997; Schauwecker, 2003), slices from these animals were used for previous studies of NMDA-triggered injury (Hoskison and Shuttleworth, 2006). The generation of GAD67-GFP (Δneo) mice was described by Tamamaki and co-workers (Tamamaki et al., 2003), and these transgenic mice were called GAD67-GFP mice for simplicity. GAD67-GFP mice were used for some studies illustrated in Figures 2&5. No more than 3 slices from a single animal were used for any single stimulation condition. All post-acquisition analyses were performed by an evaluator blinded to slice treatment conditions.
Figure 2. NMDA exposure initially produced irregular dendritic swelling followed by progressive loss of dendritic MAP2.
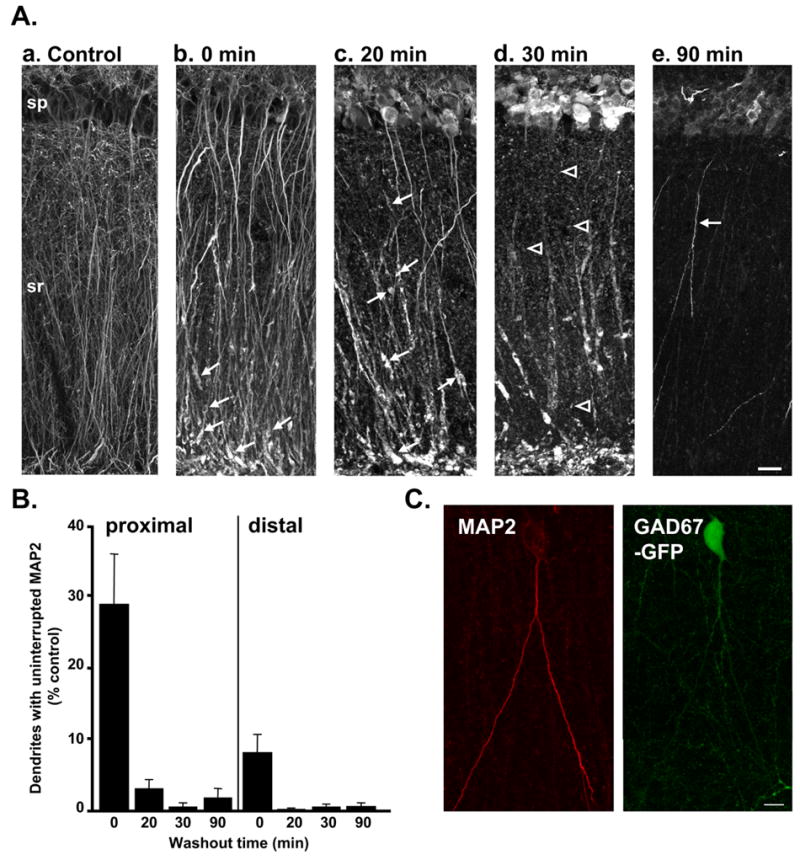
A: Representative photomontages illustrate distribution of MAP2 in a control slice (a) and in slices fixed at various timepoints (0–90 min, b–e) following transient NMDA exposure (30μM, 10 min). Dendrite swelling and loss of MAP2 labeling from fine dendritic processes was observed immediately following NMDA exposure (b). Irregular swelling and fragmentation of MAP2 labeling initially was most evident in distal apical dendrite segments (arrows in b), but by 20 min post-NMDA was also observed throughout sr (arrows in c). Most MAP2 labeling was punctate by 30 min (arrowheads in d); by 90 min post-NMDA, punctate MAP2 labeling was generally undetectable throughout dendritic regions, and brightly labeled dendrites were seen only in a very small number of neurons (arrow in e). In pyramidal cell bodies (sp), MAP2 increased over 20–30 min. Scale bar (20μm) applies to all panels. B: Mean data showing the number of continuously labeled dendrites (measured in a 20μm analysis window) in proximal (left) and distal (right) regions of sr. There was a significant loss of dendrites with continuous MAP2 labeling immediately following NMDA exposure (0 min) in both regions (p<0.01, compared with control). 20–90 min post-NMDA, there were very few dendrite segments with uninterrupted MAP2 labeling in either region (p<0.001, compared with controls n=4–6 for each condition). C: Example of a neuron with preserved dendritic MAP2 labeling (left panel) 90 min post-NMDA in a slice from a GAD67-GFP animal. The GFP fluorescence in this neuron (right panel) suggests that this is an interneuron. Scale bar: 10μm.
Figure 5. Majority of beaded dendrites appear to be interneurons.
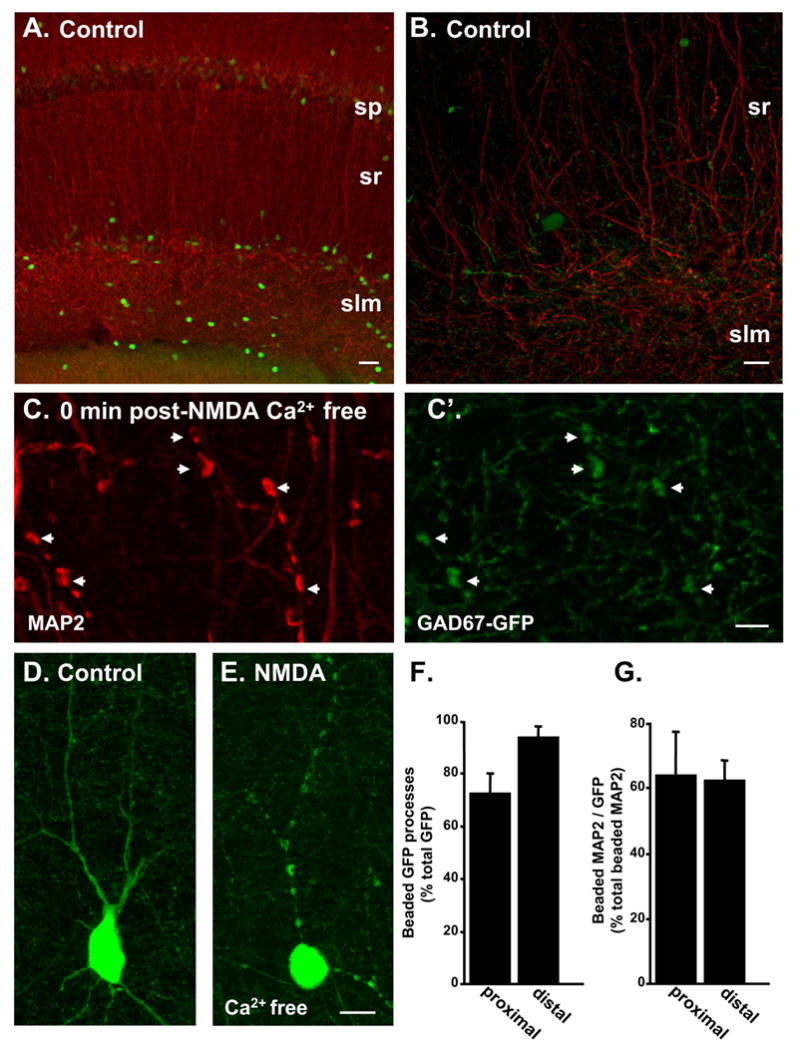
A: Control slice from GAD67-GFP animal processed for MAP2 immunoreactivity (red). This representative example shows a low density of GFP-positive neurons (green) in the hippocampal CA1 region. B: Higher power image of control slice on left. C: MAP2-positive beaded dendrites (arrowheads) observed immediately following NMDA exposure in 0 Ca2+. C’: GFP fluorescence in the same preparation illustrated in C, showing co-localization with MAP2 in dendrite beads (arrowheads) D: Representative GFP-positive neuron observed under control conditions. E: Extensive dendritic beading in a GFP-positive neuron, observed immediately following NMDA exposure in 0 Ca2+. F: Mean data illustrating that in CA1 stratum radiatum most GFP-positive processes were beaded under these stimulation conditions. G: GFP-positive dendrites also accounted for the majority of beaded dendrites detected with MAP2 under these conditions (n=6). Scale bars A: 50μm; B, D, E: 10μm; C: 5μm.
Slices were warmed to 37°C for 30 min before stimulation. Drug exposures were made by complete exchange of the bathing medium with pre-warmed (37°C) solutions. Time-matched controls were subjected to identical numbers of solution changes as stimulated slices. At appropriate timepoints, individual slices were removed and immediately placed into 4% paraformaldehyde.
Immunohistochemistry
Localization of MAP2 or GABA-like immunoreactivity was performed on vibratome slices that were not re-sectioned, as described previously (Hoskison & Shuttleworth 2006). Extended incubation periods were required for adequate penetration of reagents (see also Hamam and Kennedy, 2003). Briefly, slices were washed (3x15 min) in PBS, blocked using normal goat serum (10% final, 120 min; Jackson Laboratories, Westgrove, PA) and then exposed to primary antiserum for 48 hours at room temperature (anti-MAP2 (M-1406) 1:500, anti-GABA (A0310) 1:1000, both from Sigma-Aldrich, St. Louis, MO). Primary antiserum was exchanged after 24 hours incubation. Slices were then washed in PBS (8x30min), blocked again (10% goat serum, 120 min) and then primary antiserum was localized using Cy3 goat anti-mouse serum (1:150, Jackson Laboratories). PBS containing triton X-100 (0.15%) was used as the diluent. After extensive washing (PBS; 8x30min) preparations were mounted with ProLong Antifade Gold medium (Molecular Probes, Eugene, OR) and examined using a Zeiss 510 confocal microscope.
Image Acquisition and Analysis
Specific labeling was detectable up to ~75μm deep on both sides of slices. Minimal surface damage was observed, likely due to the time allowed for recovery from cutting procedure prior to stimulation. Confocal images were taken at an identical distance below the slice surface to allow comparison of regions receiving similar NMDA exposure across slices and also to minimize contribution of neurons damaged at the slice surface by the slicing procedure. Control and stimulated preparations from each animal were processed and analyzed in parallel and similar exposure settings were used across experimental groups, but small adjustments were sometimes necessary to utilize the full range of detectors. For all analyses in which the distribution of fluorescence intensity was compared, intensity data were normalized in order to account for variations that occur between different batches of processing.
All analyses were performed on area CA1, in the region most directly centered above the apex of the upper blade of the dentate gyrus. The analyses concentrate on disruption of apical dendrites in stratum radiatum, since deregulation of Ca2+ homeostasis following exposure to glutamate receptor agonists initiates in apical dendritic processes in these preparations (Shuttleworth and Connor, 2001). Low power images were used to assess the general distribution of MAP2 (10x, 0.3NA; 20μm stacks with 2.51μm steps). Analysis of fluorescence intensity was made from a sub-region centered in the field (90x450μm; extending from stratum oriens to stratum lacunosum-moleculare) using NIH ImageJ software (http://rsb.info.nih.gov/ij). A series of higher-power images (63X, 1.25NA, 10μm stackwith 0.5μm step interval, beginning 10μm below the slice surface) were obtained from 3 regions of interest; stratum pyramidale (sp), proximal stratum radiatum (proximal sr; analyzed 150μm from sp) and distal stratum radiatum (distal sr; analyzed adjacent to border of stratum radiatum and stratum lacunosum-moleculare, ~210–260 μm from sp). A sub-region of the projection image (20x147μm) was used for dendrite analysis using the LSM Image Browser (Zeiss, version 3,2,0,115) and all analyses were carried out on un-thresholded images. For figure presentation, images were exported from the LSM image browser as TIFF images, without post-acquisition manipulation. For photomontages, minimal adjustments of brightness were made to match background fluorescence levels between overlapping components of the figure.
Ballistic loading of lipophilic indicator (DiO)
In some experiments, visualization of discrete subpopulations of neurons was achieved by ballistic loading with the lipophilic indicator, DiO (Molecular Probes, Eugene, OR) (see Gan et al., 2000). The methods were as described by the laboratory of R.O. Wong, Washington University School of Med. (http://thalamus.wustl.edu/wonglab/doc/DiOlistic_Protocol1.doc) with minor modifications. DiO was evaporated onto tungsten particles (1.7μm; BioRad, Hercules, CA) and used to prepare cartridges for a “Gene Gun” (BioRad). Dye-coated particles were delivered to the slice through a filter (3.0 μm, VWR Brisbane, CA) with a helium pressure pulse (70–100 psi). Initial live imaging studies showed that the ballistic approach produced dendritic damage in acute slices, presumably due to the pressure pulse associated with the labeling (Wellmann et al., 1999). Reducing delivery pressures and/or increasing shooting distance did not significantly reduce the degree of damage and provide sufficient labeling. Therefore slices were fixed at various post-NMDA timepoints and then subsequently ballistically labeled with DiO (see Wu et al., 2004). Confocal microscopy was used to visualize DiO-labeled dendrite segments (Ex 488 nm, Em >505nm).
Solutions
Slice cutting solution contained (in mM) : 220 sucrose, 3 KCl, 6 MgSO4, 0.2 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose and 0.43 ketamine bubbled with 95% O2/5% CO2. ACSF contained (in mM): 126 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 1 MgSO4, 10 glucose and 2 CaCl2 continuously bubbled with 95% O2/5% CO2. Cutting and recording solutions were both 300–305 mOsmol/l. PBS, contained in mM: 137 NaCl, 2.68 KCl, 10.14 Na2HPO4, 1.76 KH2PO4; pH 7.4). N-Methyl-D-aspartic acid (NMDA) was applied in modified ACSF lacking added Mg2+. Ca2+-free ACSF was supplemented with 0.5mM ethylene-bis(oxyethylenenitrilo)tetraacetic acid tetrasodium (EGTA). Except where noted, the NMDA receptor antagonist MK-801 was included during NMDA washout periods. Some experiments utilized Gd3+ as a putative inhibitor of non-selective cation channels. It is acknowledged that Gd3+ binds with high avidity to components of ACSF, particularly phosphate (forming a readily-observable precipitate) and bicarbonate anions, however it is not yet known whether such binding prevents blocking of channels and it is possible that pore blocking activity remains (Caldwell et al., 1998). Therefore all experiments utilizing Gd3+ were performed in phosphate-free modified ACSF (but containing bicarbonate, pH 7.4). All reagents were from Sigma-Aldrich (St Louis, MO), unless otherwise indicated.
Statistical Analysis
All data appear as means ± S.E.M. Number (n) in the text refers to the number of slices in each treatment condition. Statistical analysis was carried out using unpaired Student’s t-tests. Significance criterion was set at p<0.05, and Bonferroni’s adjustment was used to correct for multiple comparisons where appropriate.
RESULTS
Dendrite swelling and MAP2 loss following NMDA exposure in normal ACSF
Acute hippocampal slices, prepared as routinely used for electrophysiological studies, were transiently exposed to NMDA and changes in dendritic morphology initially were investigated utilizing MAP2 immunohistochemistry. The first sets of studies involved stimulation in buffer containing 2mM Ca2+. Following transient exposure (10 min, in modified Mg2+-free ACSF), NMDA was removed by incubation in ACSF (with 1mM Mg2+, 90 min) before fixation and processing for MAP2 immunoreactivity. Figure 1A shows that time-matched control slices exhibited the expected dendritic localization of MAP2, with little labeling in CA1 cell bodies. As has been previously shown in rat hippocampal slices with OGD (Buddle et al., 2003) and in mouse slices (Hoskison and Shuttleworth, 2006), 90 min after transient NMDA exposure MAP2 was prominent in the somata in sp and was almost absent in proximal and distal dendritic regions of s. radiatum (sr) (Figure 1B). Population data for this MAP2 redistribution is illustrated in Figure 1C. These data show normalized MAP2 intensity (normalized against mean values in sp in each slice) plotted as a function of distance across slices. This analysis controls for variability in raw fluorescence intensity between experiments, and emphasizes the shift in MAP2 immunofluorescence from dendritic to somatic compartments 90 min following NMDA. Analysis of raw fluorescence from paired preparations processed in parallel confirmed a significant increase in somatic MAP2 immunofluorescence following NMDA exposure (data not shown). Figure 1C also shows that the degree of redistribution was indistinguishable whether or not MK-801 (50μM) was added to ACSF for the duration of NMDA washout, implying that the degree of redistribution is not dependent on persistent NMDA receptor activation.
Figure 1. MAP2 redistribution following transient NMDA exposure in acute slices.
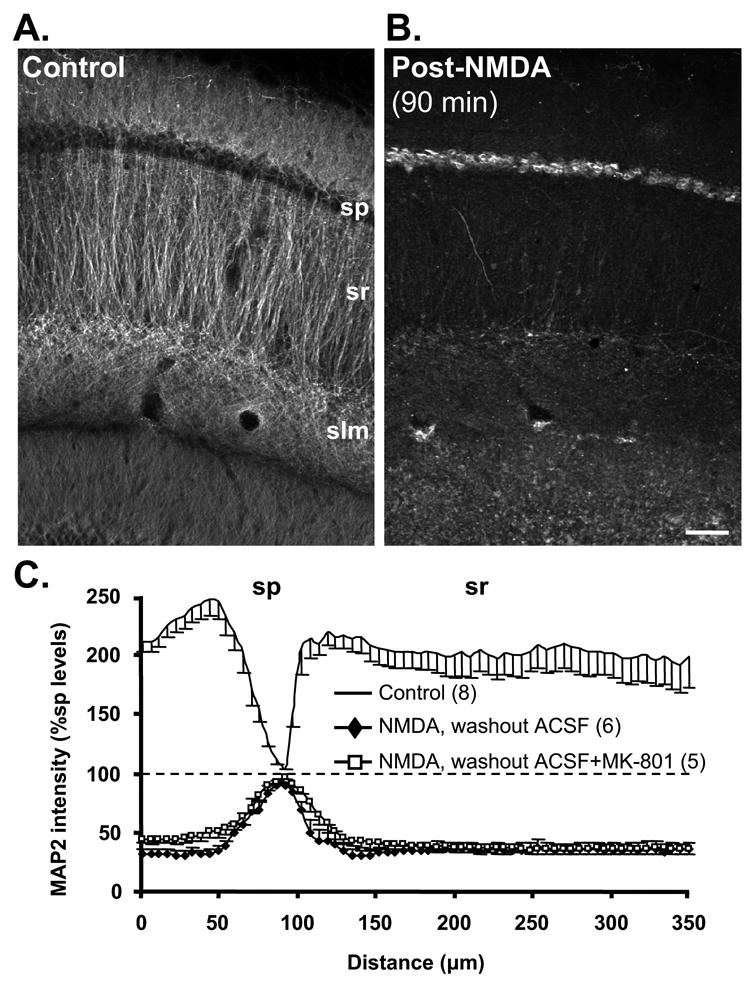
A: Control slice showing extensive MAP2 labeling in dendritic compartments and negligible labeling in somata. sp: stratum pyramidale, sr: stratum radiatum, slm: stratum lacunosum-moleculare. B: Transient NMDA exposure (30μM, 10 min) greatly reduced MAP2 fluorescence in dendrites and increased signal in somata. Following NMDA, slice was returned to normal ACSF for 90 min before processing. Scale bar: 50μm. C: MAP2 fluorescence intensity (normalized against mean values in sp) plotted as a function of distance across slices. This analysis controls for variability in raw immunofluorescence intensity between experiments, and emphasizes the extent of MAP2 redistribution from dendritic to somatic compartments 90 min following NMDA (filled diamonds). The inclusion of MK-801 (50μM) during the 90 min NMDA washout period (open squares) did not influence the degree of redistribution. Control and test slices were prepared from the same animals and run in parallel; numbers in parentheses refer to numbers of slices.
Figure 2 illustrates experiments in which the NMDA stimulus was held constant (10 min, 30μM), and slices were removed and fixed either immediately after the 10 min exposure (0 min washout), or after 20–90 min washout. These higher-power images demonstrate that post-NMDA, MAP2 immunoreactivity initially reveals extensive disorganized dendrite swelling (Figure 2Ab-d) before dendritic labeling is lost (Figure 2Ae). Figure 2Aa shows MAP2 localization in a control slice, illustrating continuous linear MAP2 labeling of apical dendrites, including clear labeling of fine secondary and tertiary dendrite branches throughout sr. In slices fixed immediately following NMDA exposure, most dendrites appeared swollen, particularly in most distal processes in sr. Dendrite swellings were irregular and elongated (Figure 2Ab-d) with a range of lengths (up to ~15μm). Organized spherical dendritic beading with regular inter-bead constrictions was rarely observed under these conditions.
In the fine tertiary dendrites, MAP2 immunoreactivity was generally undetectable after NMDA exposure (Figure 2Ab), and the morphology of these processes could not be clearly distinguished. Irregular dendrite swelling (described above) in larger primary and secondary dendrites could be visualized with MAP2 immunoreactivity ≤ 30 min following NMDA exposure (Figure 2Ad), though intermittent loss of detectable MAP2 resulted in a fragmented pattern of labeling. Dendrite segments were scored as uninterrupted unless gaps in MAP2 labeling were greater than 1.5 μm. Figure 2B shows quantification of results from multiple such experiments and reveals that immediately following NMDA there was a significant loss of dendrites with continuous MAP2 labeling in sr. This loss was more pronounced in distal sr at this early timepoint, and persisted in both proximal and distal sr throughout the washout period. At later timepoints (20–30min), visible MAP2 either was part of a remaining, damaged dendrite segment (arrows, Figure 2Ac), or appeared punctuate and did not belong to a visibly discernable structure (arrowheads, Figure 2Ad). The progressive increase in somatic MAP2 immunoreactivity after NMDA exposure is also illustrated in Figure 2Ab-d, and appears to decrease by 90 min post-NMDA (Figure 2Ae; see also (Arias et al., 1997)).
A small number of neurons showed relatively well preserved MAP2 labeling throughout dendritic processes, 90 min following NMDA washout (e.g. arrow in Figure 2Ae). The possibility that these cells were GABA-releasing interneurons was investigated using slices prepared from GAD67-GFP mice expressing GFP under the endogenous GAD67 promoter (Tamamaki et al., 2003). We first assessed whether GFP-positive neurons were also immunoreactive for GABA, in the same analysis regions. In a set of 3 slices, 22/24 GFP-positive somata were also clearly positive for GABA-like immunoreactivity. 5 additional neurons were positive for GABA-like immunoreactivity, but lacked demonstrable GFP in the somata. It is possible that this discrepancy may be because it is difficult to detect low levels of GFP in some neurons within the neuropil (Tamamaki et al., 2003). One possible explanation is that the expression level of GAD65, but not GAD67 is high in these cells. Figure 2C shows an example of a rare neuron with well preserved MAP2 labeling observed 90 min following NMDA washout, and this neuron also strongly expressed GFP, consistent with the hypothesis that interneurons may be responsible, at least in part, for the low level of preserved dendritic MAP2 labeling in stratum radiatum at this late timepoint.
Morphological dendritic changes observed with DiO support MAP2 results
Since NMDA exposure produced a dramatic loss of dendritic MAP2, in a separate set of experiments we assessed labeling with a lipophilic indicator. Figure 3A shows that ballistic delivery of DiO to fixed slices (see Methods) did not cause dendritic damage. In control slices (time controls for 0 min recovery and 90 min recovery post-NMDA), the majority of labeled dendrites (81.0 ± 12.0%, n=10) appeared normal, with little or no detectable dendritic swelling or beading, and spines were readily observed (arrowheads, Figure 3A). Spherical dendritic beading in control slices was rare (2/53 dendrites, n=10) and not increased by NMDA exposure (2/52 dendrites, n=10), suggesting that punctate MAP2 labeling observed throughout sr after NMDA (e.g. Figure 2Ad) was not due to MAP2 accumulation in fine dendritic beads. In slices fixed immediately following NMDA exposure, the majority of dendrites (74.3 ± 14.7%, n=5) displayed large irregular swellings (arrows, Figure 3B). Some DiO-labeled CA1 dendrites (22.9±14.9%, n=5) continued to exhibit spines and appeared generally less affected, but the shafts of these often appeared thinned, compared to control dendrites (arrowheads, Figure 3B). In preparations fixed 90 min post-NMDA, substantial dendrite swelling, observed as large irregular varicosities throughout, was evident in most dendritic processes (arrows, Figure 3C, 94.3±5.7%, p<0.01, compared with time controls n=5). Disorganized swellings seen with DiO appeared morphologically similar to those visualized with MAP2, and DiO labeling at 90 min post-NMDA revealed that dendrite segments were still intact despite the fragmented appearance and eventual loss of MAP2 labeling.
Figure 3. Persistence of irregular dendritic swelling visualized using DiO.
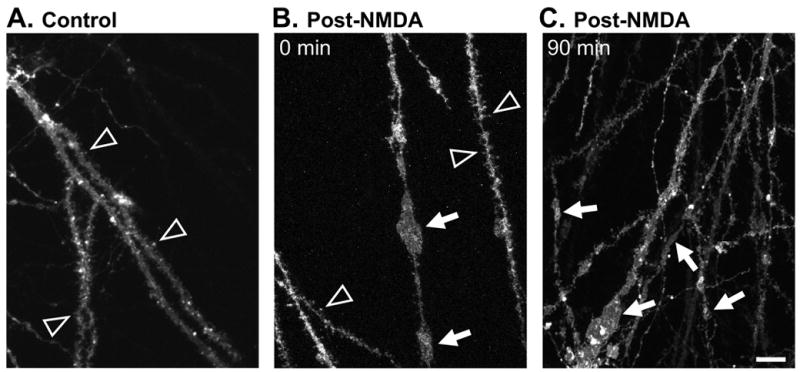
The lipophilic indicator DiO was loaded ballistically following slice fixation (see Methods) to label a small percentage of neurons. Images include regions in stratum radiatum centered ~100μm from stratum pyramidale. A: Representative control preparation showing dendritic spines (arrowheads) and no dendrite swelling. B: Preparation fixed immediately post-NMDA, showing some large irregular swellings (arrows) and some dendrite segments retaining fine dendritic spines (arrowheads). C: Preparation fixed 90 min after transient NMDA exposure, showing irregular swellings (arrows) in branches of dendrites with a range of diameters. Each image is representative of 5 slices analyzed per condition. Scale bar: 10μm.
Ca2+ removal during NMDA reduced irregular swelling and increased dendritic beading
Figure 4 illustrates a different pattern of dendrite morphology labeled by MAP2 when NMDA was applied in modified Ca2+-free ACSF. Removing extracellular Ca2+during NMDA exposure resulted in a significant number of dendrites with a series of smaller, organized spherical varicosities separated by clear dendrite constrictions (arrowheads, Figure 4B). The degree of this type of dendrite beading in a population of slices is quantified in Figure 4C and shows that Ca2+ removal throughout NMDA exposure significantly increased the number of beaded dendrites immediately post-stimulus by approximately 10 and 25 fold in proximal and distal sr, respectively (compared to slices exposed in parallel to NMDA in 2mM Ca2+). These values shown in Figure 4C correspond to 8.9±4.4% (proximal sr) and 12.3±4.9% (distal sr) of the total number of dendrite segments counted under conditions of Ca2+ removal. After 90 min post-NMDA washout, the number of beaded dendrites returned to the low baseline levels observed in a population of slices not exposed to NMDA (naïve).
Figure 4. Irregular dendritic swelling is Ca2+–dependent and Ca2+ removal reveals organized spherical beading.
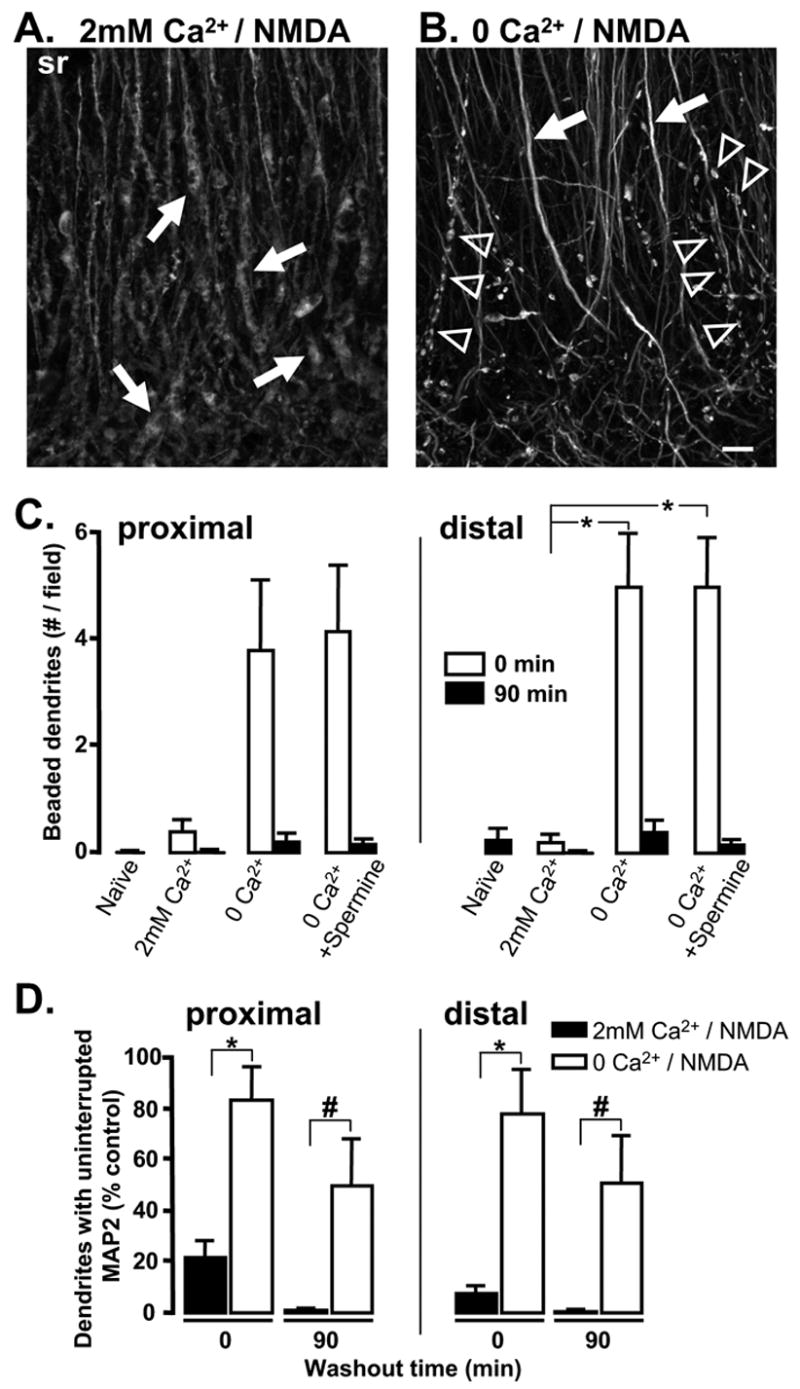
A&B: Paired slices fixed immediately following 10 min NMDA exposure in either 2mM Ca2+ (A) or Ca2+-free solution (B). Distal regions of stratum radiatum are shown. Arrows in A indicate irregular swelling in distal apical dendrites. Arrowheads in B identify more spherical and organized dendritic “beading”. In panel B, dendrites that were not regularly beaded exhibited uniform, uninterrupted MAP2 labeling (arrows). Scale bar (10μm) applies to both panels. C: Mean number of dendrites exhibiting spherical, regular beading. Results from proximal (left) and distal (right) sr are shown. Ca2+ removal increased the number of beaded dendrites observed immediately after NMDA exposure (open bars, 0 Ca2+, n=5, *p<0.01). Addition of spermine during 0 Ca2+/NMDA exposure did not reduce the number of beaded dendrites observed following stimulation in Ca2+-free solution. D: Mean data from 5 such experiments, showing the significant increase in numbers of dendrites (% control) with uninterrupted MAP2 labeling following stimulation in Ca2+-free solution. *p<0.01; #p<0.03.
As shown in Figure 4B spherical dendritic beading was observed only in a subpopulation of dendrite segments, while dendrites that were not beaded instead exhibited control-like uninterrupted MAP2 labeling (arrows, Figure 4B). Figure 4D summarizes the Ca2+-dependent difference in fragmentation of dendritic MAP2 labeling in a population of slices. Ca2+ removal significantly attenuated NMDA-induced loss of dendritic MAP2 immediately post-stimulus (open bars) and this protection persisted at 90 min post-NMDA. However, a significant decrease in the number of continuously labeled MAP2 dendrites was still detected when these slices were compared with naïve controls (proximal sr: 50.0±16.9%, p<0.01; distal sr: 49.4±18.3%, p<0.03; n=5).
Figure 5 summarizes experiments to address the question of whether the low proportion of dendrite segments that showed dendritic beading were processes of interneurons. The distribution of MAP2 and GFP is shown in control slices from GAD67-GFP animals (Figure 5A&B) and extensive beading of apical dendrites of GPF-positive neuron was evident immediately following after NMDA exposure in Ca2+-free ACSF (Figure 5E). The population data in Figure 5F shows that the great majority of GFP-positive processes were beaded (~70–90%). In stratum radiatum, the majority of beaded dendrites observed with MAP2 (immediately following NMDA exposure in Ca2+-free ACSF) were also positive for GFP (Figure 5C,G), suggesting that most of the dendritic beading observed with MAP2 may be restricted to interneurons.
Contribution of Ca2+ to MAP2 redistribution
Previous work showed that Ca2+ removal improved functional outcome and was associated with preservation of organized dendritic MAP2 at late timepoints following NMDA exposure (Hoskison and Shuttleworth, 2006). Figure 6 shows the contribution of Ca2+-dependent mechanisms on MAP2 redistribution at both early and late timepoints. Although some MAP2 redistribution did persist in slices exposed to NMDA in Ca2+-free ACSF, this was substantially less than that seen when Ca2+ remained present throughout NMDA exposure (compare bottom panels in Figures 6A & B). Mean data shown in Figure 6C reveal that, even by 90 min, MAP2 labeling in sp was not greater than in dendritic regions of 0 Ca2+/NMDA-slices (open squares), but instead appeared more evenly distributed across dendritic and somatic compartments.
Figure 6. Ca2+ removal during NMDA exposure attenuated MAP2 redistribution.
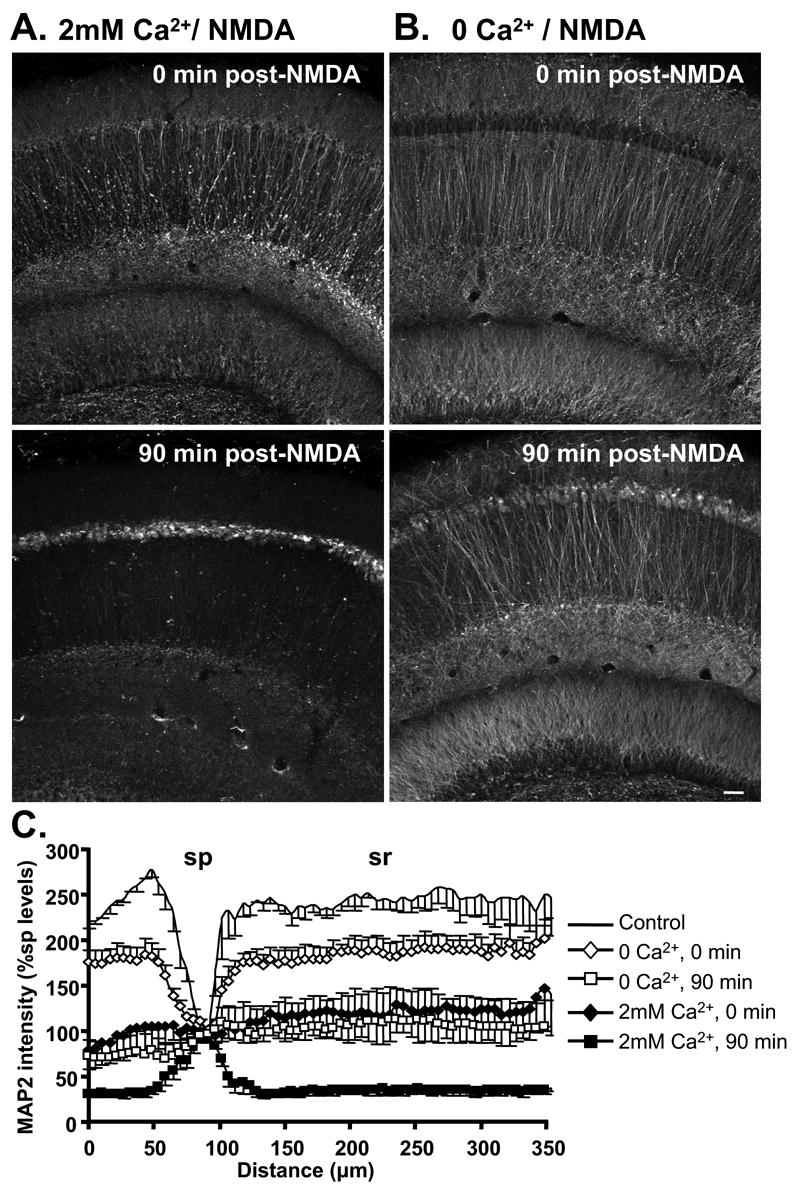
MAP2 distribution following NMDA exposure in either 2mM Ca2+ (A) or Ca2+-free solution (B). Top panels are immediately following 10 min NMDA, and bottom panels are after 90 min washout (2mM Ca2+ present throughout washout in both cases). When Ca2+ was removed during NMDA exposure, redistribution of MAP2 immunofluorescence from dendritic to somatic compartments was substantially decreased immediately following exposure (top panels). At 90 min post-NMDA, some somatic MAP2 accumulation was still evident despite Ca2+ removal, but the loss of MAP2 from dendritic compartments was greatly decreased in slices exposed to NMDA in 0 Ca2+ (bottom panels). Slices in A&B were from the same animals, and processed in parallel. Scale bar (50μm) applies to all panels. C: Normalized MAP2 fluorescence intensity, plotted as a function of distance across slices (normalized against sp levels, mean±SEM from 5 experiments). Following stimulation in Ca2+-free solution (90 min, open squares), MAP2 immunofluorescence was relatively evenly distributed across the slice, in contrast to the predominant somatic accumulation observed in 2mM Ca2+ (90 min, filled squares).
Effects of Gd3+ and spermine on beading and persistent MAP2 loss
Extracellular Ca2+ reductions can significantly increase flux through TRPM7-like non-selective cation channels, and recent work has suggested that ion flux through these channels contributes to neuronal degeneration (Aarts et al., 2003; Aarts and Tymianski, 2005). We therefore carried out an additional set of experiments to assess the potential contribution of increased conductance through these channels on (1) the degree of dendritic beading observed with 0 Ca2+/NMDA and (2) the loss of dendritic MAP2 that persisted in 0 Ca2+/NMDA. Gd3+ inhibits TRPM7 with high potency (Aarts and Tymianski, 2005), and spermine also blocks similar channels (Kozak et al., 2002; Kerschbaum et al., 2003). Since spermine may interfere with NMDA receptor activation (Sharma and Reynolds, 1999; Ferchmin et al., 2000) all experimental groups were exposed to either blocker, and the effects of Ca2+ removal on NMDA-induced beading and MAP2 loss were examined (see also Methods). This experimental design revealed that the degree of NMDA receptor stimulation was not significantly altered by spermine or Gd3+ at the concentrations used (each 30μM) since comparable MAP2 loss persisted (in normal Ca2+) in the presence of either blocker (filled bars, compare Figure 7C,D with Figure 4D).
Figure 7. In the presence of putative inhibitors of non-selective cation channels (spermine and Gd3+) MAP2 labeling was well preserved and not significantly different from naïve controls.
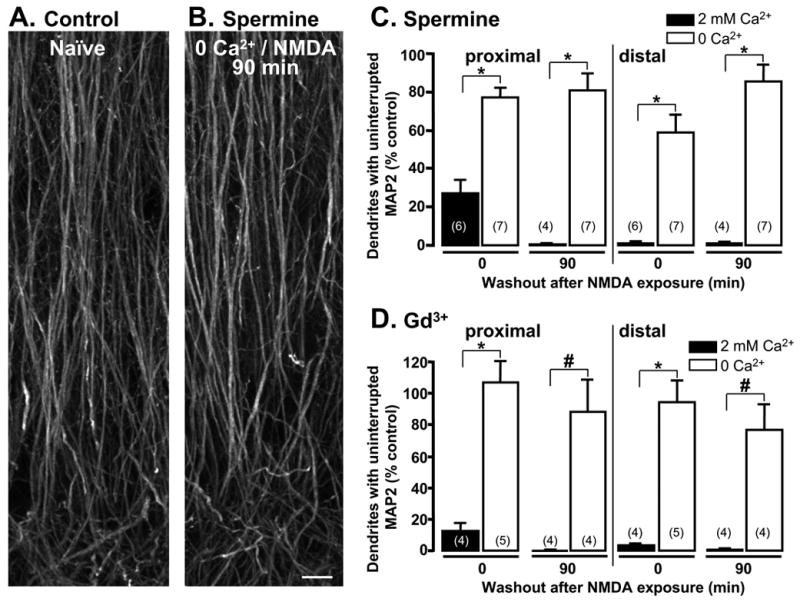
Representative photomicrographs showing MAP2 immunofluorescence in A: naïve slice and B: slice fixed 90 min after NMDA stimulation in 0 Ca2+, with the addition of 30μM spermine. Scale bar: 10 μm. Mean data from experiments testing C: spermine and D: Gd3+ exposure (each 30μM) on loss of MAP2 from dendrites. Numbers in parentheses indicate the number of slices examined in each condition. In the presence of either blocker, NMDA exposure in 2mM Ca2+ (filled bars) produced substantial MAP2 disruption, similar to that described in previous figures. In the absence of Ca2+, MAP2 labeling was significantly improved (*p<0.001; #p<0.005), and was not significantly different from naïve controls (p>0.3 for all open bars).
Dendritic beading observed in 0Ca2+/NMDA conditions was not prevented by the presence of spermine (Figure 4C) or Gd3+ (not shown). However Figure 7 shows that both inhibitors appeared to provide additional protection against NMDA-induced loss of dendritic MAP2, over and above that seen with Ca2+ removal alone (compare open bars at 90 min, Figure 7C&D with Figure 4D). Indeed, at 90 min post-NMDA, MAP2 labeling in 0Ca2+/NMDA-slices exposed in the presence of either blocker was not significantly different from the continuous MAP2 labeling observed in time-matched naïve controls (Figures 7A&B) in both proximal (spermine p=0.28, n=7; Gd3+ p=0.72, n=4) and distal sr (spermine p=0.35, n=7; Gd3+, p=0.42, n=4). These results suggest that increased conductance of these channels does not appear to contribute to beading observed following 0Ca2+/NMDA, but their activation could be involved in the residual dendritic damage detected at 90 min post-NMDA under 0 Ca2+ conditions.
DISCUSSION
This study tested the hypothesis that excitotoxic stimulation in acute slice can lead to persistent dendritic injury that is distinct from dendritic beading. Ca2+-dependent irregular, elongated dendritic swelling associated with persistent MAP2 loss was observed in most apical dendrites of CA1 pyramidal neurons and is likely to contribute to persistent loss of synaptic function. In contrast, regular, spherical beading was only observed following NMDA stimulation in Ca2+-free ACSF, and was restricted to a subpopulation of dendrites in CA1 stratum radiatum that appear to be mainly interneurons.
Irregular Dendrite Swelling
Following NMDA exposure, irregular elongated dendritic swellings were rapidly formed in the great majority of CA1 neurons. At early timepoints post-NMDA, MAP2 labeling of these processes appeared diffuse and vacuolated, but at later timepoints (>30min) MAP2 was not useful for visualization of dendrite structure, due to progressive loss of this marker from dendrites, and accumulation in somata (see below). DiO labeling was useful for demonstrating that CA1 dendrites remained intact throughout prolonged post-NMDA washout periods, despite the profound disruption of dendritic microtubules (Hoskison & Shuttleworth 2006). Indeed, large irregular dendrite swellings persisted throughout the timecourse of the experiments (up to 2 hours post-NMDA), with morphology similar to that observed at early timepoints with MAP2 immunohistochemistry. This morphology appeared very distinct from that described for dendritic beading, a noticeable difference being the lack of severe constrictions commonly observed between smooth spherical swellings (see below).
Extracellular Ca2+ was a major contributor to the irregular swelling in CA1 dendrites, suggesting a role for Ca2+-dependent proteases, or disruption of dendritic Ca2+ homeostasis in triggering catastrophic structural changes. This Ca2+-dependence also differentiates the irregular swelling described here from dendritic beading, which is generally considered to be a Ca2+-independent phenomenon, largely due to excessive Na+ accumulation (Hasbani et al., 1998; Al-Noori and Swann, 2000).
MAP2 loss
Dendritic MAP2 loss and redistribution to somata has been considered an indicator of excitotoxic damage following a range of neuronal injuries both in vivo and in vitro (Kitagawa et al., 1989; Taft et al., 1992; Pettigrew et al., 1996; Arias et al., 1997), and may predict cell death. Mechanisms of dendritic MAP2 loss were previously investigated following oxygen-glucose deprivation (OGD) in rat hippocampal slices (Buddle et al., 2003), and following NMDA in preparations identical to those described in the present study (Hoskison & Shuttleworth, 2006). Loss of MAP2 in hippocampal homogenates is Ca2+-dependent, and due (at least in part) to activation of the Ca2+-activated protease calpain (Buddle et al., 2003). However, from immunohistochemical localization of MAP2, it appears that calpain may not be responsible for the initial disruption of dendritic MAP2, but rather may contribute to removal of disrupted MAP2 from dendritic compartments, following excitotoxic microtubule disruption (Hoskison & Shuttleworth 2006).
In the present study, Ca2+-dependent MAP2 loss was first apparent in fine tertiary dendrites in stratum radiatum, followed by progressive loss from larger dendrites (Figure 2A). This observation is consistent with reports of excitotoxic injury being initiated in fine distal dendrites followed by progressive involvement of Ca2+ overload in larger distal dendrites (Shuttleworth and Connor, 2001).
Although Ca2+ removal significantly reduced MAP2 loss, some fragmentation of dendritic MAP2 labeling persisted following NMDA exposure in Ca2+-free solution (Figure 4). This residual MAP2 loss was diminished by low concentrations of Gd3+ and spermine (Figure 7). While it is acknowledged these drugs may block a range of ion channels (Hamill and McBride, 1996; Williams, 1997) and may interfere with NMDA receptor activation, these concerns were taken into account in the experimental design. Since these inhibitors did not significantly attenuate NMDA-induced damage when Ca2+ remained present, it appears unlikely that these blockers interfere with NMDA receptor activity under these conditions. Thus, in the absence of extracellular Ca2+, excessive Na+ flux through TRPM7-like or other spermine and Gd3+-sensitive channels (e.g. voltage-dependent Na+ channels (Hamill and McBride, 1996) may contribute to MAP2 loss, possibly as a consequence of localized metabolic failure.
Dendritic beading
In the present study, spherical organized dendritic beading was very rare unless the NMDA insult occurred in Ca2+-free solutions (Figure 4), and even under these conditions, the proportion of dendrites involved was low (~10%). Since previous work in rat hippocampal slice cultures showed a substantially greater incidence of dendritic beading in interneurons than in pyramidal neurons following kainic acid stimulation (Oliva et al., 2002) we examined the possibility that interneurons may preferentially bead following NMDA stimulation. The experiments with slices prepared from GAD67-GFP animals support this suggestion, since a large proportion of GFP-labeled dendrites were beaded under these conditions (NMDA in Ca2+-free ACSF), and the majority of beaded profiles (as assessed by MAP2 immunohistochemistry) were GFP-positive. These results suggest that the majority of interneurons are particularly susceptible to dendritic beading, whereas CA1 pyramidal neurons preferentially undergo irregular swelling following excitotoxic NMDA stimulation. The mechanism(s) underlying such a differential sensitivity are currently unknown, but recent work has suggested that differences in NMDA receptor currents may explain differential sensitivity of interneurons and pyramidal cells in acutely-prepared slices exposed to 30μM NMDA (Avignone et al., 2005).
It is of interest that while the great majority of neurons showed almost complete loss of MAP2 by 90 min post-NMDA, a very small number of dendrites exhibited control-like MAP2 labeling (Figure 2Ae). From co-localization with GFP in GAD67-GFP slices, it appears that interneurons contribute to this small population of neurons that maintain dendritic morphology following NMDA (Figure 2C). Hippocampal interneurons, in general, appear more resistant to cell death than pyramidal neurons following ischemia (Lipton, 1999), although following seizure activity, somatostatin-expressing interneurons appear selectively vulnerable (Sloviter, 1987). It was not possible to track the fate of individual neurons in this study, so it is not known whether neurons that showed beading following NMDA went on to recover dendritic structure.
The observation that extracellular Ca2+ reduction increased the incidence of beading (Figure 4B&C) is consistent with previous reports of NMDA-induced beading in both cortical cultures (Hasbani et al., 1998) and hippocampal slice culture (Ikegaya et al., 2001). This may be due to increased Na+ influx, as a consequence of Ca2+ removal (Hasbani et al., 1998). Extracellular Ca2+ reductions can increase Na+ conductance via activation of TRPM7-like channels (Aarts et al., 2003; Aarts and Tymianski, 2005), and since beading was selectively observed under 0 Ca2+ conditions, experiments in the current study tested the hypothesis that activation of these channels was responsible for spherical beading. Available pharmacological approaches to study such channels are not specific (Hamill and McBride, 1996; Ferchmin et al., 2000), but the agents used here (Gd3+ and spermine) are expected to inhibit TRPM7-like channels (Aarts et al., 2003; Aarts and Tymianski, 2005; Harteneck, 2005). Since these agents did not prevent the beading observed in Ca2+-free solution, it is likely that other Na+ influx pathways are involved. Investigating other channels triggered by low Ca2+ will be of interest, perhaps in studies using selective channel deletions, rather than pharmacological approaches.
It is also possible that a Na+-dependent beading mechanism may be masked by extensive Ca2+-dependent dendritic disruption and therefore spherical Na+-dependent beading may only be revealed in these mature preparations when the MAP2 loss and irregular swelling is prevented by Ca2+ removal. In neuronal cultures, NMDA primarily produces organized beading with little irregular swelling. It is possible that one reason for dissimilar results in the current study may be more rapid Ca2+ elevations in mature neurons compared with neonatal cells (e.g. Friedman and Haddad, 1993), although multiple other differences between acute slices and cultured preparations may also contribute.
Comparing dendritic injuries across studies
The two types of dendritic injuries described in this study differed in appearance and underlying mechanisms, however both types of swelling might be referred to as “dendritic beading” in the literature. Thus, distinguishing between different types of dendritic swelling might help to explain conflicting conclusions regarding the consequences of dendritic beading, and whether it contributes to irreversible slice damage (e.g. Obeidat and Andrew, 1998) or whether it may be more reversible and possibly even neuroprotective (e.g. Ikegaya et al., 2001). It is noteworthy that certain labeling methods appear better suited to detect differences in dendritic injury type. In the current study, MAP2 immunolabeling was especially useful for this purpose, whereas some distinct structural characteristics may be less evident using membranous dyes or cytoplasmic markers that do not label subcellular constituents (Matesic and Lin, 1994).
The nature of the stimulus is also likely to influence the type of the dendritic injury observed. NMDA receptor over-activation has been suggested to contribute to a range of neurodegenerative disorders (Lipton and Rosenberg, 1994; Lipton, 1999) although it is recognized that glutamate excitotoxicity may not be required for neuron death under all conditions (Obeidat et al., 2000). It is also possible that different mechanisms of dendritic injury may be triggered by oxygen and/or glucose deprivation, in addition to responses generated by receptor over-activation described in the present study.
CONCLUSION
The predominant injury to CA1 pyramidal neurons observed in the present study was persistent irregular and elongated dendritic swelling, presumably caused by excitotoxic Ca2+ overload, and this was associated with enduring loss of MAP2. This injury is different from dendritic beading, which could be demonstrated in the same preparations, but was only significant under Ca2+-free conditions, where dendritic MAP2 was preserved after termination of the NMDA stimulus. If these two types of dendritic structural modification translate into differential recovery from injury, then distinguishing between these morphological consequences could help focus potential targets for neuroprotective treatments.
Acknowledgments
Supported by NIH grants NS43458, NS51288 & RR15636 and by Grant-in-Aids for Scientific Research from the MEXT and the MHLW, Japan. Images were generated in the UNM Cancer Center Fluorescence Microscopy Facility, which in turn received support from NCRR 1 S10 RR14668, NSF MCB9982161, NCRR P20 RR11830, NCI R24 CA88339, NCRR S10 RR19287, NCRR S10 RR016918, the University of New Mexico Health Sciences Center, and the University of New Mexico Cancer Center. The authors thank Dr R. O. Wong (Washington University School of Medicine) for advice on DiO labeling techniques, and Dr Michael Wilson and Lawrence Tafoya for providing GAD67-GFP animals from their breeding colony at UNM.
Footnotes
Section Editor: Dr Constantino Sotelo, Universite Pierre et Marie Curie, Paris.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aarts M, Iihara K, Wei WL, Xiong ZG, Arundine M, Cerwinski W, MacDonald JF, Tymianski M. A key role for TRPM7 channels in anoxic neuronal death. Cell. 2003;115:863–877. doi: 10.1016/s0092-8674(03)01017-1. [DOI] [PubMed] [Google Scholar]
- Aarts MM, Tymianski M. TRPMs and neuronal cell death. Pflugers Arch. 2005;451:243–249. doi: 10.1007/s00424-005-1439-x. [DOI] [PubMed] [Google Scholar]
- Al-Noori S, Swann JW. A role for sodium and chloride in kainic acid-induced beading of inhibitory interneuron dendrites. Neuroscience. 2000;101:337–348. doi: 10.1016/s0306-4522(00)00384-5. [DOI] [PubMed] [Google Scholar]
- Andrew RD, Jarvis CR, Obeidat AS. Potential sources of intrinsic optical signals imaged in live brain slices. Methods. 1999;18:185–196. 179. doi: 10.1006/meth.1999.0771. [DOI] [PubMed] [Google Scholar]
- Arias C, Arrieta I, Massieu L, Tapia R. Neuronal damage and MAP2 changes induced by the glutamate transport inhibitor dihydrokainate and by kainate in rat hippocampus in vivo. Exp Brain Res. 1997;116:467–476. doi: 10.1007/pl00005774. [DOI] [PubMed] [Google Scholar]
- Avignone E, Frenguelli BG, Irving AJ. Differential responses to NMDA receptor activation in rat hippocampal interneurons and pyramidal cells may underlie enhanced pyramidal cell vulnerability. Eur J Neurosci. 2005;22:3077–3090. doi: 10.1111/j.1460-9568.2005.04497.x. [DOI] [PubMed] [Google Scholar]
- Buddle M, Eberhardt E, Ciminello LH, Levin T, Wing R, DiPasquale K, Raley-Susman KM. Microtubule-associated protein 2 (MAP2) associates with the NMDA receptor and is spatially redistributed within rat hippocampal neurons after oxygen-glucose deprivation. Brain Res. 2003;978:38–50. doi: 10.1016/s0006-8993(03)02758-6. [DOI] [PubMed] [Google Scholar]
- Caldwell RA, Clemo HF, Baumgarten CM. Using gadolinium to identify stretch-activated channels: technical considerations. Am J Physiol. 1998;275:C619–621. doi: 10.1152/ajpcell.1998.275.2.C619. [DOI] [PubMed] [Google Scholar]
- Faddis BT, Hasbani MJ, Goldberg MP. Calpain activation contributes to dendritic remodeling after brief excitotoxic injury in vitro. J Neurosci. 1997;17:951–959. doi: 10.1523/JNEUROSCI.17-03-00951.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferchmin PA, Perez D, Biello M. Spermine is neuroprotective against anoxia and N-methyl-D-aspartate in hippocampal slices. Brain Res. 2000;859:273–279. doi: 10.1016/s0006-8993(00)01973-9. [DOI] [PubMed] [Google Scholar]
- Friedman JE, Haddad GG. Major differences in Ca2+i response to anoxia between neonatal and adult rat CA1 neurons: role of Ca2+o and Na+o. J Neurosci. 1993;13:63–72. doi: 10.1523/JNEUROSCI.13-01-00063.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan WB, Grutzendler J, Wong WT, Wong RO, Lichtman JW. Multicolor "DiOlistic" labeling of the nervous system using lipophilic dye combinations. Neuron. 2000;27:219–225. doi: 10.1016/s0896-6273(00)00031-3. [DOI] [PubMed] [Google Scholar]
- Hamam BN, Kennedy TE. Visualization of the dendritic arbor of neurons in intact 500 microm thick brain slices. J Neurosci Methods. 2003;123:61–67. doi: 10.1016/s0165-0270(02)00341-2. [DOI] [PubMed] [Google Scholar]
- Hamill OP, McBride DW., Jr The pharmacology of mechanogated membrane ion channels. Pharmacol Rev. 1996;48:231–252. [PubMed] [Google Scholar]
- Harteneck C. Function and pharmacology of TRPM cation channels. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:307–314. doi: 10.1007/s00210-005-1034-x. [DOI] [PubMed] [Google Scholar]
- Hasbani MJ, Hyrc KL, Faddis BT, Romano C, Goldberg MP. Distinct roles for sodium, chloride, and calcium in excitotoxic dendritic injury and recovery. Exp Neurol. 1998;154:241–258. doi: 10.1006/exnr.1998.6929. [DOI] [PubMed] [Google Scholar]
- Hori N, Carpenter DO. Functional and morphological changes induced by transient in vivo ischemia. Exp Neurol. 1994;129:279–289. doi: 10.1006/exnr.1994.1170. [DOI] [PubMed] [Google Scholar]
- Hoskison MM, Shuttleworth CW. Microtubule disruption, not calpain-dependent loss of MAP2, contributes to enduring NMDA-induced dendritic dysfunction in acute hippocampal slices. Exp Neurol. 2006 doi: 10.1016/j.expneurol.2006.06.010. [DOI] [PubMed] [Google Scholar]
- Ikegaya Y, Kim JA, Baba M, Iwatsubo T, Nishiyama N, Matsuki N. Rapid and reversible changes in dendrite morphology and synaptic efficacy following NMDA receptor activation: implication for a cellular defense against excitotoxicity. J Cell Sci. 2001;114:4083–4093. doi: 10.1242/jcs.114.22.4083. [DOI] [PubMed] [Google Scholar]
- Jarvis CR, Lilge L, Vipond GJ, Andrew RD. Interpretation of intrinsic optical signals and calcein fluorescence during acute excitotoxic insult in the hippocampal slice. Neuroimage. 1999;10:357–372. doi: 10.1006/nimg.1999.0473. [DOI] [PubMed] [Google Scholar]
- Kerschbaum HH, Kozak JA, Cahalan MD. Polyvalent cations as permeant probes of MIC and TRPM7 pores. Biophys J. 2003;84:2293–2305. doi: 10.1016/S0006-3495(03)75035-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirov SA, Petrak LJ, Fiala JC, Harris KM. Dendritic spines disappear with chilling but proliferate excessively upon rewarming of mature hippocampus. Neuroscience. 2004;127:69–80. doi: 10.1016/j.neuroscience.2004.04.053. [DOI] [PubMed] [Google Scholar]
- Kitagawa K, Matsumoto M, Niinobe M, Mikoshiba K, Hata R, Ueda H, Handa N, Fukunaga R, Isaka Y, Kimura K, et al. Microtubule-associated protein 2 as a sensitive marker for cerebral ischemic damage--immunohistochemical investigation of dendritic damage. Neuroscience. 1989;31:401–411. doi: 10.1016/0306-4522(89)90383-7. [DOI] [PubMed] [Google Scholar]
- Kozak JA, Kerschbaum HH, Cahalan MD. Distinct properties of CRAC and MIC channels in RBL cells. J Gen Physiol. 2002;120:221–235. doi: 10.1085/jgp.20028601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- Matesic DF, Lin RC. Microtubule-associated protein 2 as an early indicator of ischemia-induced neurodegeneration in the gerbil forebrain. J Neurochem. 1994;63:1012–1020. doi: 10.1046/j.1471-4159.1994.63031012.x. [DOI] [PubMed] [Google Scholar]
- Obeidat AS, Andrew RD. Spreading depression determines acute cellular damage in the hippocampal slice during oxygen/glucose deprivation. Eur J Neurosci. 1998;10:3451–3461. doi: 10.1046/j.1460-9568.1998.00358.x. [DOI] [PubMed] [Google Scholar]
- Obeidat AS, Jarvis CR, Andrew RD. Glutamate does not mediate acute neuronal damage after spreading depression induced by O2/glucose deprivation in the hippocampal slice. J Cereb Blood Flow Metab. 2000;20:412–422. doi: 10.1097/00004647-200002000-00024. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Lam TT, Swann JW. Distally directed dendrotoxicity induced by kainic Acid in hippocampal interneurons of green fluorescent protein-expressing transgenic mice. J Neurosci. 2002;22:8052–8062. doi: 10.1523/JNEUROSCI.22-18-08052.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Fuller T, de Gubareff T. Acute dendrotoxic changes in the hippocampus of kainate treated rats. Brain Res. 1979;176:91–100. doi: 10.1016/0006-8993(79)90872-2. [DOI] [PubMed] [Google Scholar]
- Park JS, Bateman MC, Goldberg MP. Rapid alterations in dendrite morphology during sublethal hypoxia or glutamate receptor activation. Neurobiol Dis. 1996;3:215–227. doi: 10.1006/nbdi.1996.0022. [DOI] [PubMed] [Google Scholar]
- Pettigrew LC, Holtz ML, Craddock SD, Minger SL, Hall N, Geddes JW. Microtubular proteolysis in focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:1189–1202. doi: 10.1097/00004647-199611000-00013. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE. Genetic basis of kainate-induced excitotoxicity in mice: phenotypic modulation of seizure-induced cell death. Epilepsy Res. 2003;55:201–210. doi: 10.1016/s0920-1211(03)00115-3. [DOI] [PubMed] [Google Scholar]
- Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci U S A. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma TA, Reynolds IJ. Characterization of the effects of polyamines on [125I]MK-801 binding to recombinant N-methyl-D-aspartate receptors. J Pharmacol Exp Ther. 1999;289:1041–1047. [PubMed] [Google Scholar]
- Shuttleworth CW, Connor JA. Strain-dependent differences in calcium signaling predict excitotoxicity in murine hippocampal neurons. J Neurosci. 2001;21:4225–4236. doi: 10.1523/JNEUROSCI.21-12-04225.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloviter RS. Decreased hippocampal inhibition and a selective loss of interneurons in experimental epilepsy. Science. 1987;235:73–76. doi: 10.1126/science.2879352. [DOI] [PubMed] [Google Scholar]
- Sloviter RS, Dempster DW. "Epileptic" brain damage is replicated qualitatively in the rat hippocampus by central injection of glutamate or aspartate but not by GABA or acetylcholine. Brain Res Bull. 1985;15:39–60. doi: 10.1016/0361-9230(85)90059-0. [DOI] [PubMed] [Google Scholar]
- Taft WC, Yang K, Dixon CE, Hayes RL. Microtubule-associated protein 2 levels decrease in hippocampus following traumatic brain injury. J Neurotrauma. 1992;9:281–290. doi: 10.1089/neu.1992.9.281. [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T. Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol. 2003;467:60–79. doi: 10.1002/cne.10905. [DOI] [PubMed] [Google Scholar]
- Wellmann H, Kaltschmidt B, Kaltschmidt C. Optimized protocol for biolistic transfection of brain slices and dissociated cultured neurons with a hand-held gene gun. J Neurosci Methods. 1999;92:55–64. doi: 10.1016/s0165-0270(99)00094-1. [DOI] [PubMed] [Google Scholar]