

Abstract
Low brain levels of the inhibitory neurotransmitter γ-aminobutyric acid (GABA) leads to convulsions. Inhibition of GABA aminotransferase increases the concentration of GABA and can terminate the convulsions. Earlier we reported the synthesis of (1S,3S)-3-amino-4-difluoromethylenecyclopentanecarboxylic acid (2), which is 186 times more potent an inactivator of GABA aminotransferase than the epilepsy drug S-vigabatrin. The corresponding dichloromethylene analogue of 2 (compound 3) has been made, but it shows only weak reversible inhibition of GABA aminotransferase. However, the tetrazole isostere of 2 (compound 4) has been found to be a time-dependent inactivator of GABA aminotransferase. Although it is 20 times less potent than carboxylic acid 2, it is 2.5 times more potent than S-vigabatrin. A calculation of the ClogP values indicates that 4 is the most lipophilic of the three, being 69 times more lipophilic than 2 and 55 times more lipophilic than S-vigabatrin, indicating potential for improved bioavailability.
γ-Aminobutyric acid (GABA) is the major inhibitory neurotransmitter in the mammalian central nervous system (CNS). Low GABA levels are related to a series of neurological disorders including Parkinson’s disease,1 Huntington’s chorea,2,3 Alzheimer’s disease,4 and epilepsy.5–7 γ-Aminobutyric acid aminotransferase (GABA-AT, E.C. 2.6.1.19), a pyridoxal 5′-phosphate (PLP)-dependent enzyme that degrades GABA, is a target for antiepilepsy drugs. Inhibition of GABA-AT results in an increase in the concentration of brain GABA and has an anticonvulsant effect. Inhibitors of GABA-AT have been developed as potential anticonvulsant agents. One of these, 4-amino-5-hexenoic acid (vigabatrin or Sabril®, 1, Figure 1), is a potent inactivator of GABA-AT8 that has been marketed as an anticonvulsant drug,9 although it may cause certain vision-related side effects after long-term use.10–13
Figure 1.

Modifications of 2
Analogues of vigabatrin have been developed as drug candidates.14 One approach to optimize the antiepilepsy drug vigabatrin is to rigidify its structure by formation of a five or six-membered ring.15–18 Recently, we synthesized (1S,3S)-3-amino-4-difluoromethylenecyclopentanecarboxylic acid (2, Figure 1), a difluoro-substituted, conformationally rigid vigabatrin analogue,18 and it was found to be one of the most potent inactivators of GABA-AT; it is approximately 186 times more potent than S-vigabatrin in vitro. The simple structure and superior potency of 2 make it a desirable lead compound for further modification.
The rigid conformation of 2 minimizes the entropic penalty on binding and, therefore, could contribute to its potency; however, the same structure with hydrogen in place of the fluorines binds much less well.18 To obtain more structural information, we designed an analogue of 2 with a dichloromethylene moiety (3, Figure 1), which can be used to explore the importance of the size of the fluorine atoms in the interaction between the enzyme and the inactivator as well as the importance of the lipophilicity of those groups.
Despite its high in vitro activity, the relatively low lipophilicity of 2 caused by its carboxylic acid and ammonium groups may limit its permeability to the blood-brain barrier (BBB), and therefore, may result in lower in vivo potency.19,20 The BBB permeability of a compound, affected by its lipophilicity, hydrogen-bond desolvation potential, pKa/charge, and molecular size,21–26 is an important concern in drug delivery. Generally, uncharged, lipophilic compounds with low molecular weights are expected to cross the BBB and provide higher bioavailabilities.27 To increase the lipophilicity of the molecule and to achieve better in vivo activity, 4, a tetrazole analogue of 2, also was designed. The tetrazole group is a well-known bioisostere of a carboxylic acid with higher lipophilicity.28 In addition, our previous studies showed that the tetrazole analogue of vigabatrin is a potent inactivator of GABA-AT, which indicates that tetrazole may be a good replacement for carboxylic acid group in GABA analogues.29 In this note we report the synthesis and the results of enzymatic studies of 3 and 4.
Compound 3 was synthesized from 5 (Scheme 1). The previously synthesized intermediate 516,30 was treated with tert-butyl lithium and 7, which was synthesized from 6,31 to give 8. Deprotection of the PMB group with ceric ammonium nitrate afforded lactam 9; hydrolysis of 9 with hydrochloric acid gave 3 in the form of a hydrochloride salt.
Scheme 1.

Synthesis of 3
Compound 4 was synthesized from 2 as shown in Scheme 2. The amino group in 2 was protected with phthalic anhydride to give 10. The carboxylic acid group in 10 was converted to amide 11, which underwent dehydration to give nitrile 12. The method of Amantini and coworkers32 was used to obtain tetrazole 13. Treatment of 13 with 6 N hydrochloric acid deprotected the phthalic group and provided 4.
Scheme 2.
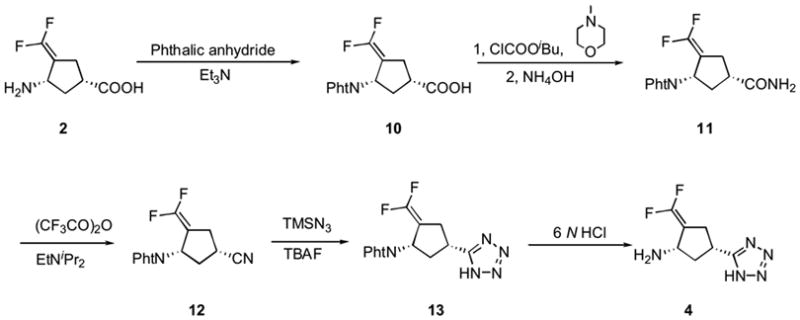
Synthesis of 4
Compound 3 was tested as a time-dependent inhibitor of GABA-AT. GABA-AT, however, showed no significant decrease in activity after up to 74 minutes of incubation with 1.7 mM concentration of 3, which indicates that 3 is not a potent inactivator. In addition, it was shown that the enzyme retains approximately 58% of its activity in the presence of 10 mM of 3, which suggests that the IC50 value for 3 is greater than 10 mM, and 3 is only a weak reversible inhibitor of GABA-AT. The decrease in activity of 3 compared with the corresponding difluoro compound 2 is quite dramatic and indicates that the chlorine atoms in 3 might be too bulky for the enzyme active site.
Compound 4 showed time- and concentration-dependent inhibition of GABA-AT. The kinetic constants for 4 at pH 8.5, the optimal pH for the enzyme, were determined using a Kitz and Wilson replot (Figure 2).33 The kinact and KI values, however, could not be determined accurately because the intercepts were very close to the origin. Instead, the kinact/KI value, which is in proportion to the slope of the plot, was calculated to be 2.48 min−1mM−1. At pH 6.5, the kinact and KI values for 4 were determined to be 1.05 min−1 and 3.75 mM, respectively, and the kinact/KI value was calculated to be 0.28 min−1mM−1. According to the kinact/KI values, 4 is less potent than its parent compound, 2 (kinact/KI = 5.7 min−1mM−1, pH 6.5),18 but it is a better inactivator of GABA-AT than S-vigabatrin (kinact/KI = 1.7 min−1mM−1 at pH 8.5, 0.11 min−1mM−1 at pH 6.5).
Figure 2.

Kitz and Wilson replot for 4 (pH 8.5, 25 °C)
Despite the decrease in activity, the replacement of a carboxylic group with a tetrazole increases the lipophilicity of the molecule and may potentially improve the in vivo bioavailability.34 The ClogP values for 2, 4, and vigabatrin were calculated to give an estimate of their lipophilicity. The ClogP for 4 (−0.48) indicated that it is approximately 69 times more lipophilic than 2 (−2.32) and 55 times more lipophilic than vigabatrin (−2.22). The considerably increased lipophilicity may result in improved permeability of the blood-brain barrier, and therefore, may compensate for the decrease in in vitro activity.
In summary, two analogues of 2, a potent inactivator of GABA-AT, were synthesized. Dichloro analogue 3 is only a weak reversible inhibitor of GABA-AT, which, presumably, is the result of the larger size of the chlorine atoms. Tetrazole analogue 4 was shown to be a time- and concentration-dependent inhibitor of GABA-AT. It is less potent than parent compound 2, although its in vitro potency is higher than that for the antiepilepsy drug vigabatrin (1). The increased lipophilicity of 4 relative to 2 and to vigabatrin makes it desirable.
Supplementary Material
Acknowledgments
The authors are grateful to the National Institutes of Health (GM 66132) for financial support of this research.
Footnotes
Supplementary data
Experimental details and spectroscopic data for compounds 3, 4, 7–13, enzymatic test for compound 4.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Perry TL, Hansen S, Lesk D, Kloster M. Adv Neurol. 1972;1:609. [Google Scholar]
- 2.Wu JY, Bird ED, Chen MS, Huang WM. Neurochem Res. 1979;4:575. doi: 10.1007/BF00964435. [DOI] [PubMed] [Google Scholar]
- 3.McGeer PL, McGeer EG. The GABA System and Function of the Basal Ganglia: Huntington’s Disease. Raven Press; New York: 1976. pp. 487–495. [Google Scholar]
- 4.Sherif FM, Ahmed SS. Clin Biochem. 1995;28:145. doi: 10.1016/0009-9120(94)00074-6. [DOI] [PubMed] [Google Scholar]
- 5.Lloyd KG, Munari C, Bossi L, Stoeffels C, Talairach J, Morseelli PL. Biochemical Evidence for the Alterations of GABA-mediated Synaptic Transmission in Pathological Brain Tissue from Epileptic Patients. Raven Press; New York: 1981. pp. 325–338. [Google Scholar]
- 6.Bakay RAE, Harris AB. Brain Res. 1981;206:387. doi: 10.1016/0006-8993(81)90539-4. [DOI] [PubMed] [Google Scholar]
- 7.Gale K. Epilepsia. 1989;30(Suppl 3):S1–11. doi: 10.1111/j.1528-1157.1989.tb05825.x. [DOI] [PubMed] [Google Scholar]
- 8.Lippert B, Metcalf BW, Jung MJ, Casara P. Eur J Biochem. 1977;74:441. doi: 10.1111/j.1432-1033.1977.tb11410.x. [DOI] [PubMed] [Google Scholar]
- 9.Gidal BE, Privitera MD, Sheth RD, Gilman JT. Annals Pharmacother. 1999;33(12):1277. doi: 10.1345/aph.18376. [DOI] [PubMed] [Google Scholar]
- 10.Arndt CF, Derambure P, Defoort-Dhellemmes S, Hache JC. Neurology. 1999;52:1201. doi: 10.1212/wnl.52.6.1201. [DOI] [PubMed] [Google Scholar]
- 11.Spence SJ, Sankar R. Drug Safety. 2001;24(5):385. doi: 10.2165/00002018-200124050-00005. [DOI] [PubMed] [Google Scholar]
- 12.Kalviainen R, Nousiainen I, Mantyjarvi M, Nikoskelainen E, Partanen J, Partanen K, Riekkinen P. Neurology. 1999;53:922. doi: 10.1212/wnl.53.5.922. [DOI] [PubMed] [Google Scholar]
- 13.Wild JM, Martinez C, Reinshagen G, Harding GF. 1999;40:1784. doi: 10.1111/j.1528-1157.1999.tb01599.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhao L-X, Park JG, Moon Y-S, Basnet A, Choi J, Kim E-k, Jeong TC, Jahng Y, Lee E-S. Il Farmaco. 2004;59(5):381. doi: 10.1016/j.farmac.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Qiu J, Pingsterhaus JM, Silverman RB. J Med Chem. 1999;42:4725. doi: 10.1021/jm990271o. [DOI] [PubMed] [Google Scholar]
- 16.Qiu J, Silverman RB. J Med Chem. 2000;43:706. doi: 10.1021/jm9904755. [DOI] [PubMed] [Google Scholar]
- 17.Choi S, Storici p, Schirmer T, Silverman RB. J Am Chem Soc. 2002;124:1620. doi: 10.1021/ja011968d. [DOI] [PubMed] [Google Scholar]
- 18.Pan Y, Qiu J, Silverman RB. J Med Chem. 2003;46:5292. doi: 10.1021/jm034162s. [DOI] [PubMed] [Google Scholar]
- 19.Koziara JM, Lockman PR, Allen DD, Mumper RJ. J Nanosci Nanotechnol. 2006;6(910):2712. doi: 10.1166/jnn.2006.441. [DOI] [PubMed] [Google Scholar]
- 20.Su Y, Sinko PJ. Exp Opin Drug Deliv. 2006;3(3):419. doi: 10.1517/17425247.3.3.419. [DOI] [PubMed] [Google Scholar]
- 21.Levin VA. J Med Chem. 1980;23:682. doi: 10.1021/jm00180a022. [DOI] [PubMed] [Google Scholar]
- 22.Young RC, Mitchell RC, Brown TH, Ganellin CR, Griffiths R, Jones M, Rana KK, Saunders D, Smith IR, Sore NE, et al. J Med Chem. 1988;31:656. doi: 10.1021/jm00398a028. [DOI] [PubMed] [Google Scholar]
- 23.Jezequel SG. Prog Drug Metab. 1992;13:141. [Google Scholar]
- 24.Chikhale EG, Ng KY, Burton PS, Borchardt RT. Pharm Res. 1994;11:412. doi: 10.1023/a:1018969222130. [DOI] [PubMed] [Google Scholar]
- 25.Atkinson F, Cole S, Green C, van de Waterbeemd H. Curr Med Chem CNS Agents. 2002;2:229. [Google Scholar]
- 26.Abraham MH. Eur J Med Chem. 2004;39:235. doi: 10.1016/j.ejmech.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 27.Bryan J. Pharm J. 2004;273:475. [Google Scholar]
- 28.Kraus JL, Faury P, Charvet AS, Camplo M. Res Commun Mol Pathol Pharmacol. 1994;83:209. [PubMed] [Google Scholar]
- 29.Yuan H, Silverman RB. Bioorg Med Chem. 2006;14:1331. doi: 10.1016/j.bmc.2005.09.067. [DOI] [PubMed] [Google Scholar]
- 30.Palmer CF, Parry KP, Roberts SM, Sik V. J Chem Soc, Perkin Trans 1. 1992;8:1021. [Google Scholar]
- 31.Carran J, Waschbuesch R, Marinetti A, Savignac P. Synthesis. 1996;12:1494. [Google Scholar]
- 32.Amantini D, Beleggia R, Fringuelli F, Pizzo F, Vaccaro L. J Org Chem. 2004;69:2896. doi: 10.1021/jo0499468. [DOI] [PubMed] [Google Scholar]
- 33.Kitz R, Wilson IB. J Biol Chem. 1962;237:3245. [PubMed] [Google Scholar]
- 34.Pardridge WM. J Neurovirol. 1999;5:556. doi: 10.3109/13550289909021285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.