

Abstract
During hematogenous cancer metastasis, tumor cells separate from a primary mass, enter the bloodstream, disperse throughout the body, migrate across vessel walls, and generate distant colonies. The later steps of metastasis superficially resemble leukocyte extravasation, a process initiated by selectin-mediated cell tethering to the blood vessel wall followed by integrin-mediated arrest and transendothelial migration. Some cancer cells express P-selectin ligands and attach to immobilized Pselectin, suggesting that these cells can arrest in blood vessels using sequential selectin- and integrin-mediated adhesion, as do leukocytes. We hypothesize that selectin binding may regulate subsequent integrin-mediated steps in metastasis. Using a model system of cultured Colo 320 human colon adenocarcinoma cells incubated with soluble P-selectin-IgG chimeric protein, we have found that P-selectin can stimulate activation of the α5β1 integrin resulting in a specific increase of adhesion and spreading of these cells on fibronectin substrates. P-selectin binding also induced activation of p38 mitogen-activated protein kinase (p38 MAPK) and phosphatidylinositol 3-kinase (PI3-K). PI3-K inhibitors blocked P-selectin-mediated integrin activation, cell attachment, and cell spreading. Inhibition of p38 MAPK activation blocked cell spreading, but not cell attachment. P-Selectin binding also resulted in formation of a signaling complex containing PI3-K and p38 MAPK. These results suggest that P-selectin binding to tumor cells can activate α5β1 integrin via PI3-K and p38 MAPK signaling pathways leading to increased cell adhesion. We propose that P-selectin ligands are important tumor cell signaling molecules that modulate integrin-mediated cell adhesion in the metastatic process.
INTRODUCTION
Tumor cell metastasis is a highly regulated and dynamic process in which cancer cells separate from a primary tumor, migrate across blood vessel walls into the bloodstream, and disperse throughout the body to generate colonies [1]. Hematogenous tumor cell dissemination depends on cell adhesive interactions that are mediated by adhesion receptors, such as selectins and integrins [2-5].
Integrins are heterodimeric, transmembrane, cell surface glycoproteins that act as receptors for extracellular matrix proteins or counter-receptors on other cells. Each integrin consists of an α subunit and a β subunit. So far, at least 18 different α subunits and eight different β integrin subunits have been identified. The combination of α and β subunits in the integrin dimers determines ligand specificity. Integrins are very highly regulated receptors that are subject to multifaceted regulatory pathways [6-10]. “Inside-out” signaling most commonly regulates the ability of integrins to bind ligands [11]. For example, protein kinase C (PKC) mediates activation of α5β1integrin [12] and phorbol esters activate αLβ2 integrin [13]. This increase in ligand binding capacity can be the result of either a change in integrin conformation or integrin clustering. The latter process increases apparent affinity as a result of multivalent cooperative binding [14].
Selectins are vascular cell-cell adhesion receptors that usually bind to certain sialyl Lewis A (sLeA)- and sialyl Lewis X (sLex)-containing mucin-type glycoproteins found on normal leukocytes and endothelium. Three members of the selectin family have been identified: L-selectin, E-selectin and P-selectin. L-Selectin is constitutively expressed on leukocytes. P-and E-selectins are expressed on activated endothelial cells. P-selectin is also expressed on thrombin-activated platelets [15]. All three members of the selectin family, E-, L-, and P-selectin, have been shown to recognize sLeA and sLeX expressed on carcinoma cell surfaces [16, 17].
Accumulation of leukocytes at sites of inflammation is initiated by selectins that mediate the capturing and rolling of leukocytes on endothelium. This step is followed by activation of leukocyte integrins, which mediate firm adhesion to the endothelial cell surface. The firmly adhering cells can then actively migrate through the endothelial cell barrier [18]. Several reports have suggested that selectins and integrins can play an important role in adhesion of tumor cells to microvascular endothelium and subsequent cell migration. Several aspects of tumor cell adhesion to the microvascular endothelial cells have been even proposed to be analogous to leukocyte recruitment [1, 5, 17, 19, 20].
E-selectin and P-selectin have been proposed as critical molecules for adhesion of some cancer cells [21, 22]. Moreover, the P-selectin ligand, CD24 (also called heat-stable antigen), has been detected on several human carcinomas [23-25], further suggesting that P-selectin could play a crucial role in metastasis. However, classical selectin-ligand binding plays a role primarily in transient adhesive processes due to a relatively high on-off rate and a requirement for less stable cell adhesion mechanisms necessary for cancer cell extravasation [26-28]. It has been suggested that changes in the affinity and avidity of integrin receptors may be required for stabilized tumor cell adhesion and subsequent cell migration into the host organ [1].
We have started to investigate a possible role of P-selectin in integrin-mediated adhesion of cultured human tumor cells. We have found that P-selectin binding to human adenocarcinoma Colo 320 cells increases their adhesion specifically to fibronectin through the α5β1 integrin. We found also that P-selectin binding to Colo 320 cells activates both p38 mitogen-activated protein kinase (p38 MAPK) and phosphotidylinositol 3-kinase (PI3-K), which form a signaling complex in a P-selectin depend manner. p38 MAPK and PI3-K are required in P-selectin-mediated cell spreading but only PI3-K is required in P-selectin-increased adhesion to fibronectin. These results suggest P-selectin ligands could be signaling molecules important in the regulation of tumor cell adhesion.
Experimental Procedures
Materials
P-selectin IgG-Fc fusion protein and E-selectin IgG-Fc fusion protein were obtained from BD Biosciences (San Jose, CA) and R&D Systems (Minneapolis, MN), respectively. P-selectin IgG-Fc fusion protein was also expressed in COS cells and purified as described [29] by the NIEHS Protein Expression Core Facility. The expression vector for the P-selectin IgG-Fc fusion protein was a generous gift from Dr. John Lowe, University of Michigan Medical Center. The recombinant E- and P-Selectin-IgG Fc are disulfide-linked homodimeric proteins, and were made by fusing the extracellular region portion of the respective selectin and the Fc portion of IgG1. These chimeric proteins have been broadly used for identifying selectin ligands and characterizing selectin biological activities [30-34]. Unlabeled control mouse IgG and human IgG-Fc were obtained from Accurate Chemical Co. (Westbury, NY). Blocking and non-blocking mAbs anti-human α5 integrin (mAb 16 and mAb 11, respectively) and blocking anti-human β1integrin, mAb 13, were characterized previously [35, 36]. Non-blocking mAb anti-human β1 integrin (K20), blocking mAbs anti-human α3 integrin (C3), and anti-human α4 integrin (HP2/1) were obtained from Immunotech (Westbrook, ME). The mAb specific for activated β1 integrin (HUTS-21) was from BD Biosciences. The p38 MAPK antibodies anti-SAPK2α/p38, anti-active p38, anti-phospho-p38 MAPK, and anti-PI3-K p85 rabbit antiserum were from Upstate Biologicals (Charlottesville, VA), Promega (Madison, WI), and Cell Signaling (Berverly, MA), respectively. The anti-PI3-K p110β rabbit polyclonal and monoclonal antibodies were obtained from Santa Cruz Biotech (Santa Cruz, CA). Anti-phosphotyrosine mAb, 4G10, was from Upstate Biologicals. The PI3-K inhibitors wortmannin and LY294002 and the specific p38 MAPK inhibitors SB203580 and SB202190 were obtained from Calbiochem (San Diego, CA). R-Phycoerythrin (PE)-conjugated Affinipure F(ab')2 fragment of goat Fc-specific anti-human IgG was from Immunotech. Alexa™ 488-coupled and Alexa™ 564-coupled goat anti-mouse IgG and anti-rabbit IgG were obtained from Molecular Probes (Eugene, OR). Fibronectin was purified from out dated, fresh-frozen human plasma as described [37, 38]. Collagen type I, collagen type IV and laminin were obtained from BD Biosciences. Gelatin and Triton X-100 were obtained from Sigma (Saint Louis, MO). Paraformaldehyde was obtained from Electron Microscopy Sciences (Ft. Washington, PA). Goat serum was obtained from Zymed Laboratories, Inc. (South San Francisco, CA). Dimethylsulfoxide (DMSO) was obtained from the American Type Culture Collection (Manassas, VA).
Cell Culture and Treatment
Colo 320 HSR human colon adenocarcinoma cells were obtained from the American Type Culture Collection (Manassas, VA) and were cultured in RPMI 1640 medium (Gibco-BRL, Grand Island, NY) with 10% FCS (Hyclone, Logan, UT), 50 u/ml penicillin and 50 μg/ml streptomycin. Subconfluent cells were harvested with 5 mM EDTA, washed twice with serum-free medium supplemented with 25 mM HEPES, resupended and incubated at 37°C in 5% CO2 for 60 min before being incubated with P-selectin-IgG Fc. Inhibitors were dissolved in DMSO and added to the cells for either 30 min (genestein, SB203580, SB202190, and wortmannin) or 60 min (LY294002) prior to treatment with P-selectin. Cells for biochemical analyses were lysed in 150 mM NaCl, 2 mM EDTA, 50 mM Hepes, 0.5% deoxycholic acid, 1% Nonidet P-40, pH 7.5 containing 2 mM sodium orthovanadate, 1 mM sodium fluoride, 2 mM phenylmethylsulfonyl fluoride, 15 μg/ml aprotinin A, 15 μg/ml leupeptin, and 15 μg/ml pepstatin (lysis buffer) for 20 min at 4°C and then cleared by centrifugation at 16,000Xg for 10 min at 4°C. All protein concentrations were determined using the BCA assay (Pierce, Rockford, IL).
Flow Cytometric Assays
Cells used for flow cytometric analysis were washed twice with ice-cold PBS, then resuspended in PBS supplemented with 1 mM CaCl2, 1 mM MgCl2, and 3% heat-denatured BSA (PBS+/BSA) at a concentration of 107 cells/ml. Aliquots (200 μl) of cell suspension were incubated with 10 μg/ml non-immune mouse IgG or a monoclonal anti-β1 integrin (K20 or HUTS-21) for 45 min at 4°C. For P-selectin cell surface-binding assays, cells were incubated with 10 μg/ml human IgG-Fc or different concentrations of P-selectin-IgG-Fc. After three washes with ice-cold PBS+/1% BSA, cells were incubated with FITC-conjugated F(ab)2 anti-mouse IgG or anti-human IgG-Fc for 30 min at 4°C. After three washes, each aliquot of cells was resuspended in 0.5 ml of PBS for immediate analysis by flow cytometry using a FACScan (BD Biosciences, Mountain View, CA).
Adhesion and Cell spreading Assays
Adhesion assays were carried out essentially as described [39]. Briefly, 96-well tissue culture clusters (Costar, Corning, NY) were coated with 5 μg/ml human fibronectin, human collagen type I, human collagen type IV, or laminin and then blocked with 30 mg/ml heat-denatured BSA (Calbiochem, San Diego, CA) for at least 2 h. Wells were preloaded with 50 μl of RPMI with or without P-selectin-IgG-Fc or control human IgG-Fc. Colo 320 HSR cells were harvested and resuspended to a density of 5×105 cells/ml in RPMI supplemented with 15 mM HEPES. Aliquots (50 μl) of cell suspension were added to each well and incubated at 37°C. After washing away unattached cells, attached cells were fixed and stained with 0.05% (w/v) crystal violet (Sigma). Values of 100%, 50% and 25% cell attachment were estimated by adding 50 μl, 25 μl, or 12.5 μl of cell suspension to wells coated with 10 μg/ml poly- D lysine and then fixed without washing, and stained. Dye solubilized with 100 μl 10% acetic acid was quantified at 575 nm using a Spectra MAX 250 ELISA reader (Molecular Devices, Sunnyvale, CA). All statistical tests were performed using Analysis of Variance based methodology using PROC GLM in the statistical software package SAS (Copyright (c) 1999-2001 by SAS Institute Inc., Cary, NC, USA).
Cell spreading assays were performed essentially as described [40]. Briefly, 24-well tissue plates (Costar) were coated with 5 μg/ml fibronectin and blocked with heat-denatured BSA as described for cell attachment assays, and pre-loaded with 150 μl of RPMI with or without P- or E-selectin-IgG-Fc, or human IgG-Fc. Aliquots (150 μl) of cell suspension at a density of 3 × 105 cells/ml were added and incubated at 37°C for 3 h. The cells were then fixed immediately and spread cells counted using phase contrast microscopy.
Immunoblotting
Whole cell lysates or immunoprecipitates were resolved by 8% or 10% SDS-Tris polyacrylamide gel electrophoresis and then electrotransferred onto polyvinylidine fluoride (PVDF) membranes (Millipore, Bedford, MA) in Tris-glycine buffer containing 20% methanol. Proteins were detected by immunoblotting as described [41]. In some cases, PVDF membranes were stripped of bound antibodies using 62.5 mM Tris-HCl, pH 6.7, 100 mM 2-mercaptoethanol, 2% SDS for 30 min 60°C and then reprobed as described in figure legends.
In vitro assays for PI3-K and p38 MAPK activity
Activity of p38 MAPK was determined using a commercially available kit for kinase activity (Cell Signaling). In vitro assays for PI3-K were performed essentially as described [41]. Briefly, PI3-K was immunoprecipitated by incubating with 1 μg of anti-PI3-K p85 antibody and protein-A agarose. Immunoprecipitates were washed twice with ice-cold PBS containing 1% NP-40 and 1 mM sodium orthovanadate, twice with cold 100 mM Tris pH 7.4, 5 mM LiCl, and twice with cold TNE buffer (10 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA and 10 mM sodium orthovanadate), then resuspended in 70 μl of kinase assay buffer (10 mM Tris pH 7.4, 150 mM NaCl, 5 mM EDTA, 10 mM MgCl2 and 0.88 mM ATP), containing 15 μCi of [γ-32P] (3000 Ci/mmol; Amersham Pharmacia Biotech, Piscataway, NJ) and 10 μg of phosphatidylinositol (Avanti Polar-Lipids, Alabaster, AL). The mixture was incubated at 37°C for 30 min and the reaction stopped by adding 20 μl of 6 N HCl. Lipids were extracted by adding 200 μl of CHCl3: CH3OH (1:1 v/v), mixing vigorously, and then centrifuging at 14,000Xg for 5 min in a microcentrifuge. The lower organic phase (50 μl) was applied to an axylated silica gel plate (Sigma) and lipids were resolved by thin-layer chromatography using a solvent mixture of CHCl3: CH3OH: H2O: NH4OH (60:47:11.3:2 v/v). Phosphorylated lipids were detected by autoradiography. To evaluate the amount of protein immunoprecipitated, an aliquot of the sample was separated and immunoblotted with anti-PI3-K antibodies.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed with Colo 320 HSR cells on poly-D-lysine- or fibronectin-coated 18-mm diameter glass cover slips in the presence of serum-free medium with or without P-selectin. Cells were fixed in 3% paraformaldehyde in PBS containing 1 mM CaCl2 and 1 mM MgCl2 (PBS+) for 30 min at room temperature, permeabilized for 4 min in PBS+ containing 0.1% Triton X-100, and washed three times with PBS+. Nonspecific binding sites were blocked for 30 min with 0.5% gelatin and 1% goat serum in PBS+, and the fixed cells were incubated for 60 min with primary antibody. Cells were washed three times and incubated with the appropriate secondary antibody for 45 min. All washes and antibody dilutions were carried out using PBS+ containing 0.1% gelatin and 1% goat serum. After washing, the cover slips were mounted on glass slides with ProLong Antifade (Molecular Probes) according to the manufacturer's directions to inhibit photobleaching. Immunofluorescence was documented with a LSM 410 inverted confocal laser-scanning microscope (Carl Zeiss, Oberkochen, Germany) equipped with an Omnichrome argon-krypton laser. Images were obtained with a Zeiss Plan-Apo 100X oil immersion objective (1.4 NA).
Results
P-Selectin induces an increase in cell adhesion to fibronectin through activation of α5β1 integrin
In a previous study, it was shown that human colon carcinoma cells can attach to immobilized P-selectin substrates, and some tumor cells, such as Colo 320 cells, attach preferentially to P-selectin, rather than to E-selectin [16, 42]. To test whether the Colo 320 cells have the ability to bind to soluble P-selectin, these cells were incubated with different concentrations a soluble recombinant P-selectin-IgG-Fc fusion protein. P-selectin-IgG-Fc, but not human IgG-Fc or E-selectin-IgG-Fc, bound to Colo 320 cells as showed by flow cytometry (Fig. 1A and B). Maximal staining was observed at ∼15 μg/ml P-selectin. Similarly, immunofluorescence confocal microscopy clearly indicated that P-selectin chimera bound at the plasma membrane of cells (Fig. 1C and D). These observations suggest that P-selectin can bind to the surface of Colo 320 cells in a specific and saturable manner.
Figure 1.
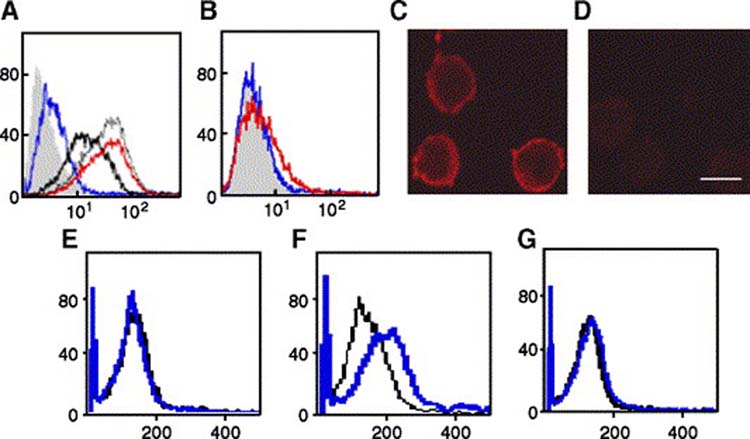
P-Selectin binding induces β1 integrin activation. (Panel A) Colo 320 cells were incubated with 5 μg/ml (blue line), 10 μg/ml (thick black line), 15 μg/ml (thin gray line), or 20 μg/ml (red line) P-Selectin-IgG Fc or with 20 μg/ml human IgG-Fc (solid gray histogram), followed by FITC-conjugated anti-human IgG-Fc F(ab)2, and binding was assessed by flow cytometry. (Panel B) Colo 320 cells were incubated with 5 μg/ml (blue line) or 20 μg/ml (red line) E-selectin-IgG Fc, or with 20 μg/ml human IgG-Fc (solid gray histogram), followed by FITC-conjugated anti-human IgG-Fc F(ab)2, and binding was assessed by flow cytometry. (Panels C and D) Colo 320 cells on poly-lysine coated cover slips were incubated with P-selectin-IgG-Fc (C) or human IgG Fc (D), which was then visualized by indirect immunoflourescence. Bar, 5 μm. (Panels E and F) Colo 320 cells were stimulated in suspension with 10 μg/ml P-selectin-IgG Fc (blue line) or left unstimulated (black line) for 5 min(E) or 30 min (F). After quenching with ice-cold PBS, cells were incubated with R-PE-conjugated mouse anti-activated β1 integrin mAb HUTS-21 mAb, and analyzed by flow cytometry.
Previous studies suggested that P-selectin could mediate the initial attachment between tumor cells and microvascular endothelial cells [42-44]. In leukocytes, after selectin-mediated cell tethering to the blood vessel wall, integrin involvement is required to produce the stable adhesion for their extravasation [18]. Therefore, we tested the hypothesis that P-selectin binding could induce activation of β1 integrin, a mechanism by which increased cell adhesion may occur through a change in integrin conformation increasing its ligand affinity or/and through integrin clustering [11, 45, 46]. The ability of P-selectin to induce a conformational change resulting in β1-integrin activation was analyzed by flow cytometry using two different anti-β1 integrin mAbs: HUTS-21, which recognizes the activation-dependent epitope of β1 integrin [47] and K20 which reacts with total β1 integrin. Only cells stimulated with P-selectin expressed the HUTS-21 epitope after 30 min stimulation (Fig. 1E and F) without changing the level of total cell surface β1 integrin expression (not shown). To test whether P-selectin binding induced β1 integrin activation by clustering, cells stained with HUTS-21 or K20 antibodies were also analyzed by confocal microscopy. The integrins on P-selectin-stimulated cells did not appear to be appreciably clustered (data not shown). We also observed that the staining with HUTS-21 mAb on P-selectin-stimulated cells detected only a fraction of total β1 integrin (not shown). These results suggest that P-selectin binding induces β1 integrin activation of a subpopulation of cell surface β1 integrins probably by inducing a conformational change without appreciable integrin clustering or any obvious increased expression of total β1 integrin.
To test if the β1 integrin conformational change induced by P-selectin resulted in functional changes, and to identify the specific α-containing integrin heterodimers activated by P-selectin binding, we investigated the effect of P-selectin-IgG-Fc on Colo 320 cell adhesion to different adhesive proteins that function as β1 integrin ligands. As shown in Figure 2A, P-selectin binding to these cells stimulated a significant (P<0.0001) increase of the adhesion of Colo 320 cells specifically to fibronectin without affecting adhesion to collagen type I, collagen type IV, or laminin. This increase was both time- and dose-dependent (data not shown). The β1 integrin subfamily members that most commonly function as fibronectin receptors are α3β1, α4β1, and α5β1 [9]. To identify which of these integrins are responsible for P-selectin-induced adhesion, we assessed the effect of α-integrin specific inhibitory monoclonal antibodies on Colo 320 adhesion to fibronectin (Fig. 2B). P-Selectin-induced adhesion was completely abrogated when the cells were pre-treated with function blocking mAbs to β1 or α5 integrin (P<0.0001) whereas blocking mAbs to α3 or α4 integrin did not show a significant decrease (P<0.604 and P<0.02, respectively). These results suggest that the P-selectin-induced adhesion to fibronectin is primarily through activation of the α5β1 integrin.
Figure 2.
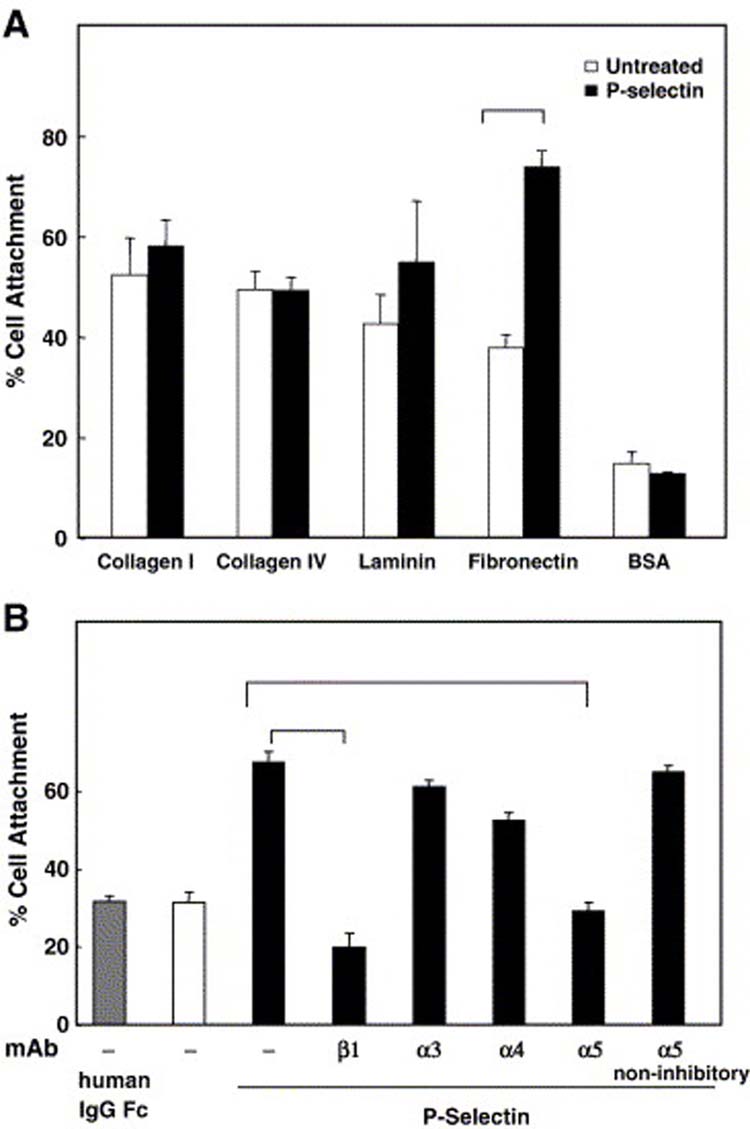
P-Selectin binding increases cell adhesion to fibronectin throughα5β1 integrin.(A) Colo 320 cells were allowed to attach to substrates prepared with the indicated β1 integrin ligands or BSA for 90 min in the absence (white bars) or presence (black bars) of 10 μg/ml P-selectin-IgG Fc. (B) P-selectin stimulated cells were pretreated with anti-β1 integrin (β1), anti-α3 integrin (α3), anti-α4 integrin (α4) or anti-α5 integrin (α5) inhibitory antibodies or anti-α5 integrin non-inhibitory antibody for 45 min at 4°C before incubating on immobilized fibronectin. Data are representative of three independent experiments; error bars indicate standard deviation. The data were analyzed by Student's t-test. There was a statistically significant increase between Pselectin stimulated cells and unstimulated cells (p ≤0.0001). P-selectin stimulated cells show a significant decrease when were pretreated with anti-β1 integrin (p ≤−7) or anti-α5 integrin (α5) inhibitory (p ≤10−5) antibodies.
P-Selectin binding induces p38 MAPK and PI3-K activation
The results that P-selectin induced β1 integrin activation suggested that an intracellular signaling process might be involved. Therefore, we examined the possibility that P-selectin binding leads to changes in the tyrosine phosphorylation state of proteins. Anti-phosphotyrosine immunoblotting of whole cell lysates from Colo 320 cells stimulated with P-selectin-IgG-Fc for different times at 37°C showed a marked, but transient, increase in tyrosine phosphorylation of several proteins, including one broad band centered around ∼40 kDa (Fig. 3A). The increase in tyrosine phosphorylation was readily detected within 3 min of P-selectin stimulation, reached a maximal level at 10 min, then declined.
Figure 3.
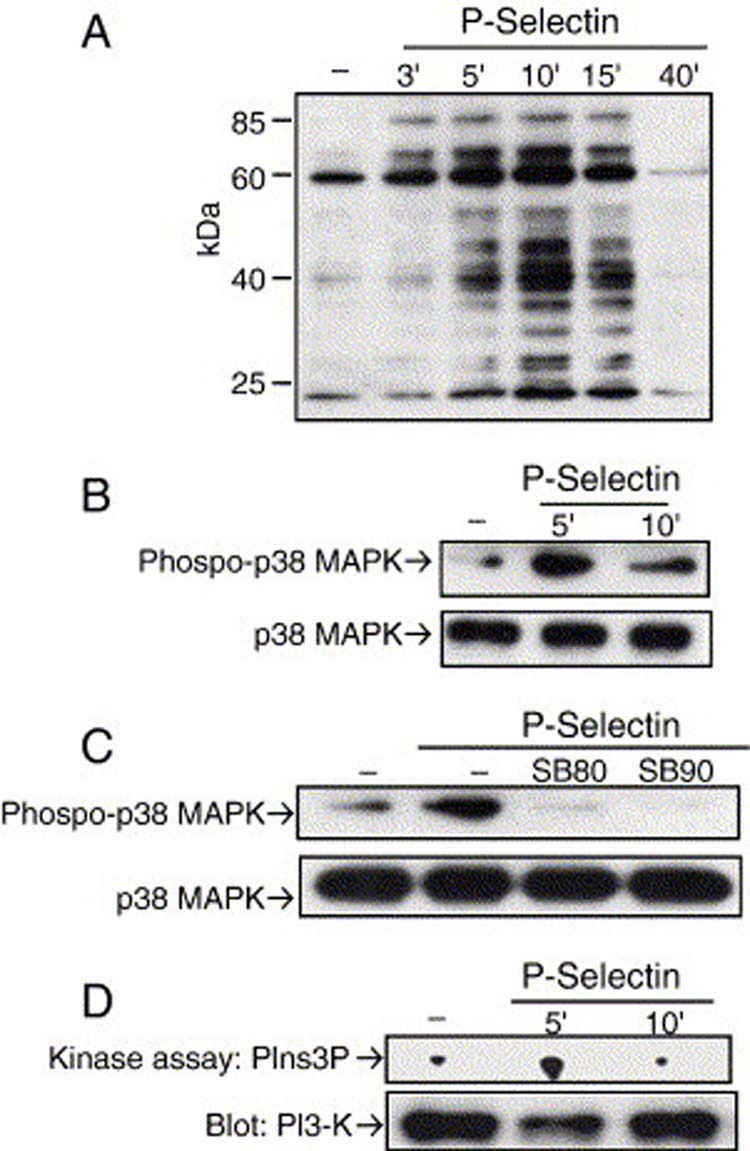
P-Selectin binding activates p38 MAPK and PI3-K. Colo 320 cells were stimulated in suspension with 10 μg/ml P-selectin-IgG Fc and lysed at the indicated times. Proteins from whole cell lysates or p38 MAPK immunoprecipitates were analyzed by immunoblotting with (A) anti-phosphotyrosine or (B) anti-phospho-p38 MAPK. In panel B, the same membranes were probed for total p38 MAPK (lower panels) after stripping bound antibodies. (C) Cells were pretreated with or without one of two specific p38 MAPK inhibitors, 10 μM SB203580 (SB80) or 5 μM SB202190 (SB90), for 30 min at 37°C prior to P-selectin stimulation. p38 MAPK immunoprecipitates from cell lysates were analyzed by immunoblotting for activated p38 MAPK. After stripping bound antibodies, the same membranes were reprobed for total p38 MAPK (lower panels). (D) PI3-K activity in immune complexes from cell lysates was measured by an in vitro kinase assay. The lower panel is an immunoblot of PI3-K that shows the same amount of protein was immunoprecipitated with each complex. Data are representative of three separate experiments. PIns3P, phosphatidylinositol 3-phosphate.
Because p38 MAPK has been implicated in arachidonic acid-regulated adhesion of breast cancer cells to collagen type IV [48] and the band of approximately 40 kDa that showed a marked increase in tyrosine phosphorylation could be p38 MAPK, we decided to look directly at the activation of this kinase in response to P-selectin binding. Colo 320 cells showed a marked increase of p38 MAPK activation after 5 min of P-selectin stimulation (Fig. 3B). This activation was completely blocked with two specific p38 MAPK inhibitors (Fig. 3C). p42/44 MAPK , another protein that might be the band of 40 kDa, was not found to be activated in response P-selectin stimulation (data not shown).
Recent reports have shown that PI3-K can also play a crucial role in up regulation of integrin activity [49-51]. Therefore, we used an in vitro kinase assay to examine whether P-selectin could induce PI3-K activation. We observed an increase in PI3-K activation after 5 min of P-selectin stimulation (Fig. 3D). PI3-K activation was completely blocked when the Colo 320 cells were pretreated with LY294902, a specific PI3-K inhibitor (Fig. 6A). These results support our hypothesis that P-selectin engagement to Colo 320 cells induces signal transduction pathways that could result in integrin activation.
Figure 6.
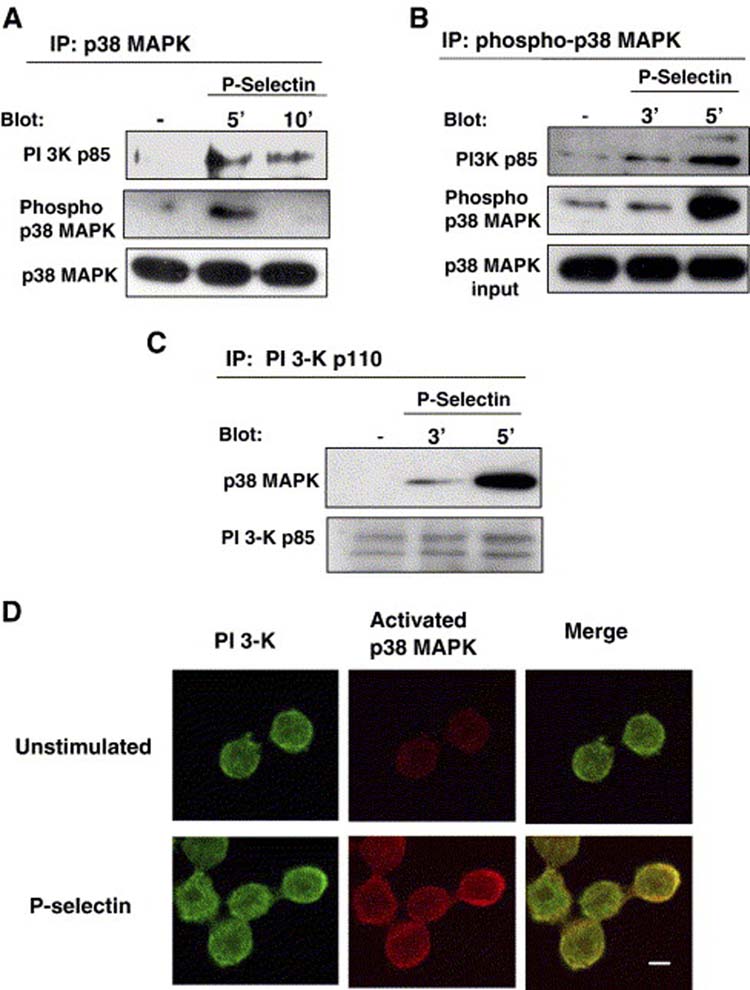
PI 3-K and p38 MAPK co-immunoprecipate and co-localize in a P-selectin dependent manner. Colo-320 cells were either left untreated (−) or stimulated with P-selectin-IgG Fc in suspension and lysed at the indicated times. p38 MAPK, phospho-p38 MAPK, or PI3-K were immunoprecitated. (A) Immunoprecitated total p38 MAPK was analyzed by immunoblotting with anti-PI 3-K p85. The same membrane was then stripped and reprobed with anti-activated p38 MAPK or anti-total p38 MAPK. (B) Immunoprecipitated phospho-p38 MAPK was analyzed by immunoblotting with anti-PI3-K p85. After the stripping, the same membrane was probed for phospho-p38 MAPK. The lowest panel depicts a blot for total p38 MAPK in an aliquot of total cell lysates taken before phospho p38 MAPK was immunoprecipitated. (C) Immunoprecipitated PI3-K p110 was analyzed by immonoblotting with anti-p38 MAPK. The stripped membrane was reprobed with anti-PI 3K p110. Data are representative of two separate experiments. (D) Colo-320 cells were plated on fibronectin coated cover slips in the presence or absence of P-selectin-IgG-Fc and incubated for 5 min at 37°C. After stimulation, the distribution of PI 3-K and activated p38 MAPK was detected with primary antibodies against anti-phospho p38 MAPK and anti-PI3-K, respectively. Anti-activated p38 MAPK was localized with Alexa 565 goat anti-rabbit IgG Fc (red) and PI3-K was localized with Alexa 488 goat anti-mouse IgG Fc (green). Fluorescent images were overlayed to determine colocalization as indicated by the yellow color. Bar, 5 μM.
P-Selectin-induced β1 integrin activation depends on PI3-K, but not p38 MAPK activation
The results above show that P-selectin can activate two specific kinases: p38 MAPK and PI3-K. To test whether these signaling molecules participate in P-selectin-induced β1 integrin activation, we assessed the effect of specific PI3-K and p38 MAPK inhibitors on cell attachment to fibronectin and binding of HUTS-21 antibody (specific for activated β1 integrin) induced by P-selectin. The PI3-K specific inhibitor, LY294002, blocked both P-selectin-induced cell adhesion and cell binding to HUTS-21 (Fig. 4 A and B). However, specific p38 MAPK inhibitors had no affect on P-selectin-induced HUTS-21 epitope expression or cell attachment to fibronectin (Fig. 4C and D). These data suggest that PI3-K is required for P-selectin dependent activation of β1 integrin activation, and that P-selectin-mediated p38 MAPK activation might participate in events subsequent to β1 integrin activation.
Figure 4.
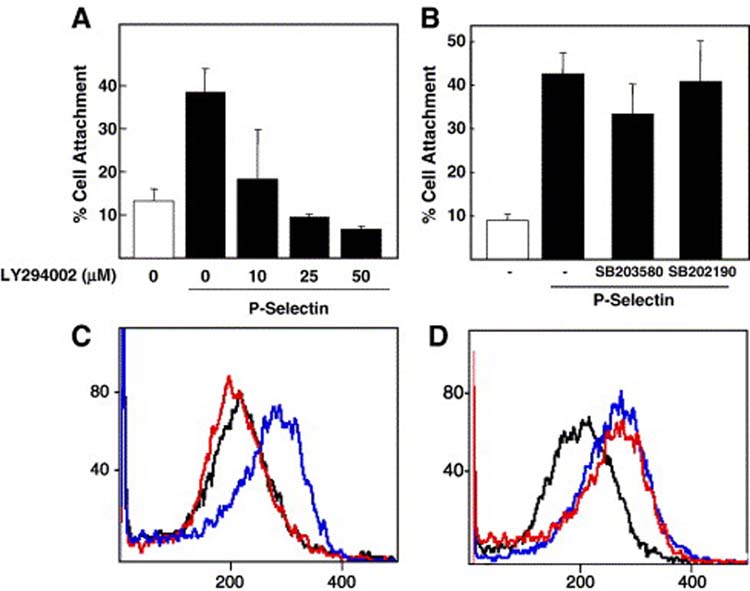
PI3-K, but not p38 MAPK, is required for P-selectin-induced β1 integrin activation. Colo 320 human carcinoma cells were allowed to attach to fibronectin substrates for 90 min at 37°C in the absence or presence of 10 μg/ml P-selectin. Cells were pretreated with the indicated concentrations of (A) PI 3-K inhibitor, LY294002 (LY) for 60 min or (B) 10 μM SB203580 (SB80) or 5 μM SB202190 (SB90) for 30 min before plating on immobilized fibronectin. Error bars indicate standard deviation for triplicate assays. Cells were stimulated in suspension with 10 μg/ml P-selectin-IgG Fc (blue line) or left unstimulated (black line). Before P-selectin stimulation, cells were pretreated with vehicle or with (C) PI3-K inhibitor (red line), or (D) p38 MAPK inhibitor 5 μM SB202190 for 5 min (red line). After stopping the stimulation with ice-cold PBS, cells were incubated with R-PE-conjugated mouse anti-activatedβ1 integrin mAb HUTS-21 mAb and analyzed by flow cytometry.
P-selectin binding induces cell spreading through PI3-K and p38 MAPK
Studies in vivo of metastasis by video microscopy have shown that after entering blood vessels, cancer cells adhere to and spread along the vessel walls before extravasating into the organ parenchyma to proliferate and establish a micrometastasis [52-54]. We therefore examined whether P-selectin could also regulate cell spreading on fibronectin. As shown in Fig. 5A, P-selectin binding stimulated approximately a twofold increase of the number of cells spread on fibronectin. In control experiments, neither E-selectin nor human-IgG-Fc increased cell spreading (Fig. 5A). We then examined whether p38 MAPK or PI3-K was required in P-selectin-mediated increased cell spreading. Both p38 MAPK and PI3-K inhibitors blocked the cell spreading induced by P-selectin stimulation (Fig. 5B). These results suggested that both PI3-K and p38 MAPK are required for P-selectin-activated cell spreading.
Figure 5.
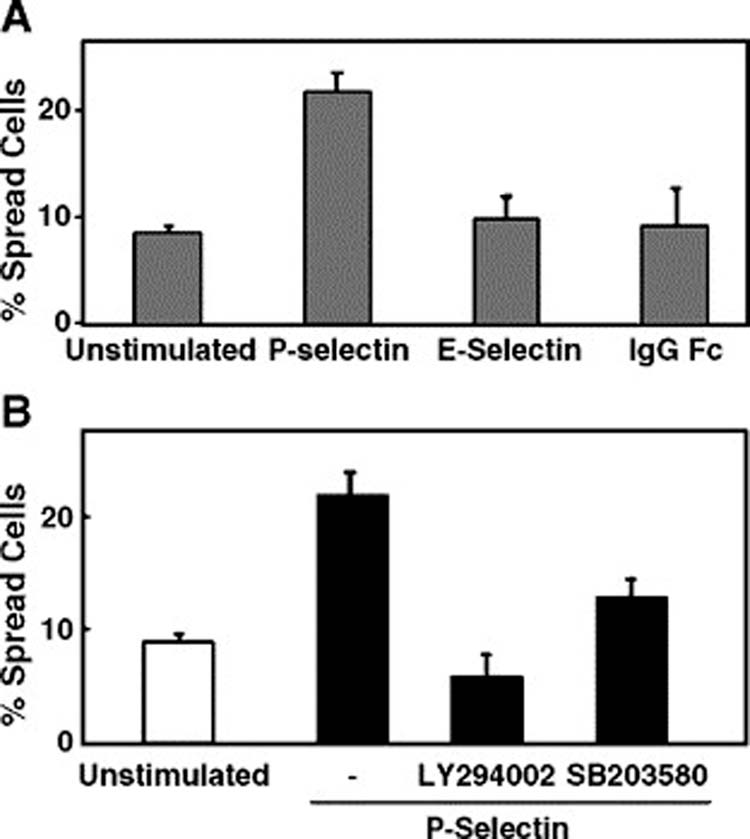
P-Selectin binding induces cell spreading through PI3-K and p38 MAPK. (A) Colo 320 cells were allowed to spread on fibronectin for 3 h in the absence or presence of 10 μg/ml P-selectin-IgG Fc, E-selectin-IgG Fc or human IgG Fc. (B) Cells were pretreated with 25 μM LY294009 for 60 min, or 10 μM SB203580 for 30 min, or the solvent alone, before incubation on immobilized fibronectin. Data are representative of one of three independent experiments; error bars indicate standard deviation.
p38 MAPK and PI3-Kinase form a complex following P-selectin stimulation
In order to identify a mechanistic connection between the p38 MAPK and PI3-K signaling pathways activated by P-selectin binding, we investigated whether these molecules could be co-immunoprecipited as result of P-selectin binding to Colo 320 cells. The p38 MAPK was immunoprecipitated from cell lysates using two different antibodies, anti-p38 MAPK (Fig. 6A) and anti-phospho p38 MAPK (Fig. 6B), and the resulting precipitates were immunoblotted with antibodies that recognize the p85 subunit of PI3-K. Prior to P-selectin binding, PI3-K was not detected in p38 MAPK immunoprecipitates (Fig. 6A and 6B). However, after P-selectin treatment, PI3-K was identified in the material immunoprecipitated from cell lysates with p38 MAPK or phospho-p38 MAPK. Similarly, the p110 subunit of PI3-K was immunoprecipitated from cell lysates, and the resulting precipitates were immunoblotted with anti-p38 MAPK antibodies. Prior to P-selectin binding, p38 MAPK was not detected in PI3-K immunoprecipitates (Fig. 6C). However, after P-selectin treatment, p38 MAPK was identified in the material immunoprecipitated from cell lysates with PI3-K. (Fig. 6C). These results suggest that p38 MAPK and PI3-K can form a complex, but only after cells are treated with P-selectin. To support this idea, we examined whether PI3-K and p38 MAPK co-localizated in intact cells, as detected by confocal microscopy. As shown in figure 6, we observed that PI3-K and activated p38 MAPK were co-localizated only in the cells stimulated with P-selectin (Fig. 6D). Thus, PI3-K and p38 MAPK appear to form a signaling complex that is induced by P-selectin binding in Colo 320 cells.
P-selectin-induced p38 MAPK activation is independent of PI3-K
Our results suggest that PI3-K, but not p38 MAPK, is required for P-selectin-induced β1 integrin activation, but both PI3-K and p38 are required for cell spreading and form a signaling complex. One simple interpretation of these results is that p38 MAPK could be downstream from PI3-K in the P-selectin-activated signaling pathway, suggesting the hypothesis that PI3-K activation would not be affected by inhibition of p38 MAPK. To test this idea, we investigated the effects of the general tyrosine kinase inhibitor, genestein, and the two specific p38 MAPK inhibitors, SB203580 and SB202190, on the activity of PI3-K after P-selectin binding. We found that PI3-K activity was inhibited by genestein, but not by either of two different p38 MAPK inhibitors (Fig. 7A). To test whether PI3-K activity was required for P-selectin-induced p38 MAPK activation, we evaluated the effect of the specific PI3-K inhibitor, LY294002, on the activity of p38 MAPK after P-selectin binding. p38 MAPK immunoprecipitates from Colo 320 cells were assessed by immunoblot anti-phospho-p38 MAPK (Fig. 7B) and an in vitro kinase assay (Fig. 7C). The specific PI3-K inhibitor did not block P-selectin-mediated p38 MAPK phosphorylation, but rather, may have induced an increase p38 MAPK activation (Fig. 7B and C). Although the mechanism of this increased p38 MAPK is unknown, these results suggest that PI3-K activation is not required for P-selectin-induced p38 MAPK phosphorylation.
Figure 7.
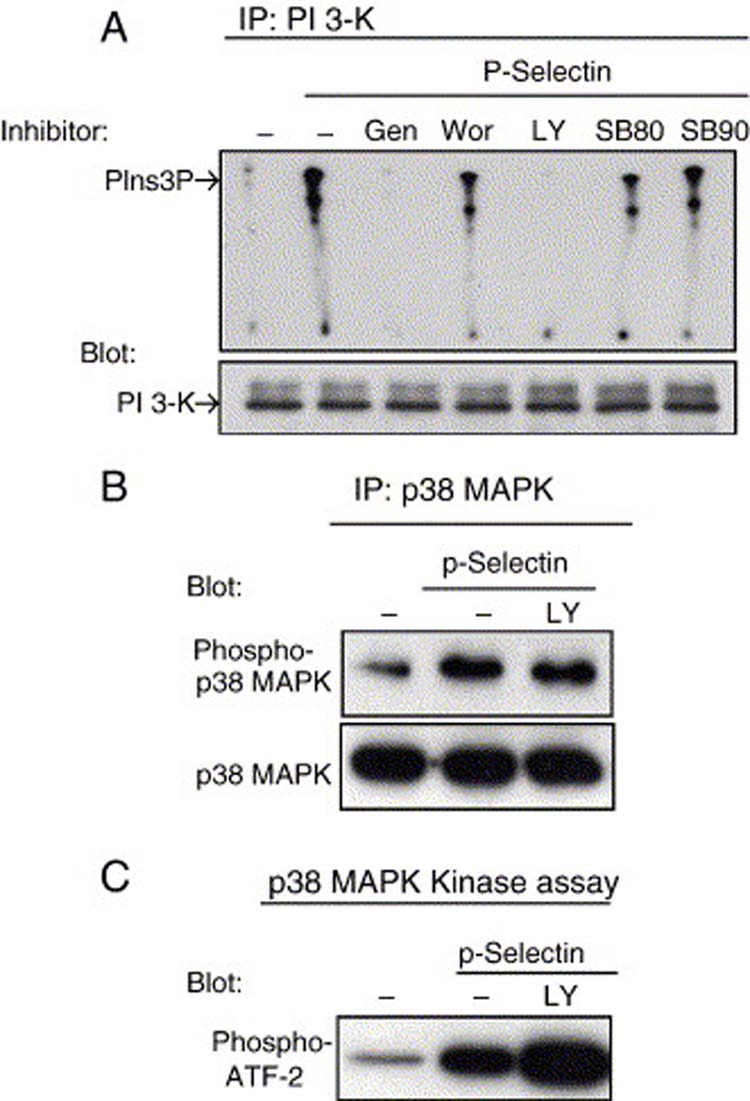
P-Selectin-induced PI3-K and p38 MAPK activation are independent events. Colo 320 cells were left unstimulated or stimulated with 10 μg/ml P-selectin-IgG Fc in suspension for 5 min and then lysed. Prior to stimulation, some cells were pretreated with 100 μM genestein (Gen) for 60 min, 25 nM wortmannin (Wor) for 30 min, 25 μM LY294009 (LY) for 60 min, 10 μM SB203580 (SB80), 5 μM SB202190 (SB90) for 30 min, or with the solvent alone. (A) PI3-K activity was then analyzed using in vitro kinase assays. The lower panel is a immunoblot of PI3-K showing equal amounts immunoprecipitated in each complex. PIns3P, phosphatidylinositol 3-phosphate. (B) Total p38 MAPK was immunoprecipitated from cell lysates, resolved by 10% SDS-PAGE and then immunoblotted for anti-active p38 MAPK. After stripping bound antibodies, the same membrane was reprobed with antibody that recognizes total p38 MAPK (lower panel). (C) Activated p38 MAPK was immunoprecipitated from cell lysates and analyzed by kinase assay in vitro, using ATF-2 as the substrate. Phosphorylated ATF-2 was analyzed by immunoblotting with anti-phosphorylated ATF-2 (p-ATF-2). Data are representative of two separate experiments.
DISCUSSION
In this study, we have used an in vitro model system consisting of cultured Colo 320 human carcinoma cells incubated with soluble P-selectin-IgG chimeric protein to characterize the regulation by selectins of integrin function in cancer cells. We have shown that P-selectin binding to Colo 320 cells can specifically activate the α5β1 integrin, which results in increased cell attachment and cell spreading specifically on fibronectin in a concentration- and time-dependent manner. This selectin-mediated integrin activation results from at least two distinct intracellular signaling pathways—the p38 MAPK pathway and the PI3-K pathway—that are linked by a p38 MAPK-PI3-K signaling complex.
During the hematogenous phase of cancer metastasis, cells that reach the blood stream must eventually gain the ability to attach firmly to the blood vessel endothelium prior to transendothelial migration and colonization of host organs [1, 54]. The selectins and integrins have both been shown to play roles in this process. Selectins have been suggested to participate in the initial tethering of circulating tumor cells to host organ endothelium, probably due to abnormal glycosylation of tumor cell surface proteins can result in expression of carbohydrate moieties that mimic ligands for selectins [55, 56]. We observed that Colo 320 cells express at least two glycoproteins reported to function as P-selectin ligands -- CD24 and CD44 (Reyes and Akiyama, unpublished data), but we have not yet determined the role of these molecules as P-selectin ligands.
All three members of the selectin family have been shown to bind to certain transformed human cells [4, 16, 57-61]. E-Selectin has been the most intensely studied because it was the first member of this family found to mediate adhesion of colon cancer cells to vascular endothelium. The role of L-selectin in the metastatic process has remained less clear. Current experimental evidence suggests that P-selectin can promote initial attachment of cancer cells to the blood vessel endothelium and can mediate the interactions between cancer cells and platelets to form emboli, both of which facilitate the arrest of cancer cells [2, 4, 17, 24, 42, 62].
We present several lines of evidence suggesting that P-selectin may specifically activate the α5β1 integrin on Colo 320 cells. Binding of P-selectin induced the expression of an activation-dependent epitope of β1 integrin, as judged by increased binding of the activation-specific HUTS-21 mAb (40), but did not appear to affect total β1 integrin expression on the cell surface. Similarly, we found no evidence for integrin clustering (Reyes, Styslinger, and Akiyama, unpublished data) nor can we observe any direct interaction of integrins with P-selectin (Reyes and Akiyama, unpublished data). Thus, we propose that selectin-induced integrin activation occurs by a mechanism involving a conformational change of β1 integrins in the absence of observable clustering.
We have observed that P-selectin binding induces a significant increase of adhesion of Colo 320 cells specifically to fibronectin without significantly affecting adhesion to collagen type I, collagen type lV, or laminin. Experiments with function blocking antibodies indicate that the increase in cell adhesion can be completely accounted for by the α5β1 integrin. Although our results cannot completely rule out participation of other fibronectin-binding integrins (e.g., the αvβ1 and αvβ6 integrins), the complete inhibition by either the β1 or α5 blocking antibody suggests that it is highly unlikely that integrins complexes other than the α5β1 play a meaningful role in Pselectin-mediated activation of cell adhesion to fibronectin.
Fibronectin receptors are associated with a tumorigenic phenotype and are important for metatasis [63-65]. Several integrins have been reported to function as fibronectin receptors (α3β1, α4β1, α5β1, α8β1, αvβ1, αvβ3, and αvβ6) [9]. Among these, the α5β1 integrin is generally regarded as the classical fibronectin receptor due to its ligand specificity. Normal colon has been reported to express α3β1, α5β1, and αvβ1 as fibronectin receptors but do not express αvβ3 or αvβ6 integrins [66-70]. Tumor cells display altered constitutive expression patterns of integrins that bind to fibronectin compared to normal cells [69-71] . Colon tumor samples and several colon carcinoma cells can present an elevated expression of the αvβ6 integrin [66, 67, 72]. The elevated expression αvβ6 integrin has been associated with a significantly reduced survival time of patients with colon cancer in comparison with tumors with no or low αvβ6 expression [67] A significant loss of tissue staining for α5β1 and α3β1 has also been observed in colon cancer, suggesting that αvβ6 integrin may be a major fibronectin-binding receptor for colon cancer cells. However, in a systematic study of integrin expression in normal colon, adenomas, and carcinomas within the same patients, it was observed that the expression of α5β1 integrin was not modified in 25% of the colon carcinomas analyzed [68]. It will be important to examine whether P-selectin binding can modulate the function of β3 and β6 integrins in the future.
The role of the α5β1 integrin during colon cancer progression in vivo is unclear. Some reports suggest that loss of α5β1 expression could be correlated with an increase of malignancy in colon cancer. However, it has been shown that the loss of fibronectin or α5β1 integrin does not contribute to tumorigenesis or metastasis [73]. Other reports have suggested that α5β1 integrin expression could contribute to malignant progression in colon carcinoma [74, 75]. Our observation that P-selectin could activate α5β1 integrin-mediated adhesion suggests that α5β1 integrin could contribute to malignant progression, consistent with a previous study showing that inhibition of the α5β1 integrin with monoclonal antibodies inhibited the experimental metastasis of MDA-MB-231 cells in athymic nude mice [76].
The ability of selectins and/or selectin ligands to regulate integrin-mediated cell-adhesive events has previously been observed in leukocytes. Cross-linking of selectins induces signals that can regulate activation of β2 integrins [77-79]. Integrin activation can also be triggered by selectin ligands. The E-selectin ligand, endothelial-leukocyte molecule-1, was reported to stimulate αMβ2 integrin binding to C3bi-coated erythrocytes and β2 integrin-mediated binding to endothelial cells [79, 80]. Similarly, the ligation of P-selectin glycoprotein ligand-1 to P-selectin was shown to activate β2 integrin-mediated cell attachment to intracellular adhesion molecule 1 [30]. These studies suggest that selectin ligands can induce intracellular signaling to stimulate the higher-affinity β2 integrin-mediated adhesion necessary for extravasation. However, this is the first report showing evidence that P-selectin and their ligands could regulate β1 integrin-mediated adhesion and spreading by cancer cells from solid tumors, thus suggesting a mechanism by which stable tumor cell adhesion can be regulated.
Our finding that P-selectin binding to Colo 320 cells results in activation of both the PI3-K and p38 MAPK signaling pathways suggests that selectin ligands can function as signaling molecules. Both the p38 MAPK and PI3-K pathways have been previously reported to modulate integrin activity in other systems. In mast cells, PI3-K activation by FcεRI, c-kit or PDGF increases the affinity of α5β1 [81-83] and in carcinoma cells, PI3-K activation by HGF increases integrin avidity [51]. In breast cancer cells, p38 MAPK is involved in the regulation of arachidonic acid-mediated adhesion and spreading on collagen type IV [48].
Interestingly, we found that only the PI3-K, and not the p38 MAPK, pathway participates in P-selectin-induced adhesion of Colo 320 cells to fibronectin. In contrast, we found both PI3-K and p38 MAPK regulate P-selectin-induced cell spreading. We also found that that a general tyrosine inhibitor, genistein, blocks completely the P-selectin-mediated PI3K activity. Since specific p38 inhibitors did not detectably inhibit PI3-K, these results suggest strongly that at least one as yet unidentified tyrosine kinase and PI3-K, but not p38 MAPK, regulate the P-selectin-induced α5β1 integrin activation. In a different system, E-selectin binding to HT-29 colon carcinoma cells has been shown to mediate transendothelial migration in a p38 MAPK-dependent manner [59]. This result, taken together with our observations, suggests that p38 MAPK regulates more complex cell adhesion events such as cell spreading and migration, which may play a roles in cancer cell extravasation. It would be very interesting to determine whether E-selectin binding can also activate PI3-K and activate integrin-mediated adhesiveness of HT-29 cells.
We initially expected that PI 3-K would be upstream from p38 MAPK activation because the latter was not required for increased cell adhesion to fibronectin. However, a PI3-K inhibitor had no significant effect on p38 MAPK activation. Conversely, a p38 MAPK inhibitor did not affect P-selectin-induced PI 3-K activation. These data suggest that the activation of PI 3K and p38 MAPK may be independent events in P-selectin-mediated signaling pathway, consistent with previous results [84-89].
We found that P-selectin stimulation causes the formation of a protein complex containing both PI3-K and activated p38 MAPK as judged by co-immunoprecitation experiments and the co-localization of both proteins at the cells periphery. The colocalization of p38 MAPK and PI3-K in intact P-selectin treated, but not untreated, cells suggest that complex formation is not an artifact of cell lysis. This is the first report, to our knowledge, demonstrating the formation of a specific, stimulus-dependent PI3-K-p38 signaling complex, and suggests a mechanism for crosstalk between PI3-K and p38 MAPK pathways.
Previous studies have shown that multiple stimuli can activate both the PI3-K and p38 MAPK signaling pathways leading to a variety of outcomes. In breast cancer cells, heregulin-mediated-PI3K and -p38 MAPK activation has been reported to regulate the activation the matrix metalloproteinase-2 (MMP-2) [84] and MMP-9 [88], proteins that play important roles in cancer cell invasion [85]. Insulin-like growth factor can activate PI3K and p38 MAPK leading to increased breast cancer migration [89]. Both pathways are activated in a Src family kinase-dependent manner via the epidermal growth factor (EGF) receptor and are important for EGF-mediated vascular epidermal growth factor production and promotion of the angiogenic potential of pancreatic cancer cells [86]. Hypoxia can also activate both PI3-K and p38 MAPK pathways, which have been implicated in the regulation of IL-8 expression and angiogenesis by ovarian carcinoma cells [87]. Out results suggest the hypothesis that formation of a P-selectin-mediated PI3-K-p38 signaling complex could lead to the specific activation of integrins as a result of P-selectin stimulation. We also propose that formation of a PI3-K-p38 signaling complex could provide a mechanism to regulate the activity of both molecules.
From analysis of protein sequences (http://www.elm.eu.org/) and the activation mechanisms of PI3-K and p38 MAPK [90-93], we suggest that the interaction between PI3-K and p38 MAPK may be through as yet unidentified adaptor proteins. Analysis of the amino acid sequences of PI3-K p85 and p38 MAPK yields possible protein binding sites —e.g.,SH2 ligand domains, SH3 ligand domains, PDZ ligands domains--that can be recognized by adaptor proteins. Interestingly, both PI3-K p85 and p38 MAPK protein sequences have functional domains that could bind to adaptor proteins such as Grb2 and the 14-3-3 protein family. Grb2 has been reported to complex with PI3K and regulate PI3K activity in several systems [94-96]. The formation of a signaling complex containing p38 MAPK and Grb2 protein has not been reported. However, Grb2 has been implicated in regulation p38 MAPK activity [97-101]. For example, it has been reported that the inhibition of T-cell antigen receptor-mediated Shc-Grb2 signaling complex or the decrease of Grb2 expression in mice result in a defective TCR-induced p38 MAPK activation [97, 100]. The 14-3-3 protein has also been reported to participate in the regulation of p38 MAPK activation [102-104] and to form a signaling complex with PI3 K [105, 106]. Clearly it will be important to determinate other components of the PI3-K-p38 signaling complex to understand the mechnanism of P-selectin-mediated β1 integrin activation.
The role of selectins and their counter-receptors as signaling molecules in cancer cells has not been well-explored. Most studies implying selectins as signaling molecules have been carried out with leukocytes [30, 77, 79, 80, 107, 108]. The only selectin family member analyzed as a signaling molecule in cancer cells has been E-selectin. The binding of E-selectin to colon carcinoma HT-29 cells was reported to increase tyrosine phosphorylation of various proteins including c-Src [34] and p38 MAPK [59]. The role of P-selectin as a signaling molecule in cancer cells is less clear. Studies with mutant mice have shown that the absence of P-selectin results in a significant reduction in primary tumor growth and metastasis compared to P-selectin wild-type controls [60, 109], suggesting that P-selectin binding to cancer cells may induce activation of signals that effect cell proliferation.
An understanding of the mechanisms that regulate the attachment of circulating tumor cells to vascular endothelium of the target organ and their subsequent transendothelial migration may be clinically significant. It has already been shown that the dissociation rate for bonds between P-selectin and tumor cells is faster by an order of magnitude than between P-selectin and normal cells, such as polymorphonuclear leukocytes [110]. Thus, selectin-mediated tethering of carcinoma cells to vascular endothelium [1, 24, 44, 58, 111] and integrin activation is a particularly attractive step for possible therapeutic intervention for the control of metastatic disease. The identification of the role of each member of the selectin family and the identification of the P-selectin ligands that participate in the hematogenous spread of cancer could be valuable to determine whether these proteins could be used as potential therapeutic targets to inhibit cancer metastasis.
Acknowledgements
We thank our colleagues at the NIEHS for critically reviewing this manuscript and Dr. Robert Petrovich for working out the large scale expression and purification of the P-selectin-IgG-Fc chimeric protein. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
References
- 1.Haier J, Nicolson G. Tumor cell adhesion under hydrodynamic conditions of fluid flow. APMIS. 2001;109:241–262. doi: 10.1034/j.1600-0463.2001.d01-118.x. [DOI] [PubMed] [Google Scholar]
- 2.Goetz D, Ding H, Atkinson W, Vachino G, Camphausen R, Cumming D, Luscinskas F. A human colon carcinoma cell line exhibits adhesive interactions with P-selectin under fluid flow via a PSGL-1-independent mechanism. Am J Pathol. 1996;149:1661–1673. [PMC free article] [PubMed] [Google Scholar]
- 3.Hood J, Cheresh D. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2:91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 4.Kaytes P, Geng J. P-selectin mediates adhesion of the human melanoma cell line NKI-4: identification of glycoprotein ligands. Biochemistry. 1998;37:10514–10521. doi: 10.1021/bi9730846. [DOI] [PubMed] [Google Scholar]
- 5.Weiss L. Biomechanical interactions of cancer cells with the microvasture during hematoenous metastasis. Cancer Metastasis Rev. 1992;11:227–235. doi: 10.1007/BF01307179. [DOI] [PubMed] [Google Scholar]
- 6.Hynes R. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 7.Lee J, Juliano R. Mitogenic signal transduction by integrin- and growth factor receptor-mediated pathways. Mol Cell Biol. 2004;17:188–202. [PubMed] [Google Scholar]
- 8.Schwartz M, Ginsberg M. Networks and crosstalk: integrin signalling spreads. Nat Cell Biol. 2002;4:E65–68. doi: 10.1038/ncb0402-e65. [DOI] [PubMed] [Google Scholar]
- 9.van der Flier A, Sonnenberg A. Function and interactions of integrins. Cell Tissue Res. 2001;305:285–238. doi: 10.1007/s004410100417. [DOI] [PubMed] [Google Scholar]
- 10.Giancotti F, Ruoslahti E. Integrin signaling. Science. 1999;285:1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- 11.Liddington R, Ginsberg M. Integrin activation takes shape. J Cell Biol. 2002;158:833–839. doi: 10.1083/jcb.200206011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vuori K, Ruoslahti E. Activation of protein kinase C precedes alpha 5 beta 1 integrin-mediated cell spreading on fibronectin. J Biol Chem. 1993;268:21459–21462. [PubMed] [Google Scholar]
- 13.van Kooyk Y, van de Wiel-van Kemenade P, Weder P, Kuijpers T, Figdor C. Enhancement of LFA-1-mediated cell adhesion by triggering through CD2 or CD3 on T lymphocytes. Nature. 1989;342:811–813. doi: 10.1038/342811a0. [DOI] [PubMed] [Google Scholar]
- 14.Bazzoni G, Hemler M. Are changes in integrin affinity and conformation overemphasized? TIBS. 1998;23:30–34. doi: 10.1016/s0968-0004(97)01141-9. [DOI] [PubMed] [Google Scholar]
- 15.Vestweber D, Blanks J. Mechanisms that regulate the function of the selectins and their ligands. Physiol Rev. 1999;79:181–213. doi: 10.1152/physrev.1999.79.1.181. [DOI] [PubMed] [Google Scholar]
- 16.Mannori G, Crottet P, Cecconi O, Hanasaki K, Aruffo A, Nelson R, Varki A, Bevilacqua M. Differential colon cancer cell adhesion to E-, P-, and L-selectin: Role of mucin type glycoproteins. Cancer Res. 1995;55:4425–4431. [PubMed] [Google Scholar]
- 17.Stone J, Wagner D. P-Selectin mediates adhesion of platelets to neuroblastoma and small cell lung cancer. J Clin Invest. 1993;92:804–813. doi: 10.1172/JCI116654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Springer T. Traffic signals for lymphocytes recirculation and leukocyte emigration: the multistep paradigm. Cell. 1994;76:301–314. doi: 10.1016/0092-8674(94)90337-9. [DOI] [PubMed] [Google Scholar]
- 19.Haier J, Nasralla M, Nicolson G. 1-integrin mediated dynamic adhesion of colon carcinoma cells to extracellular matrix under laminar flow. Clin Exp Metastasis. 1999;17:377–387. doi: 10.1023/a:1006658414040. [DOI] [PubMed] [Google Scholar]
- 20.Mould A, Askari J, Craig S, Garratt A, Clements J, Humphries M. Integrin alpha(4)beta(1)-mediate melanoma cell adhesion and migration on vascular cell adhesion molecule-1 (VCAM-1) and the altenatively splice IIICS region of fibronectin. J Biol Chem. 1994;269:27224–27230. [PubMed] [Google Scholar]
- 21.Sawada R, Tsuboi S, Fukuda M. Differential E-selectin-dependent adhesion efficiency in sublines of a human colon cancer exhibiting distinct metastatic potentials. J Biol Chem. 1994;269:1425–1431. [PubMed] [Google Scholar]
- 22.Walz G, Aruffo A, Kolanus W, Bevilacqua M, Seed B. Recognition by ELAM-1 of the sialyl-Lex determinant on myeloid and tumor cells. Science. 1990;250:1132–1135. doi: 10.1126/science.1701275. [DOI] [PubMed] [Google Scholar]
- 23.Sammar M, Aigner S, Hubbe M, Schirrmacher V, Schachner M, Vestweber D, Altevogt P. Heat-stable antigen (CD24) as ligand for mouse Pselectin. Int Immunol. 1994;6:1027–1036. doi: 10.1093/intimm/6.7.1027. [DOI] [PubMed] [Google Scholar]
- 24.Aigner S, Ramos C, Hafezi-Moghadam A, Lawrence M, Friederichs J, Altevogt P, Ley K. CD24 mediates rolling of breast carcinoma cells on P-selectin. FASEB J. 1998;12:1241–1251. doi: 10.1096/fasebj.12.12.1241. [DOI] [PubMed] [Google Scholar]
- 25.Aigner S, Sthoeger Z, Fogel M, Weber E, Zarn J, Ruppert M, Zeller Y, Vestweber D, Stahel R, Sammar M, Altevogt P. CD24, a mucin-type glycoprotein, is a ligand for P-selectin on human tumor cells. Blood. 1997;89:3385–3395. [PubMed] [Google Scholar]
- 26.Labadia M, Jeanfavre D, Caviness G, Morelock M. Molecular regulation of the interaction between leukocyte function-associated antigen-1 and soluble ICAM-1 by divalent metal cations. J Immnol. 1998;161:836–842. [PubMed] [Google Scholar]
- 27.Nicholson M, Barclay A, Singer M, Rosen S, van der Merwe P. Affinity and kinetic analysis of L-selectin (CD62L) binding to glycosylation-dependent cell-adhesion molecule-1. J Biol Chem. 1998;273:763–770. doi: 10.1074/jbc.273.2.763. [DOI] [PubMed] [Google Scholar]
- 28.Rinko L, Lawrence M, Guilford W. The molecular mechanics of P- and L-selectin lectin domains binding to PSGL-1. Biophys J. 2004;86:544–554. doi: 10.1016/S0006-3495(04)74133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maly P, Thall A, Petryniak B, Rogers C, Smith P, Marks R, Kelly R, Gersten K, Cheng G, Saunders T, Camper S, Camphausen R, Sullivan F, Isogai Y, Hindsgaul O, von Andrian U, Lowe J. The alpha(1,3)fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell. 1996;86:643–653. doi: 10.1016/s0092-8674(00)80137-3. [DOI] [PubMed] [Google Scholar]
- 30.Blanks J, Moll T, Eytner R, Vestweber D. Stimulation of P-selectin glycoprotein ligand-1 on mouse neutrophils activates beta 2-integrin mediated cell attachment to ICAM-1. Eur J Immunol. 1998;28:433–443. doi: 10.1002/(SICI)1521-4141(199802)28:02<433::AID-IMMU433>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 31.Haller H, Kunzendorf U, Sacherer K, Lindschau C, Walz G, Distler A, Luft F. T cell adhesion to P-selectin induces tyrosine phosphorylation of pp125 focal adhesion kinase and other substrates. J Immunol. 1997;158:1061–1067. [PubMed] [Google Scholar]
- 32.Levinovitz A, Muhlhoff J, Isenmann S, Vestweber D. Identification of a glycoprotein ligand for E-selectin on mouse myeloid cells. J Cell Biol. 1993;121:449–459. doi: 10.1083/jcb.121.2.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sako D, Chang X, Barone K, Vachino G, White H, Shaw G, Veldman G, Bean K, Ahern T, Furie B, Cumming D, Larsen G. Expression cloning of a functional glycoprotein ligand for P-selectin. Cell. 1993;75:1179–1186. doi: 10.1016/0092-8674(93)90327-m. [DOI] [PubMed] [Google Scholar]
- 34.Soltesz S, Powers E, Geng J, Fisher C. Adhesion of HT-29 colon carcinoma cells to E-selectin results in increased tyrosine phosphorylation and decreased activity of c-src. Int J Cancer. 1997;71:645–653. doi: 10.1002/(sici)1097-0215(19970516)71:4<645::aid-ijc22>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 35.Akiyama S, Yamada S, Chen W, Yamada K. Analysis of fibronectin receptor function with monoclonal antibodies: roles in cell adhesion, migration, matrix assembly, and cytoskeletal organization. J Cell Biol. 1989;109:863–875. doi: 10.1083/jcb.109.2.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LaFlamme S, Thomas L, Yamada S, Yamada K. Single subunit chimeric integrins as mimics and inhibitors of endogenous integrin function in receptor localization, cell spreading and migration, and matrix assembly. J Cell Biol. 1994;126:1287–1298. doi: 10.1083/jcb.126.5.1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miekka S, Ingham K, Menache D. Rapid methods for isolation of human plasma fibronectin. Thromb Res. 1982;27:1–14. doi: 10.1016/0049-3848(82)90272-9. [DOI] [PubMed] [Google Scholar]
- 38.Akiyama S, Yamada K. The interaction of plasma fibronectin with fibroblastic cells in suspension. J Biol Chem. 1985;260:4492–4500. [PubMed] [Google Scholar]
- 39.Whittard J, Akiyama S. Activation of beta1 integrins induces cell-cell adhesion. Exp Cell Res. 2001;263:65–76. doi: 10.1006/excr.2000.5099. [DOI] [PubMed] [Google Scholar]
- 40.Akiyama S, Yamada S, Yamada K. Characterization of a 140-kD avian cell surface antigen as a fibronectin-binding molecule. J Biol Chem. 1986;102:442–448. doi: 10.1083/jcb.102.2.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reyes-Reyes M, Mora N, Zentella A, Rosales C. Phosphatidylinositol 3-kinase mediates integrin-dependent NF-kappaB and MAPK activation through separate signaling pathways. J Cell Sci. 2001;114:1579–89. doi: 10.1242/jcs.114.8.1579. [DOI] [PubMed] [Google Scholar]
- 42.Arruffo A, Dietsch M, Wan H, Hellstrom K, Hellstrom I. Granule membrane protein 140 (GMP140) binds to carcinomas and carcinoma-derived cell lines. Proc Natl Acad Sci USA. 1992;89:2292–2296. doi: 10.1073/pnas.89.6.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dardik R, Kaufmann Y, Savion N, Rosenberg N, Shenkman B, Varon D. Platelets mediate tumor cell adhesion to the subendothelium under flow conditions: involvement of platelet GPIIb-IIIa and tumor cell alpha(v) integrins. Int J Cancer. 1997;70:201–207. doi: 10.1002/(sici)1097-0215(19970117)70:2<201::aid-ijc11>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 44.Dardik R, Savion N, Kaufmann Y, Varon D. Thrombin promotes platelet-mediated melanoma cell adhesion to endothelial cells under flow conditions: role of platelet glycoproteins P-selectin and GPIIb-IIIA. Br J Cancer. 1998;77:2069–2075. doi: 10.1038/bjc.1998.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sims P, Ginsberg M, Plow E, Shattil S. Effect of platelet activation on the conformation of the plasma membrane glycoprotein IIb-IIIa complex. J Biol Chem. 1991;266:7345–7352. [PubMed] [Google Scholar]
- 46.Woodside D, Liu S, Ginsberg M. Integrin activation. Thromb Res. 2001;86:316–323. [PubMed] [Google Scholar]
- 47.Luque A, Gomez M, Puzon W, Takada Y, Sanchez-Madrid F, Cabanas C. Activated conformations of very late activation integrins detected by a group of antibodies (HUTS) specific for a novel regulatory region of the common beta 1 chain. J Biol Chem. 1996;271:11067–11075. doi: 10.1074/jbc.271.19.11067. [DOI] [PubMed] [Google Scholar]
- 48.Paine E, Palmantier R, Akiyama S, Olden K, Robert J. Arachidonic acid activates mitogen-activated protein (MAP) kinase-activated protein kinase and mediates adhesion of a human breast carcinoma cell line to collagen type IV through a p38 MAP kinase-depent pathway. J Biol Chem. 2000;275:11284–11290. doi: 10.1074/jbc.275.15.11284. [DOI] [PubMed] [Google Scholar]
- 49.Danilkovitch-Miagkova A, Zbar B. Dysregulation of Met receptor tyrosine kinase activity in invasive tumors. J Clin Invest. 2002;109:863–867. doi: 10.1172/JCI15418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Katagiri K, Hattori M, Minato N, Irie S, Takatsu K, Kinashi T. Rap1 is a potent activation signal for leukocyte function-associated antigen 1 distinct from protein kinase C and phosphatidylinositol-3-OH kinase. Mol Cell Biol. 2000;20:1956–1969. doi: 10.1128/mcb.20.6.1956-1969.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Trusolino L, Cavassa S, Angelini P, Ando M, Bertotti A, Comoglio P, Boccaccio C. HGF/scatter factor selectively promotes cell invasion by increasing integrin avidity. FASEB J. 2000;14:1629–1640. doi: 10.1096/fj.14.11.1629. [DOI] [PubMed] [Google Scholar]
- 52.Morris V, Koop S, MacDonald I, Schmidt E, Grattan M, Percy D, Chambers A, Groom A. Mammary carcinoma cell lines of high and low metastatic potential differ not in extravasation but in subsequent migration and growth. Clin Exp Metastasis. 1994;12:357–367. doi: 10.1007/BF01755879. [DOI] [PubMed] [Google Scholar]
- 53.Morris V, Schmidt E, Koop S, MacDonald I, Grattan M, Khokha R, McLane M, Niewiarowski S, Chambers A, Groom A. Effects of the disintegrin eristostatin on individual steps of hematogenous metastasis. Exp Cell Res. 1995;19:571–578. doi: 10.1006/excr.1995.1266. [DOI] [PubMed] [Google Scholar]
- 54.MacDonald I, Groom A, Chambers A. Cancer spread and micrometastasis development: quantitative approaches for in vivo models. Bioessays. 2002;24:885–893. doi: 10.1002/bies.10156. [DOI] [PubMed] [Google Scholar]
- 55.Irimura T, Nakamori S, Matsushita Y, Taniuchi Y, Todoroki N, Tsuji T, Izumi Y, Kawamura Y, Hoff S, leary K. Colorectal cancer metastasis determined by carbohydrate-mediated cell adhesion: role of sialyl-LeX antigens. Semin Cancer Biol. 1993;4:319–324. [PubMed] [Google Scholar]
- 56.Takada A, Ohmori K, Yoneda T, Tsuyuoka K, Hasegawa A, Kiso M, Kannagi R. Contribution of carbohydrate antigens sialyl Lewis A and sialyl Lewis X to adhesion of human cancer cells to vascular endothelium. Cancer Res. 1993;53:354–361. [PubMed] [Google Scholar]
- 57.Dejana E, Martin-Padura D, Lauri S, Bernasconi M, Bani A. Endothelial leukocyte adhesion molecule-1-dependent adhesion of colon carcinoma cells to vascular endothelium is inhibited by an anti-body to Lewis fucosylated type I carbohydrate chain. Lab Invest. 1992;66:324–330. [PubMed] [Google Scholar]
- 58.Iwai K, Ishikura H, Kaji M, Sugiura H, Ishizu A, Takahashi C, Kato H, Tanabe T, Yoshiki T. Importance of E-selectin (ELAM-1) and sialyl Lewis(a) in the adhesion of pancreatic carcinoma cells to activated endothelium. Int J Cancer. 1993;54:972–977. doi: 10.1002/ijc.2910540618. [DOI] [PubMed] [Google Scholar]
- 59.Laferriere J, Houle F, Taher M, Valerie K, Huot J. Transendothelial migration of colon carcinoma cells requires expression of E-selectin by endothelial cells and activation of stress-activated protein kinase-2 (SAPK2/p38) in the tumor cells. J Biol Chem. 2001;276:33762–33772. doi: 10.1074/jbc.M008564200. [DOI] [PubMed] [Google Scholar]
- 60.Ludwig R, Boehme B, Podda M, Henschler R, Jager E, Tandi C, Boehncke W, Zollner T, Kaufmann R, Gille J. Endothelial P-selectin as a target of heparin action in experimental melanoma lung metastasis. Cancer Res. 2004;64:2743–2750. doi: 10.1158/0008-5472.can-03-1054. [DOI] [PubMed] [Google Scholar]
- 61.Majuri M, Mattila P, Renkonen R. Recombinant E-selectin-protein mediates tumor cell adhesion via sialyl-Le(a) and sialyl-Le(x) Biochem Biophys Res Commun. 1992;182:1376–1382. doi: 10.1016/0006-291x(92)91885-t. [DOI] [PubMed] [Google Scholar]
- 62.Karpatkin S, Pearlstein E, Salk P, Yogeeswaran G. Role of platelets in tumor cell metastases. Ann N Y Acad Sci. 1981;370:101–118. doi: 10.1111/j.1749-6632.1981.tb29726.x. [DOI] [PubMed] [Google Scholar]
- 63.Danen E, Yamada K. Fibronectin, integrins, and growth control. J Cell Physiol. 2001;189:1–13. doi: 10.1002/jcp.1137. [DOI] [PubMed] [Google Scholar]
- 64.Jaskiewicz K, Chasen M, Robson S, Jaskiewicz K, Chasen MR, Robson SC. Differential expression of extracellular matrix proteins and integrins in hepatocellular carcinoma and chronic liver disease. Anticancer Res. 1993;13:2229–2237. [PubMed] [Google Scholar]
- 65.Kemperman H, Driessens M, La Riviere G, Meijne A, Roos E. Adhesion mechanisms in liver metastasis formation. Cancer Surv. 1995;24:67–79. [PubMed] [Google Scholar]
- 66.Agrez M, Bates R. Colorectal cancer and the integrin family of cell adhesion receptors: current status and future directions. Eur J Cancer. 1994;30A:2166–2170. doi: 10.1016/0959-8049(94)00473-i. [DOI] [PubMed] [Google Scholar]
- 67.Bates R, Bellovin D, Brown C, Maynard E, Wu B, Kawakatsu H, Sheppard D, Oettgen P, Mercurio A. Transcriptional activation of integrin beta6 during the epithelial-mesenchymal transition defines a novel prognostic indicator of aggressive colon carcinoma. J Clin Invest. 2005;115:339–347. doi: 10.1172/JCI23183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Stallmach A, von Lampe B, Matthes H, Bornhoft G, Riecken E. Diminished expression of integrin adhesion molecules on human colonic epithelial cells during the benign to malign tumour transformation. Gut. 1992;33:342–346. doi: 10.1136/gut.33.3.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choy M, Richman P, Horton M, MacDonald T. Expression of the VLA family of integrins in human intestine. J Pathol. 1990;160:35–40. doi: 10.1002/path.1711600109. [DOI] [PubMed] [Google Scholar]
- 70.Koretz K, Schlag P, Boumsell L, Moller P. Expression of VLA-alpha 2, VLA-alpha 6, and VLA-beta 1 chains in normal mucosa and adenomas of the colon, and in colon carcinomas and their liver metastases. Am J Pathol. 1991;138:741–750. [PMC free article] [PubMed] [Google Scholar]
- 71.Lindmark G, Gerdin B, Pahlman L, Glimelius B, Gehlsen K, Rubin K. Interconnection of integrins alpha 2 and alpha 3 and structure of the basal membrane in colorectal cancer: relation to survival. Eur J Surg Oncol. 1993;19:50–60. [PubMed] [Google Scholar]
- 72.Agrez M, Bates R, Mitchell D, Wilson N, Ferguson N, Anseline P, Sheppard D. Multiplicity of fibronectin-binding alpha V integrin receptors in colorectal cancer. Br J Cancer. 1996;73:887–892. doi: 10.1038/bjc.1996.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Taverna D, Ullman-Cullere M, Rayburn H, Bronson R, Hynes R. A test of the role of alpha5 integrin/fibronectin interactions in tumorigenesis. Cancer Res. 1998;58:848–853. [PubMed] [Google Scholar]
- 74.Gong J, Wang D, Sun L, Zborowska E, Willson J, Brattain M. Role of alpha 5 beta 1 integrin in determining malignant properties of colon carcinoma cells. Cell Growth Differ. 1997;8:83–90. [PubMed] [Google Scholar]
- 75.Murillo C, Rychahou P, Evers B. Inhibition of alpha5 integrin decreases PI3K activation and cell adhesion of human colon cancers. Surgery. 2004;136:143–149. doi: 10.1016/j.surg.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 76.Newton S, Reeves E, Gralnick H, Mohla S, Yamada K, Olden K, Akiyama S. Inhibition of experimental metastasis of human breast carcinoma cells in athymic nude mice by anti-α5β1 fibronectin receptor integrin antibodies. Int. J. Oncol. 1995;6:1063–1070. doi: 10.3892/ijo.6.5.1063. [DOI] [PubMed] [Google Scholar]
- 77.Crockett-Torabi E, Sulenbarger B, Smith C, Fantone J. Activation of human neutrophils through L-selectin and Mac-1 molecules. J Immnol. 1995;154:2291–2302. [PubMed] [Google Scholar]
- 78.Simon S, Burns A, Taylor A, Gopalan P, Lynam E, Sklar L, Smith C. L-selectin (CD62L) cross-linking signals neutrophil adhesive functions via the Mac-1 (CD11b/CD18) beta 2-integrin. J Immnol. 1995;155:1502–1514. [PubMed] [Google Scholar]
- 79.Simon S, Hu Y, Vestweber D, Smith C. Neutrophil tethering on E-selectin activates beta 2 integrin binding to ICAM-1 through a mitogen-activated protein kinase signal transduction pathway. J Immnol. 2000;164:4348–4358. doi: 10.4049/jimmunol.164.8.4348. [DOI] [PubMed] [Google Scholar]
- 80.Lo S, Lee S, Ramos R, Lobb R, Rosa M, Chi-Rosso G, Wright S. Endothelial-leukocyte adhesion molecule 1 stimulates the adhesive activity of leukocyte integrin CR3 (CD11b/CD18, Mac-1, alpha m beta 2) on human neutrophils. J Exp Med. 1991;173:1493–1500. doi: 10.1084/jem.173.6.1493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Serve H, Yee N, Stella G, Sepp-Lorenzino L, Tan J, Besmer P. Differential roles of PI3-kinase and Kit tyrosine 821 in Kit receptor-mediated proliferation, survival and cell adhesion in mast cells. EMBO J. 1995;14:473–483. doi: 10.1002/j.1460-2075.1995.tb07023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kinashi T, Asaoka T, Setoguchi R, Takatsu K. Affinity modulation of very late antigen-5 through phosphatidylinositol 3-kinase in mast cells. J Immnol. 1999;162:2850–2857. [PubMed] [Google Scholar]
- 83.Kinashi T, Escobedo J, Williams L, Takatsu K, Springer T. Receptor tyrosine kinase stimulates cell-matrix adhesion by phosphatidylinositol 3 kinase and phospholipase C-gamma 1 pathways. Blood. 1995;86:2086–2090. [PubMed] [Google Scholar]
- 84.Ke Z, Lin H, Fan Z, Cai T, Kaplan R, Ma C, Bower K, Shi X, Luo J. MMP-2 mediates ethanol-induced invasion of mammary epithelial cells over-expressing ErbB2. Int J Cancer. 2006;119:8–16. doi: 10.1002/ijc.21769. [DOI] [PubMed] [Google Scholar]
- 85.Klein G, Vellenga E, Fraaije M, Kamps W, de Bont E. The possible role of matrix metalloproteinase (MMP)-2 and MMP-9 in cancer, e.g. acute leukemia. Crit Rev Oncol Hematol. 2004;50:87–100. doi: 10.1016/j.critrevonc.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 86.Summy J, Trevino J, Baker C, Gallick G. c-Src regulates constitutive and EGF-mediated VEGF expression in pancreatic tumor cells through activation of phosphatidyl inositol-3 kinase and p38 MAPK. Pancreas. 2005;31:263–274. doi: 10.1097/01.mpa.0000178280.50534.0c. [DOI] [PubMed] [Google Scholar]
- 87.Xu L, Pathak P, Fukumura D. Hypoxia-induced activation of p38 mitogen-activated protein kinase and phosphatidylinositol 3′-kinase signaling pathways contributes to expression of interleukin 8 in human ovarian carcinoma cells. Clin Cancer Res. 2004. 2004;10:701–707. doi: 10.1158/1078-0432.ccr-0953-03. [DOI] [PubMed] [Google Scholar]
- 88.Yao J, Xiong S, Klos K, Nguyen N, Grijalva R, Li P, Yu D. Multiple signaling pathways involved in activation of matrix metalloproteinase-9 (MMP-9) by heregulin-beta1 in human breast cancer cells. Oncogene. 2001;20:8066–8074. doi: 10.1038/sj.onc.1204944. [DOI] [PubMed] [Google Scholar]
- 89.Zhang X, Lin M, van Golen K, Yoshioka K, Itoh K, Yee D. Multiple signaling pathways are activated during insulin-like growth factor-I (IGF-I) stimulated breast cancer cell migration. Breast Cancer Res Treat. 2005;93:159–168. doi: 10.1007/s10549-005-4626-8. [DOI] [PubMed] [Google Scholar]
- 90.Mittelstadt P, Salvador J, Fornace AJ, Ashwell J. Activating p38 MAPK: new tricks for an old kinase. Cell Cycle. 2005;4:1189–1192. doi: 10.4161/cc.4.9.2043. [DOI] [PubMed] [Google Scholar]
- 91.Schieven G. The biology of p38 kinase: a central role in inflammation. Curr Top Med Chem. 2005;5:921–928. doi: 10.2174/1568026054985902. [DOI] [PubMed] [Google Scholar]
- 92.Wymann M, Marone R. Phosphoinositide 3-kinase in disease: timing, location, and scaffolding. Curr Opin Cell Biol. 2005;17:141–149. doi: 10.1016/j.ceb.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 93.Wymann M, Pirola L. Structure and function of phosphoinositide 3-kinases. Biochim Biophys Acta. 1998;1436:127–150. doi: 10.1016/s0005-2760(98)00139-8. [DOI] [PubMed] [Google Scholar]
- 94.Goudreau N, Cornille F, Duchesne M, Parker F, Tocque B, Garbay C, Roques B. NMR structure of the N-terminal SH3 domain of GRB2 and its complex with a proline-rich peptide from Sos. Nat Struct Biol. 1994;1:898–907. doi: 10.1038/nsb1294-898. [DOI] [PubMed] [Google Scholar]
- 95.Ong S, Dilworth S, Hauck-Schmalenberger I, Pawson T, Kiefer F. ShcA and Grb2 mediate polyoma middle T antigen-induced endothelial transformation and Gab1 tyrosine phosphorylation. EMBO J. 2001;20:6327–6336. doi: 10.1093/emboj/20.22.6327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shen T, Guan J. Differential regulation of cell migration and cell cycle progression by FAK complexes with Src, PI3K, Grb7 and Grb2 in focal contacts. FEBS Lett. 2001;499:176–181. doi: 10.1016/s0014-5793(01)02545-5. [DOI] [PubMed] [Google Scholar]
- 97.Gong Q, Cheng A, Akk A, Alberola-Ila J, Gong G, Pawson T, Chan A. Disruption of T cell signaling networks and development by Grb2 haploid insufficiency. Nat Immunol. 2001;2:29–36. doi: 10.1038/83134. [DOI] [PubMed] [Google Scholar]
- 98.Hayashi K, Shibata K, Morita T, Iwasaki K, Watanabe M, Sobue K. Insulin receptor substrate-1/SHP-2 interaction, a phenotype-dependent switching machinery of insulin-like growth factor-I signaling in vascular smooth muscle cells. J Biol Chem. 2004;279:40807–40818. doi: 10.1074/jbc.M405100200. [DOI] [PubMed] [Google Scholar]
- 99.Kallin A, Demoulin J, Nishida K, Hirano T, Ronnstrand L, Heldin C. Gab1 contributes to cytoskeletal reorganization and chemotaxis in response to platelet-derived growth factor. J Biol Chem. 2004;279:17897–17904. doi: 10.1074/jbc.M312996200. [DOI] [PubMed] [Google Scholar]
- 100.Patrussi L, Savino M, Pellegrini M, Paccani S, Migliaccio E, Plyte S, Lanfrancone L, Pelicci P, Baldari C. Cooperation and selectivity of the two Grb2 binding sites of p52Shc in T-cell antigen receptor signaling to Ras family GTPases and Myc-dependent survival. Oncogene. 2005;24:2218–2228. doi: 10.1038/sj.onc.1208384. [DOI] [PubMed] [Google Scholar]
- 101.Zhang S, Weinheimer C, Courtois M, Kovacs A, Zhang C, Cheng A, Wang Y, Muslin A. The role of the Grb2-p38 MAPK signaling pathway in cardiac hypertrophy and fibrosis. J Clin Invest. 2003;111:833–841. doi: 10.1172/JCI16290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gurusamy N, Watanabe K, Ma M, Zhang S, Muslin A, Kodama M, Aizawa Y. Dominant negative 14-3-3 promotes cardiomyocyte apoptosis in early stage of type I diabetes mellitus through activation of JNK. Biochem Biophys Res Commun. 2004;320:773–780. doi: 10.1016/j.bbrc.2004.06.023. [DOI] [PubMed] [Google Scholar]
- 103.Santoro M, Gaudino G, Marchisio P. The MSP receptor regulates alpha6beta4 and alpha3beta1 integrins via 14-3-3 proteins in keratinocyte migration. Dev Cell. 2003;5:257–271. doi: 10.1016/s1534-5807(03)00201-6. [DOI] [PubMed] [Google Scholar]
- 104.Zhang S, Ren J, Zhang C, Treskov I, Wang Y, Muslin A. Role of 14-3-3-mediated p38 mitogen-activated protein kinase inhibition in cardiac myocyte survival. Circ Res. 2003;93:1026–1028. doi: 10.1161/01.RES.0000104084.88317.91. [DOI] [PubMed] [Google Scholar]
- 105.Kosaki A, Yamada K, Suga J, Otaka A, Kuzuya H. 14-3-3beta protein associates with insulin receptor substrate 1 and decreases insulin-stimulated phosphatidylinositol 3′-kinase activity in 3T3L1 adipocytes. J Biol Chem. 1998;273:940–944. doi: 10.1074/jbc.273.2.940. [DOI] [PubMed] [Google Scholar]
- 106.Xiang X, Yuan M, Song Y, Ruderman N, Wen R, Luo Z. 14-3-3 facilitates insulin-stimulated intracellular trafficking of insulin receptor substrate 1. Mol Endocrinol. 2002;16:552–562. doi: 10.1210/mend.16.3.0790. [DOI] [PubMed] [Google Scholar]
- 107.Celi A, Pellegrini G, Lorenzet R, De Blasi A, Ready N, Furie B, Furie B. Pselectin induces the expression of tissue factor on monocytes. Proc Natl Acad Sci USA. 1994;91:8767–8771. doi: 10.1073/pnas.91.19.8767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cooper D, Butcher C, Berndt M, Vadas M. P-selectin interacts with a beta 2-integrin to enhance phagocytosis. J Immnol. 1994;153:3199–3209. [PubMed] [Google Scholar]
- 109.Kim Y, Borsig L, Varki N, Varki A. P-selectin deficiency attenuates tumor growth and metastasis. Proc Natl Acad Sci USA. 1998;95:9325–30. doi: 10.1073/pnas.95.16.9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hanley W, McCarty O, Jadhav S, Tseng Y, Wirtz D, Konstantopoulos K. Single molecule characterization of P-selectin/ligand binding. J Biol Chem. 2003;278:10556–10561. doi: 10.1074/jbc.M213233200. [DOI] [PubMed] [Google Scholar]
- 111.Tozeren A, Kleinman H, Grant D, Morales D, Mercurio A, Byers S. E-selectin-mediated dynamic interactions of breast- and colon-cancer cells with endothelial-cell monolayers. Int J Cancer. 1995;60:426–431. doi: 10.1002/ijc.2910600326. [DOI] [PubMed] [Google Scholar]