

Abstract
Neurogenesis in the adult hippocampus has been implicated in regulating long-term memory and mood, but its integrity in Alzheimer’s disease (AD) is uncertain. Studies of neurogenesis in transgenic mouse models of familial AD are complicated by ectopic overexpression restricted to terminally differentiated neurons, while AD cases have been studied only at the pre-senile or end-stage of disease. To investigate further the fidelity of adult neurogenesis, we examined mice carrying targeted mutations in amyloid precursor protein (APP), presenilin-1 (PS-1), or both APP and PS-1, in which FAD-causing mutations have been inserted into their endogenous genes. The latter “double knock-in” mice developed aging- and region-dependent amyloid deposition starting around 6 months, and by 9 months exhibited microglial activation associated with the amyloid. In the 9 month old dentate gyrus, the double knock-in mutations reduced the numbers of MCM2-positive neural stem and progenitor cells by 3-fold and doublecortin-positive neuroblasts by 2-fold. The reduction in dentate neuroblasts persisted at 18 months of age. The impairment in neurogenesis was confirmed by quantitative Western blot analysis of doublecortin content, and was restricted to the hippocampal but not the olfactory bulb neurogenic system. In contrast, neither mutant PS-1 nor APP alone led to amyloid deposition or significant alterations in the two markers. These results demonstrate long-lasting and selective impairment in adult hippocampal neurogenesis in a knock-in mutant mouse model of FAD, and suggest a novel mechanism by which amyloid and its attendant microglia-mediated neuroinflammation could contribute to the cognitive and behavioral abnormalities of AD.
Keywords: amyloid, presenilin, neurogenesis, familial Alzheimer’s disease, neuroinflammation, neural plasticity
Introduction
Although the majority of cases of Alzheimer’s disease (AD) do not have a known direct genetic cause and are considered sporadic, a subset are triggered by inherited mutations in either the amyloid precursor protein (APP), presenilin-1 (PS-1), or presenilin-2 (PS-2) gene (St. George-Hyslop, 2000; Tanzi and Bertram, 2005). Excepting its early age of onset, familial Alzheimer’s disease (FAD) resembles sporadic forms of the disease in its clinical signs, as well as its slow progression and characteristic neuropathologies, which include regionally restricted amyloid deposition in the brain parenchyma and vasculature, intraneuronal neurofibrillary tangles, amyloid-associated gliosis and neuroinflammation, and the loss of neurons and synapses (Braak et al., 1998; Lleo et al., 2004). The pathogenic mechanisms by which APP, PS-1, and PS-2 mutations cause AD have been the subject of extensive study. When expressed in cellular systems, mice, and humans, the FAD-linked mutants invariably increase production of the Aβ42 variant (Scheuner et al., 1996; Hardy and Selkoe, 2002) that is the major component of parenchymal amyloid plaques (Yang et al., 1994; Savage et al., 1995). In transgenic mice mutant APP overexpression recapitulates the amyloid-associated abnormalities, effects that are accelerated markedly by the co-expression of mutant PS-1 and lead to behavioral dysfunction (Games et al., 1995; Hsaio et al., 1996; Holcomb et al., 1998; Ashe, 2001).
The adult mammalian brain contains two major neurogenic systems: the subgranular zone of the dentate gyrus, which provides new neurons for the hippocampus, and the subventricular zone along the lateral ventricle, which supplies the olfactory bulb. In particular, adult hippocampal neurogenesis has been implicated in the regulation of cognition (Rola et al., 2004; Schaffer and Gage, 2004) and mood (Jacobs et al., 2000), behaviors that are a signature abnormality of AD or a frequent co-morbid occurrence, respectively. Thus it is critically important to assess the integrity of this form of neural plasticity in the AD brain and discern the contribution made by altered neurogenesis to the disease process. Presenilins are expressed in neural progenitors and their genetic deletion disrupts developmental neurogenesis (Handler et al., 2000; Wen et al., 2002). Transgenic overexpression of mutant PS-1 in the adult mouse reportedly impairs adult hippocampal neurogenesis (Wen et al., 2004; Chevallier et al., 2005), whereas mutant APP either enhances (Jin et al., 2004a) or impairs it (Haughey et al., 2002). Unfortunately, these studies were all conducted using heterologous promoters that preferentially drive supraphysiological expression in terminally differentiated neurons. As a consequence, the mutant transgenes are not expressed in the neural stem and progenitor cells themselves, and the relevance of these findings for the AD brain are uncertain. Neuroblast numbers are reportedly increased in hippocampus of AD patients (Jin et al., 2004b), but this end-stage analysis leaves unexplored the role of impaired neurogenesis in the onset and progression of the disease, and is not substantiated by a study of pre-senile cases of probable AD (Boekhoorn et al., 2006).
Here we investigated the integrity of adult brain neurogenesis using the APP/PS-1 double knock-in mutant mouse, in which FAD-causing mutations were targeted into their endogenous genes (Reaume et al., 1996; Siman et al., 2000; Siman and Salidas, 2004). The resulting mice express mutant APP and PS-1 without the complications of ectopic expression and overexpression inherent to transgenic approaches, and their brains exhibit AD-type amyloid deposition and microgliosis. Using markers for neural stem and progenitor cells, along with differentiating neuroblasts, we evaluated the impact of mutant APP, PS-1, and the double knock-in mutations on neurogenesis in the adult brain.
Materials and Methods
Gene-targeted mouse lines
The APP knock-in, PS-1 knock-in, and APP/PS-1 double knock-in mouse lines were created using gene targeting in embryonic stem cells, and have been described and characterized previously (Reaume et al., 1996; Siman et al., 2000; Flood et al., 2002; Siman and Salidas, 2004). For the APP line, the Swedish double point mutation was introduced into the APP gene of the CD-1 outbred mouse strain by changing the sequence in exon 17 encoding the Lys-Met at codons 670–671 to one encoding Asn-Leu, and the 3 variant codons within the Aβ domain changed from the mouse to the human sequence. The neomycin selection cassette reduced transcription of the APP gene, and so the cassette was removed from the intron upstream of the targeted exon 17 using the Cre recombinase, and the resulting APP knock-in mutant mice were bred to homozygosity (APP KI/KI line). A similar strategy was used to introduce the FAD-linked P264L mutation into the mouse PS-1 gene and remove the neomycin selection cassette, also in the CD-1 background. The mRNA and protein expression of APP and PS-1 were confirmed by Northern and Western blot analyses as being equivalent between the targeted and control lines. The APP homozygous mutant mice were crossed with PS-1 heterozygous mutant mice to generate offspring of 3 different genotypes: APP KI/KI + PS-1 WT/WT, APP KI/KI + PS-1 KI/WT, and APP KI/KI + PS-1 KI/KI. The latter line is referred to as the APP/PS-1 double knock-in mouse. APP WT/WT + PS-1 KI/WT mice were crossed with one another to generate offspring of two additional genotypes for experimentation: APP WT/WT + PS-1 WT/WT, and APP WT/WT + PS-1 KI/KI. The mice were given free access to food and water, and maintained under veterinary supervision in strict compliance with all standards for animal care and investigation established in the “Guide For the Care and Use of Laboratory Animals” (National Academy Press ISBN# 0-309-05377-3).
The genotypes of the APP and PS-1 alleles were determined by a PCR strategy. For PS-1, the following primers were used: forward – GCT GGA GCA ATG CTG TGT TA; reverse – GAG ATG GCT TAC GGG TTG AG. The amplified product is 190 bp for the wild-type PS-1 allele and 280 bp for the mutant knock-in allele. For APP, the following primers were used: forward – CAC ACC AAG AAG TAC AAT AGA; reverse – CCT GGG TTG TAG GGA CTG TAC TTG. In this case, the amplified product is 214 bp for the wild-type APP allele and 298 bp for the mutant knock-in allele.
Immunocytochemistry and cell counting
Male and female mice of either 8–9 months or 18–24 months of age were anesthetized deeply with an overdose of pentobarbital, then perfused transcardially with ice cold 0.1 M sodium phosphate (pH 7.4; PB) followed by freshly prepared 4% paraformaldehyde in PB. Brains were carefully dissected, left in fixative for 4 hours at 4°C, cryoprotected in 20% sucrose in PB overnight, blocked, and then frozen in dry ice/2-methylbutane at −40°C and stored at −80°C. Sagittal sections 40 μm thick were prepared with a sliding microtome and collected sequentially into ten series, with each series starting from the medial border of hippocampus and extending laterally to the start of the ventral hippocampus. The total number of sections between these anatomical boundaries did not vary as a function of APP or PS-1 genotype. A minimum of two series of sections each were immunostained for MCM2, a marker for neural stem and progenitor cells (Maslov et al., 2004), or doublecortin, a marker for immature neuroblasts (Nacher et al., 2001; Rao and Shetty, 2004). Thus, each marker was evaluated on a minimum of 10 sections per mouse. Goat antibodies to MCM2 and doublecortin were obtained from Santa Cruz Biotechnologies (Santa Cruz, CA) and used at 1/1,000 and 1/3,000 dilution respectively. The following antibodies stained one additional series each: anti-amyloid Aβ1–28 (rabbit antibody Ab1153 at 1/5,000; Savage et al., 1995; Siman and Salidas, 2004); the macrophage lineage markers F4/80 (mouse antibody at 1/500; Serotec) or anti-CD45 (rat monoclonal antibody at 1/2000; Accurate Chemical), or the activated microglial marker anti-FCγ receptor 2/3 (rat monoclonal antibody at 1/1000; BD Biosciences). Sections were rinsed in TBS (20 mM Tris-HCl, pH 7.4 + 150 mM NaCl), permeabilized and blocked in TBS containing 0.05% Triton X-100 and 3% of either normal goat serum (for labeling with all mouse and rabbit primary antibodies) or normal donkey serum (for labeling with goat primary antibodies). Sections were incubated in primary antibodies in their respective blocking solutions 24–48 hr at 4°C, then washed 3 times in TBS. Next, the sections were treated with species-specific biotinylated secondary antibodies (obtained from Vector Laboratories and used at 1/800 dilution) for 1 hr at 22°C, and then washed 3 times in TBS. Sections were incubated with avidin-biotin-horseradish peroxidase complex (prepared from the Vector ABC kit) in TBS/1% bovine serum albumin for 1 hr at 22°C, and then washed 4 times in TBS. Diaminobenzidine plus hydrogen peroxide was used as chromagen. Stained sections were slide-mounted, dehydrated, delipidated, and coverslipped. Finally, to discern any possible overt cytoarchitechtonic differences among the genotypes, one series of sections was stained with cresyl violet. Photomicrographs were taken under brightfield and Nomarski illumination on a Nikon Eclipse E600 microscope outfitted with a Nikon DXM-1200 12 megapixel digital camera.
The numbers of MCM2-positive neural stem and progenitor cells and doublecortin-positive immature neuroblasts were counted by an observer blind to age and genotype during bright field observation at 400X magnification. The subgranular zone adjacent to the granule cell layer for both blades of the dentate gyrus was evaluated along its entire rostro-caudal axis, starting at the medial border of dorsal hippocampus and extending laterally to the start of ventral hippocampus. Total numbers of immunopositive cells within these anatomical boundaries were counted, and represented as the percentage of immunostained cells per dorsal hippocampus compared with APP/PS-1 wild type mice. A minimum of 100 immunopositive cells were counted for each marker and mouse brain. Between 5–6 mice were analyzed at 8–9 months of age for each of the following genotypes: APP WT/WT + PS-1 WT/WT; APP KI/KI + PS-1 WT/WT; APP KI/KI + PS-1 KI/WT; APP KI/KI + PS-1 KI/KI. An additional 3 mice were analyzed for the APP WT/WT + PS-1 KI/KI genotype. Between 3–4 mice were examined at 18–24 months of age for the following genotypes: APP WT/WT + PS-1 WT/WT; APP KI/KI + PS-1 KI/WT; APP KI/KI + PS-1 KI/KI; APP WT/WT + PS-1 KI/KI.
Quantitative immunoblotting
Doublecortin content of the hippocampus and olfactory bulb was measured for APP KI/KI + PS-1 WT/WT, APP KI/KI + PS-1 KI/WT, and APP KI/KI + PS-1 KI/KI mouse lines, using methods described previously (Siman et al., 2001; Siman and Salidas, 2004). Briefly, brain regions were homogenized by brief sonication in electrophoresis sample buffer, heated to 90°C for 5 min, and stored at −80°C. Protein concentration was determined by the Bradford method. For each sample, 30 μg hippocampal protein or 6 μg olfactory bulb protein was fractionated on 10% SDS gels and the proteins transferred to PVDF membranes. The membranes were probed with goat anti-doublecortin (1/1000), followed by donkey anti-goat-horseradish peroxidase. Proteins were visualized by enhanced chemiluminescence using Kodak Bio-Max film. Doublecortin was detected as a characteristic Mr~42 kDa polypeptide band. Doublecortin band densities were determined from digitized images of the films using Image Quant software. Serial dilution of a sample confirmed that band densities varied in linear proportion with the amount of doublecortin loaded. Some of the blots were reprobed for neurofilament 68 (rabbit anti-NFL, Sigma; 1/30,000), and band densities were measured using the same method. Between 5–6 mice of each genotype for used for the quantitative analysis.
Statistical Analyses
The data are shown as mean +/− SEM. The numbers of MCM2-positive neural stem and progenitor cells and doublecortin-positive immature neurons were compared across the different genotypes using a one-way ANOVA followed by post hoc Scheffe test. The Western blot analysis of doublecortin content across different genotypes was performed using both a one-way ANOVA and a t-test.
Results
Effects of FAD-linked APP and PS-1 knock-in mutations on hippocampal neuroblasts
We investigated whether FAD-linked knock-in mutations in APP (Swedish KM670/671NL mutation), PS-1 (P264L mutation), or both genes influence the fidelity of neurogenesis in the adult hippocampus using cell type-specific markers. These mouse lines are especially well-suited for evaluating the impact of FAD-linked mutations on neurogenesis. The mutant genes and protein products are expressed at physiological levels, and with all endogenous controls regulating cell type- and developmental stage-specific expression. Furthermore, both knock-in mutations are phenotypically active, and the APP/PS-1 double knock-in mutation leads to brain region- and aging-dependent Alzheimer-type amyloid neuropathology (Reaume et al., 1996; Siman et al., 2000; Flood et al., 2002; Siman and Salidas, 2004).
To label differentiating neurons in the dentate gyrus, sections from 8–9 month old mice were immunostained for the immature neuronal marker doublecortin. This protein is expressed only in developing neuroblasts in the adult brain, as evidenced by cellular morphology and electrophysiologic properties, and confirmed by multiple labeling studies incorporating the mitotic marker bromodeoxyuridine and the neuron-specific marker NeuN (Rao and Shetty, 2004; Overstreet-Wadich and Westbrook, 2006). As shown in Figure 1, in hippocampus these immature neurons were confined to the subgranular zone immediately adjacent to both the superior and inferior blades of the dentate granule cell layer. Doublecortin immunoreactivity extended from the neuroblast perikarya to the developing dendritic arbors in the molecular layer. The dorsal hippocampus extending from its medial border laterally to the start of the ventral hippocampus contained, on average, about 1,400 doublecortin-positive differentiating neuroblasts. Their number did not differ overtly between wild-type mice and those carrying the APP knock-in mutation. The PS-1 knock-in mutant hippocampus exhibited only a small decrease in the number of doublecortin-positive immature neurons. In sharp contrast, doublecortin-positive neuroblasts were markedly reduced in the subgranular zone of the APP/PS-1 double knock-in mutant mouse. Quantitative cell counting analysis demonstrated that mutant APP alone caused <5% change in neuroblast number (Figure 4; N=5–6/genotype), while the addition of one mutant PS-1 allele led to an ~20% decline (N=3; not significant). In the dentate gyrus of the APP/PS-1 double knock-in mutant mouse, the number of differentiating neuroblasts was reduced by 50% (N=6; p<0.01).
Figure 1.
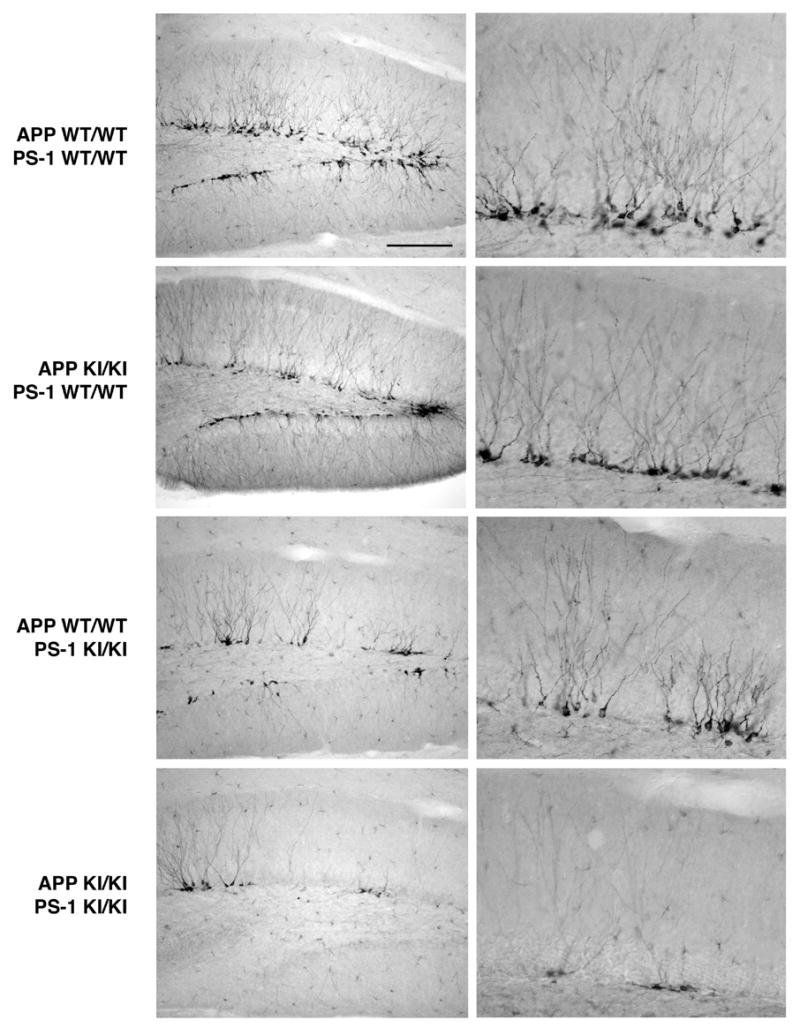
Developing neurons in adult hippocampus: effects of FAD-linked knock-in mutations in APP, PS-1, and both genes. Sagittal sections from 8–9 month old mice of the indicated genotypes were immunostained for the neuroblast marker doublecortin. Low (left panels) and high (right panels) magnification views of the dentate gyrus at a mid-mediolateral level. Note the abundance of immature neurons in the subgranular zone adjacent to the granule cell layer of both the suprapyramidal and infrapyramidal blades of the dentate gyrus. The homozygous APP Swedish knock-in mutation had no effect on immunopositive neuroblasts, whereas the homozygous PS-1 P264L mutation led to a small decrease in doublecortin-positive neuroblast number. In contrast to the effects of the mutations alone, the combined homozygous mutations in both APP and PS-1 caused a marked reduction in the number of developing neurons. Scale bar = 200 μm (left panels), 50 μm (right panels).
Figure 4.
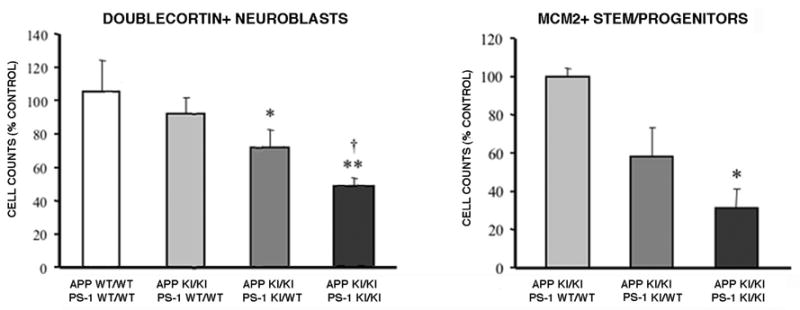
Quantitative analysis of neuroblasts and neural stem/progenitors as functions of APP and PS-1 genotype. The numbers of doublecortin-positive immature neurons and MCM2-positive neural stem and progenitors were measured for the entire dorsal extent of the hippocampal dentate gyrus, as described in Materials and Methods. When combined with the homozygous APP Swedish knock-in mutation, one PS-1 P264L mutant allele caused a small decline in both types of cell populations. Significant decreases in the neuroblast and neural stem/progenitor populations of greater magnitude were observed for the APP/PS-1 double homozygous knock-in mutation. (A) *p<0.05, **p<0.01 versus wild type mice, †p<0.01 versus APP KI/KI + PS-1 WT/WT mice; (B) *p<0.05 versus APP KI/KI + PS-1 WT/WT mice, ANOVA post hoc comparison.
The impairment in neurogenesis is long-lasting in the APP/PS-1 double knock-in mouse hippocampus
During aging, there is a substantial decline in neurogenesis in the hippocampal dentate gyrus, as evidenced both by doublecortin immunostaining and bromodeoxyuridine incorporation (Cameron and McKay, 1999; Jin et al., 2003). This was readily apparent in our own hands as well, as the number of doublecortin-positive neuroblasts in the 18–24 month old mouse brain declined by ~60% when compared with the 8–9 month old hippocampus (Figure 2). Consistent with our observations of the younger mice, at 18–24 months of age the APP and PS-1 knock-in mutations by themselves had only small effects on the number of doublecortin-positive immature neurons. On the other hand, the APP/PS-1 double knock-in mutant mouse exhibited a marked decrease in the number of immunopositive neuroblasts. This provides evidence that the decrement in hippocampal neurogenesis in the double knock-in mutant mouse genetic model of FAD is long-lasting.
Figure 2.
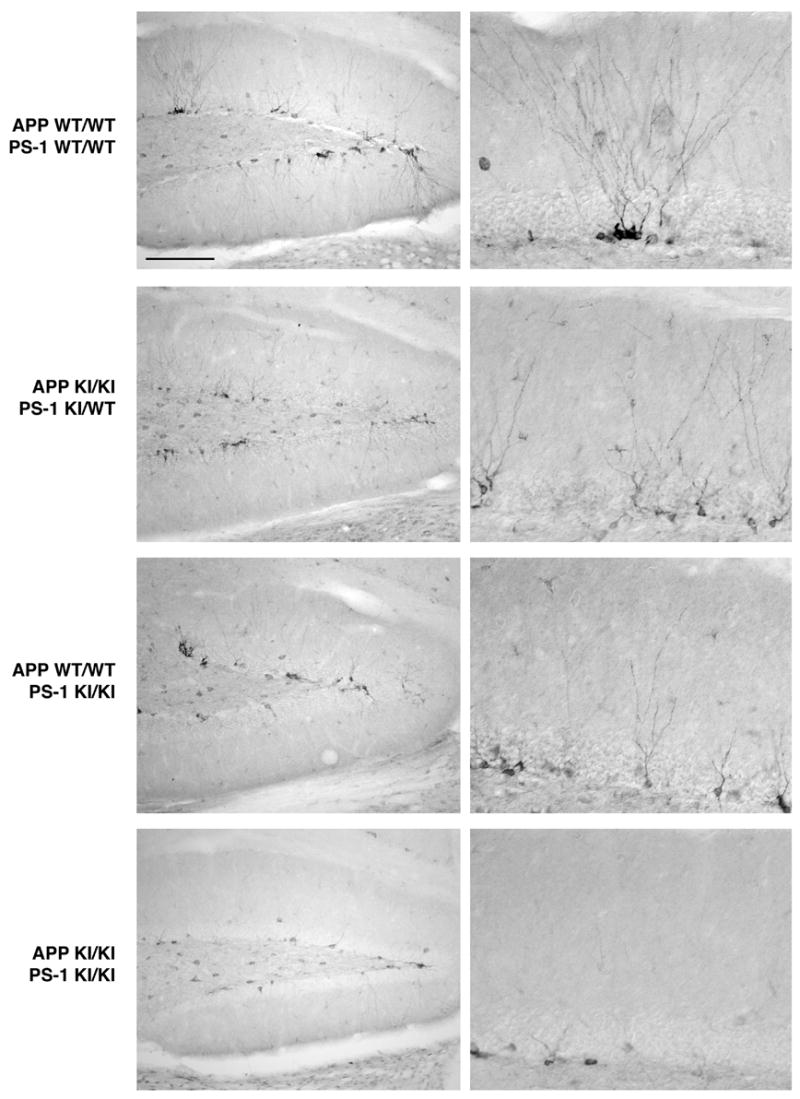
Defective neurogenesis in the APP/PS-1 double knock-in mutant mouse hippocampus persists during aging. Sagittal sections from 18–20 month old mice of the indicated genotypes immunostained for doublecortin. With aging, there is a reduction in the number of developing neurons in the subgranular zone when compared with the 8–9 month age. As seen for the earlier age, the APP knock-in mutation does not affect neuroblast number and the PS-1 knock-in mutation causes only a small decrease. On the other hand, the APP/PS-1 double knock-in mutation led to a large reduction in the number of developing neurons. Scale bar = 200 μm (left panels), 50 μm (right panels).
Evidence that the APP/PS-1 double knock-in mutant mouse hippocampus has reduced numbers of neural stem/progenitor cells
Differentiating neurons in the adult hippocampus arise from a population of neural progenitor cells with relatively rapid doubling times that reside in the subgranular zone. This pool is, in turn, populated by relatively slowly dividing multipotential stem cells that are co-resident (Maslov et al., 2004). Both the stem and progenitor cell populations in hippocampus can be detected by immunostaining for MCM2, a marker for dividing cells (Maiorano et al., 2006). The decrement in immature neurons of the APP/PS-1 double knock-in mutant mouse could arise from a decline in the progenitor and/or stem cell pools, a failure of the progenitor pool to undergo neuronal differentiation, or a decreased survival of the immature neurons. To begin distinguishing between these possibilities, we used MCM2 immunoreactivity to examine the neural stem and progenitor cell population as a function of APP and PS-1 genotype. As shown in Figure 3, the 8–9 month old mouse hippocampus contained a small number of MCM2 positive cells that were concentrated in the subgranular zone known to be populated by neural stem/progenitor cells. The dorsal hippocampus extending from its medial border laterally to the start of ventral hippocampus contained, on average, about 500 MCM2-positive stem and progenitor cells. In comparison with APP knock-in mutant mice that were wild-type for PS-1, one mutant PS-1 allele had only a small effect on the number of MCM2-positive neural stem/progenitor cells, but the APP/PS-1 double knock-in mutant had substantially fewer neural stem/progenitor cells. A pilot comparison of wild-type mice with those homozygous for the APP knock-in mutation found no discernable difference in this cell population (data not shown). Quantitative analysis demonstrated that in the presence of mutant APP, one mutant PS-1 allele caused a trend toward decreased neural/stem progenitor cell numbers, but a marked and statistically significant decline of over 70% was observed for the APP/PS-1 double mutant mouse (Figure 4). These data provide evidence that a decline in the neural stem/progenitor cell population contributes to the loss of immature neurons with the APP/PS-1 double mutation.
Figure 3.
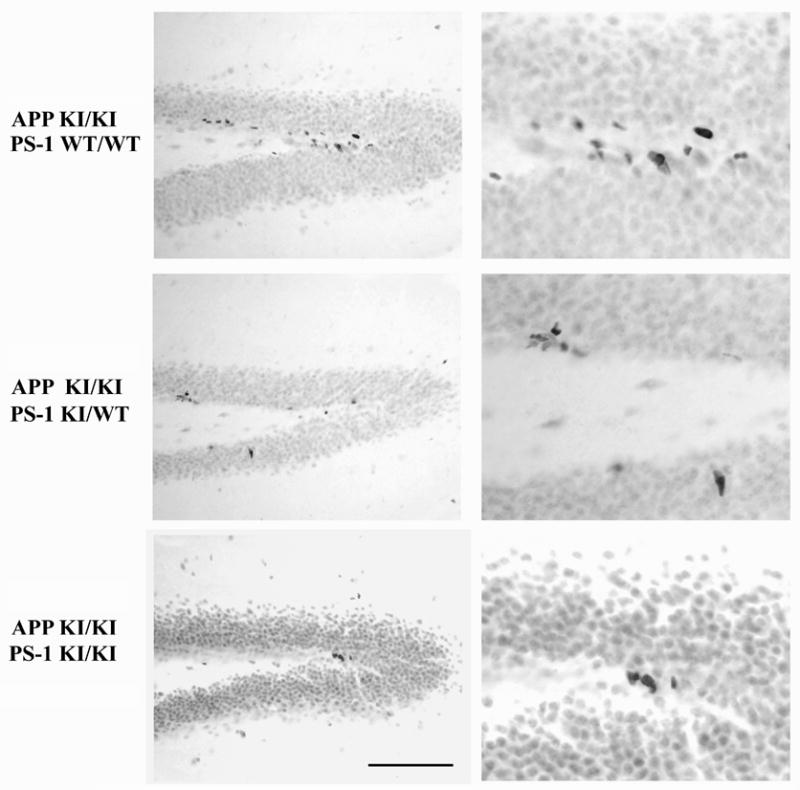
The PS-1 knock-in mutation reduces the number of neural stem and progenitor cells in the hippocampus when combined with the APP knock-in mutation. Sagittal sections from 8–9 month old mice of the indicated genotypes immunostained for the marker of cycling cells MCM2. Cells intensely stained for this marker are found along the dentate subgranular zone. For mice homozygous for the APP Swedish knock-in mutation, the addition of one mutant PS-1 allele caused a small decrease in immunopositive neural stem and progenitor cells. Two mutant PS-1 alleles led to a further decline in these cell populations. Scale bar = 100 μm (A,B), 40 μm (C,D).
Similar to the reduction in doublecortin-immunopositive differentiating neuroblasts, the number of MCM2-positive neural stem and progenitors also was markedly lower at 18–24 months of age (data not shown). The absolute numbers of immunopositive cells in the aged hippocampus was too low to permit meaningful quantitative comparisons across the genotypes.
Doublecortin content is reduced in the hippocampal but not the olfactory bulb neurogenic system by the APP/PS-1 double knock-in mutations
The abundance of doublecortin-positive neuroblasts in the adult mouse olfactory bulb made their quantitative analysis by cell counting complex. As an alternative approach for assessing neurogenesis in the subventricular-olfactory bulb system and comparing it to the subgranular-hippocampal system, we used quantitative Western blotting to measure doublecortin content. As shown in Figure 5, doublecortin was detectable in the 8–9 month old mouse hippocampus as an ~42 kDa immunostained polypeptide. In comparison with mice homozygous for mutant APP and wild-type for PS-1, one mutant PS-1 allele did not change hippocampal doublecortin content. On the other hand, hippocampal doublecortin levels were substantially lower in the APP/PS-1 double knock-in mutant. To control for sample loading, the blots were re-probed for the neuronal marker neurofilament protein NF68. There was no effect of PS-1 genotype on hippocampal NF68 content. The ratio of doublecortin/NF68 decreased by over 50% with the APP/PS-1 double knock-in mutations (p<0.05). The olfactory bulb contained much higher levels of doublecortin than the hippocampus (Figure 5; olfactory bulb samples were loaded with only 20% of the total protein of the hippcampal samples). In mice carrying the APP knock-in mutation, neither one nor two mutant PS-1 alleles caused a decline in doublecortin levels in the olfactory bulb.
Figure 5.
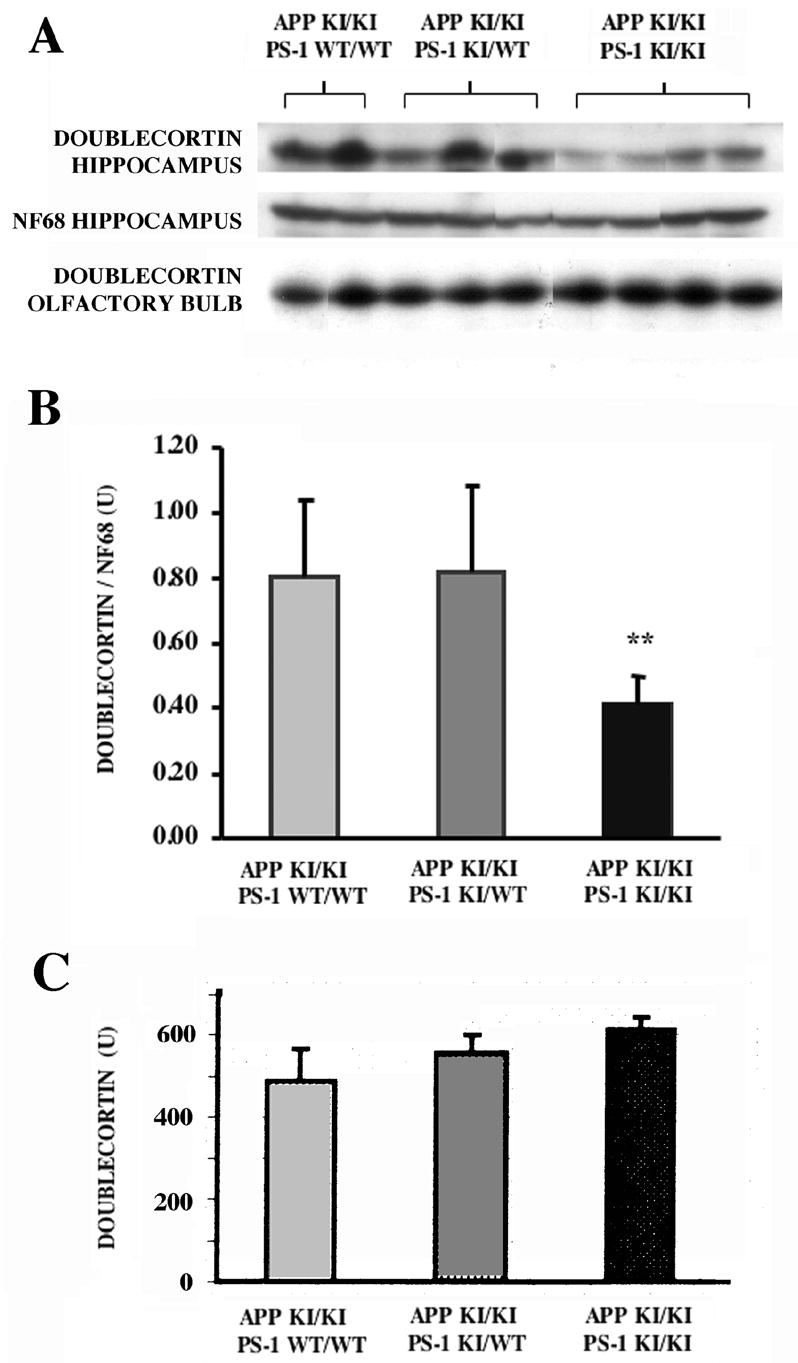
Western blot analysis of doublecortin content of hippocampus and olfactory bulb as a function of APP and PS-1 genotype. The regions were dissected from 8–9 month old mice of the indicated APP and PS-1 genotypes, and the extracts probed for the ~42 kDa doublecortin polypeptide as described in Materials and Methods. In addition, the hippocampal samples were reprobed for neurofilament L (NF68). (A) Western blots illustrating that the APP/PS-1 double knock-in mutation decreased hippocampal doublecortin content when compared with APP knock-in mutant mice bearing either zero or one mutant PS-1 allele. (B,C) Quantitative analysis of doublecortin content for hippocampus (B) and olfactory bulb (C). Hippocampal doublecortin levels are represented as the ratio of the densities of the doublecortin and neurofilament L polypeptides. Essentially identical results were obtained from analysis of doublecortin alone. The decrease in doublecortin levels in the hippocampus of the APP/PS-1 double knock-in mutant mouse is significant (p<0.05, t-test; p=.06, ANOVA). In marked contrast, there was no appreciable difference as a function of PS-1 gneotype in doublecortin content of the olfactory bulb.
Amyloid pathology is extensive in the hippocampal dentate gyrus of the APP/PS-1 double knock-in mutant mouse
To assess the temporal and spatial progression of amyloid pathology in neurogenic regions of the APP/PS-1 double knock-in mutant mouse, sections from 3,6, 9, and 18 month old mice were immunostained for Aβ. As illustrated in Figure 6, sparse Aβ-positive amyloid deposits first appeared in the hippocampal formation (and also the neocortex) at 6 months of age. In contrast, the APP and PS-1 knock-in mutations by themselves elicited no amyloid deposition in the mouse up to 20 months of age, confirming prior studies (Reaume et al., 1996; Siman et al., 2000; Flood et al., 2002). The amyloid deposition in the double mutant mouse brain progressed roughly linearly over time (Flood et al., 2002), and by 18 months of age the outer molecular layer of the dentate gyrus contained a massive amyloid burden (Figure 6C). In contrast, the striatum and corpus callosum, which border the subventricular neurogenic zone, were virtually devoid of amyloid deposition (Flood et al., 2002; Siman and Salidas, 2004; unpublished observations).
Figure 6.
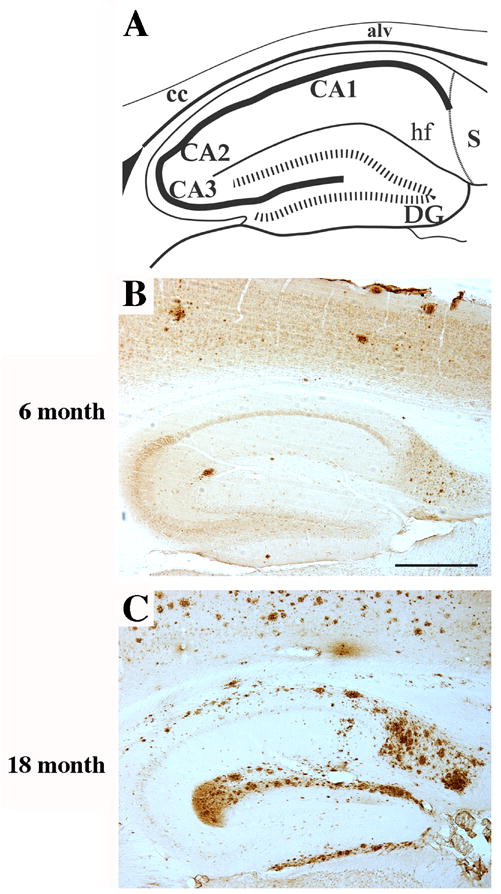
Amyloid deposition is extensive in the dentate gyrus of the APP/PS-1 double knock-in mutant mouse. Sagittal sections from either 6 (B) or 18 (C) month old APP/PS-1 double knock-in mutant mice were immunostained for amyloid Aβ. (A) Schematic diagram of mouse dorsal hippocampus in the sagittal plane at mid-mediolateral level. Amyloid deposits became detectable in the dentate gyrus starting at 6 months of age. By 18 months, amyloid plaque formation was extensive especially in the outer molecular layer of the dentate gyrus. Scale bar = 500 μm.
Amyloid-associated microgliosis as a potential contributor to altered hippocampal neurogenesis
A well established inhibitor of neurogenesis in the adult rodent hippocampus is microglial-mediated neuroinflammation (Ekdahl et al., 2003; Monje et al., 2003). Microgliosis and induction of proinflammatory cytokines have been associated with amyloid deposition in the hippocampal dentate gyrus in a number of transgenic mouse models of AD (Bornemann et al., 2001; Wilcock et al., 2003; Dudal et al., 2004), but the microglial response to amyloid deposition has not been characterized before in the APP/PS-1 double knock-in mutant mouse. Consequently, we probed sections from non-amyloid bearing APP knock-in mice and amyloid bearing APP/PS-1 double knock-in mice with markers for activated microglia. We used antibodies F4/80 and anti-CD45, which label activated cells of hematopoetic lineage (phagocytic macrophages and microglia), and have been reported to stain activated microglia in the vicinity of amyloid plaques in transgenic mouse models (Bornemann et al., 2001; Wilcock et al., 2003). There was little immunostaining in the 18–24 month old APP knock-in mouse hippocampus and neocortex using these two marker antibodies (Figure 7A; data not shown). However, regions of the neocortex and hippocampus with extensive amyloid pathology contained large numbers of activated microglia immunopositive for the two markers (Figure 7B–D). Under high magnification, labeled cells had small cell bodies and thin, highly ramified processes characteristic of microglia. Essentially identical results were obtained using antibodies to CD16/32, another marker of activated microglia. These results demonstrate microgliosis occurs in the adult APP/PS-1 double knock-in mouse in spatial proximity to amyloid deposition.
Figure 7.
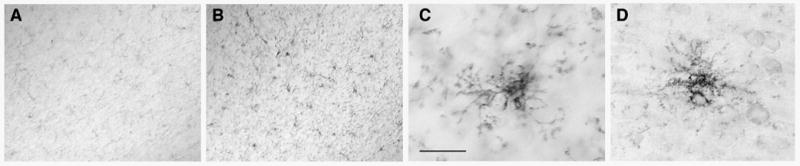
Microglial activation in association with amyloid deposition in the APP/PS-1 double knock-in mutant mouse. Sagittal sections of 18 month old mouse dentate gyrus immunostained for the activated microglial markers F4/80 (A–C) and CD45 (D). (A) APP KI/KI + PS-1 WT/WT. (B–D) APP KI/KI + PS-1 KI/KI. The numbers of cells immunostained for the markers of activated microglia were markedly higher for the double knock-in mutant mouse hippocampus. At high magnification (C,D), immunopositive cells had the small cell bodies and thin, highly ramified processes characteristic for microglia. Scale bar = 200 μm (A,B), 25 μm (C,D).
Discussion
We report here that there is a long-lasting impairment in adult hippocampal neurogenesis in the APP/PS-1 double knock-in mutant mouse model of FAD. In 8–9 month old mice, the subgranular zone of the dentate gyrus contains significantly fewer doublecortin-positive immature neurons and MCM2-positive neural stem and progenitor cells. In contrast to the APP/PS-1 double knock-in mutant, the APP knock-in mutation alone does not affect markers for adult hippocampal neurogenesis, and the effects of the PS-1 knock-in mutation are relatively small. The defects in neural plasticity in the double mutant mouse hippocampus persist to at least 18 months of age, and are measurable by both quantitative immunohistochemical and immunoblot methods. Furthermore, doublecortin content is reduced in the hippocampus but not the olfactory bulb, thereby providing evidence that the defects are specific for the subgranular-hippocampal but not the subventricular-olfactory bulb neurogenic system. Our findings provide clear evidence that an FAD-causing double gene mutation leads to selective impairment in adult hippocampal neurogenesis even in the absence of ectopic overexpression of mutant protein.
Functional studies of neurogenesis in the adult mouse hippocampus support the concept that this form of neural plasticity is an important regulator of long-term memory (Rola et al., 2004; Aimone et al., 2006). In addition, mechanistically and chemically distinct classes of antidepressant drugs stimulate adult hippocampal neurogenesis, leading to the hypothesis that the process is an important regulator of mood (Jacobs et al., 2002; Dranovsky and Hen, 2006). Given that abnormalities in cognition and mood are either a fundamental component of the behavioral syndrome of AD or a common co-morbid occurrence, respectively, it is vital to determine whether FAD-causing PS-1 and APP mutations affect neurogenesis in the adult brain as part of their pathophysiologic mechanism. PS-1 is expressed in neural stem/progenitor cells in the hippocampal subgranular zone (Wen et al., 2002), and its genetic deletion impairs neurogenesis both during development and in response to environmental cues in adulthood (Shen et al., 1997; Feng et al., 2001). Additionally, intracerebroventricular injection of either APP or the Aβ protein reportedly modulates neurogenesis (Haughley et al., 2002; Caile et al., 2004). Transgenic overexpression of mutant APP, PS-1, or both can reportedly increase or decrease neurogenesis in the adult hippocampus (Wen et al., 2002, 2004; Wang et al., 2004; Jin et al., 2004a; Chevallier et al., 2005). Unfortunately, all of the transgenic studies were conducted using heterologous promoters to drive high level expression preferentially in terminally differentiated neurons. Therefore, the mutant proteins are not present in the stem, progenitor, and neuroblast populations under study, and the relevance of the findings for neurogenesis in the AD brain is unclear. One study reported that end-stage AD patients have increased numbers of immature hippocampal neurons (Jin et al., 2004b). However, this end-stage analysis is not substantiated by a study of pre-senile cases of probable AD (Boekhoorn et al., 2006), and does not address the potential role for impaired neurogenesis in the onset and progression of disease.
The current study circumvents these complexities by studying the effects of FAD-linked mutations introduced into their endogenous genes, and evaluated in the mouse during progression of disease. Our findings demonstrate that, in the absence of ectopic overexpression, neither the Swedish double mutation in APP nor the P264L mutation in PS-1 has a major effect on adult neurogenesis. On the other hand, the co-expression of both knock-in mutations leads to extensive amyloid deposition and reduced numbers of neural stem and progenitors as well as differentiating neuroblasts in the adult dentate gyrus. Our study made use of doublecortin and MCM2 as specific and selective markers for immature neuroblasts and multipotential neural stem and progenitors, respectively. The efficacy of doublecortin as a marker for analyzing the total number of newly generated neurons in the adult rodent brain was established by Rao and Shetty (2004). They demonstrated that more than 90% of the doublecortin-expressing cells in the dentate subgranular zone are also positive for the mitotic marker bromodeoxyuridine after its long-duration administration. Furthermore, the doublecortin-positive cells co-express additional markers for neurons, but not other brain cell types. MCM2 is a G1 phase cell cycle marker and is expressed preferentially in dividing cells (Maiorano et al., 2006). It has been shown to specifically label both slowly cycling neural stem cells and rapidly proliferating progenitors in the adult mouse hippocampus (Maslov et al. 2004).
Conceivably, multiple mechanisms may contribute to lowering the number of differentiating neuroblasts in the adult hippocampus of the APP/PS-1 double knock-in mutant mouse. These include defective neural differentiation, excessive death of developing neurons, and reduction in the size of the progenitor cell pool through decreases in either their proliferation or survival. Our finding that the loss of MCM2-positive stem and progenitor cells is at least as great as the reduction in immature neurons provides evidence that a decline in the neural stem/progenitor cell pool is a major factor in the loss of newly generated neurons. Using an antibody marker for caspase substrate cleavage, we could not detect a change in the rate of apoptosis in the subgranular zone (data not shown). However, apoptotic cells are cleared rapidly and their steady-state numbers are extremely small in the adult brain, and so an additional contribution of increased neuroblast vulnerability to degeneration cannot be completely ruled out. Furthermore, our studies focused on steady-state measures of the stem/progenitor and neuroblast cell populations. Additional kinetic analyses in the APP/PS-1 double knock-in mutant mouse may be useful for distinguishing the contributions of alterations in proliferation, differentiation, and survival to the long-lasting decline in hippocampal neurogenesis. Finally, the impact of a long-lasting deficit in hippocampal neurogenesis on structure and function of the neurons, glia, and vasculature of the dentate gyrus will require additional study.
Across the genotypes we examined, the impairment in neurogenesis is paralleled by the occurrence of amyloid deposition and microgliosis. Thus, doublecortin-positive neuroblasts and MCM2-positive neural stem and progenitors are reduced markedly and significantly in mice carrying FAD-linked knock-in mutations in both their APP and PS-1 genes, whereas the PS-1 knock-in mutation alone has only small effects (see also Wang et al., 2004) and the APP knock-in mutation alone is ineffective. Similarly, amyloid deposition develops in circumscribed brain regions in the double mutant mouse but not in either single mutant line, even at an advanced age. By 8–9 months of age, amyloid deposits are detectable in the hippocampal dentate gyrus, where by later ages the amyloid burden becomes massive, particularly in the outer molecular layer. Our findings indicate that neither elevated brain Aβ42 concentrations nor mutant PS-1 alone are fully responsible for impairing adult hippocampal neurogenesis in the APP/PS-1 double knock-in mutant mouse model of FAD, and instead support the possibility that defective neurogenesis may be triggered by the amyloid deposition. Consistent with this hypothesis is the finding that doublecortin content is unaltered in the olfactory bulb (Figure 5), which receives neuroblasts originating from the subventricular zone. In contrast to the subgranular zone, which is surrounded by extensive amyloid deposits, the subventricular zone and surrounding striatum and corpus callosum are largely devoid of amyloid. Future studies evaluating our mouse model at stages presymptomatic for amyloid deposition and manipulating the pathology pharmacologically could be used to test this hypothesis.
How might amyloid deposition change the local milieu to impair neurogenesis? One possible intermediary is microglia-mediated neuroinflammation. In addition to the amyloid deposition, the APP/PS-1 double knock-in mutant mouse also exhibits microgliosis in proximity to the amyloid, as evidenced using 3 distinct markers for activated microglia. Experimental treatments that induce microgliosis and neuroinflammation interfere with neurogenesis in the adult hippocampus, and anti-inflammatory treatments prevent the defect (Ekdahl et al., 2003; Monje et al., 2003). The hypothesis that amyloid deposition and its attendant microglia-mediated neuroinflammation lead to impaired hippocampal neurogenesis may have important implications not only for the relatively rare FAD, but for all forms of the disease, for which extensive amyloid deposition and neuroinflammation in the hippocampal dentate gyrus are signature pathologies (Rogers et al., 1996; Xiang et al., 2006). Further studies are required to assess whether hippocampal neurogenesis is impaired in the AD brain during the course of the disease, evaluate the potential influences of amyloid deposition and neuroinflammation, and identify potential therapeutic strategies for stimulating neurogenesis and discerning the resulting impact on the cognitive and behavioral syndrome of AD.
Acknowledgments
This study was supported by NIH grant AG17138 to R.S. We thank Cephalon, Inc. (West Chester, PA) for the generous provision of breeding pairs of APP and PS-1 mutant knock-in mutant mice, and Sheryl Salidas for technical assistance.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aimone JB, Wiles J, Gage FH. Potential role for adult neurogenesis in the encoding of time in new memories. Nat Neurosci. 2006;9:723–727. doi: 10.1038/nn1707. [DOI] [PubMed] [Google Scholar]
- Ashe KH. Learning and memory in transgenic mice modeling Alzheimer's disease. Learn Mem. 2001;8:301–308. doi: 10.1101/lm.43701. [DOI] [PubMed] [Google Scholar]
- Bornemann KD, Wiederhold KH, Pauli C, Ermini F, Stalder M, Schnell L, Sommer B, Jucker M, Staufenbiel M. Abeta-induced inflammatory processes in microglia cells of APP23 transgenic mice. Am J Pathol. 2001;158:63–73. doi: 10.1016/s0002-9440(10)63945-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, de Vos RA, Jansen EN, Bratzke H, Braak E. Neuropathological hallmarks of Alzheimer's and Parkinson's diseases. Prog Brain Res. 1998;117:267–285. doi: 10.1016/s0079-6123(08)64021-2. [DOI] [PubMed] [Google Scholar]
- Cameron HA, McKay RD. Restoring production of hippocampal neurons in old age. Nat Neurosci. 1999;2:894–897. doi: 10.1038/13197. [DOI] [PubMed] [Google Scholar]
- Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, Koo EH. Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol. 2005;167:151–159. doi: 10.1016/S0002-9440(10)62962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranovsky A, Hen R. Hippocampal neurogenesis: regulation by stress and antidepressants. Biol Psychiatry. 2006;59:1136–1143. doi: 10.1016/j.biopsych.2006.03.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudal S, Krzywkowski P, Paquette J, Morissette C, Lacombe D, Tremblay P, Gervais F. Inflammation occurs early during the Abeta deposition process in TgCRND8 mice. Neurobiol Aging. 2004;25:861–871. doi: 10.1016/j.neurobiolaging.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Ekdahl CT, Claasen JH, Bonde S, Kokaia Z, Lindvall O. Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A. 2003;100:13632–13637. doi: 10.1073/pnas.2234031100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng R, Rampon C, Tang YP, Shrom D, Jin J, Kyin M, Sopher B, Miller MW, Ware CB, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32:911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Flood DG, Reaume AG, Dorfman KS, Lin YG, Lang DM, Trusko SP, Savage MJ, Annaert WG, De Strooper B, Siman R, Scott RW. FAD mutant PS-1 gene-targeted mice: increased A beta 42 and A beta deposition without APP overproduction. Neurobiol Aging. 2002;23:335–348. doi: 10.1016/s0197-4580(01)00330-x. [DOI] [PubMed] [Google Scholar]
- Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- Handler M, Yang X, Shen J. Presenilin-1 regulates neuronal differentiation during neurogenesis. Development. 2000;127:2593–2606. doi: 10.1242/dev.127.12.2593. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Liu D, Nath A, Borchard AC, Mattson MP. Disruption of neurogenesis in the subventricular zone of adult mice, and in human cortical neuronal precursor cells in culture, by amyloid beta-peptide: implications for the pathogenesis of Alzheimer's disease. Neuromolecular Med. 2002;1:125–135. doi: 10.1385/NMM:1:2:125. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Holcomb L, Gordon MN, McGowan E, Yu X, Benkovic S, Jantzen P, Wright K, Saad I, Mueller R, Morgan D, Sanders S, Zehr C, O'Campo K, Hardy J, Prada CM, Eckman C, Younkin S, Hsiao K, Duff K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat Med. 1998;4:97–100. doi: 10.1038/nm0198-097. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Jacobs BL, Praag H, Gage FH. Adult brain neurogenesis and psychiatry: a novel theory of depression. Mol Psychiatry. 2002;5:262–269. doi: 10.1038/sj.mp.4000712. [DOI] [PubMed] [Google Scholar]
- Jin K, Sun Y, Xie L, Batteur S, Mao XO, Smelick C, Logvinova A, Greenberg DA. Neurogenesis and aging: FGF-2 and HB-EGF restore neurogenesis in hippocampus and subventricular zone of aged mice. Aging Cell. 2003;2:175–183. doi: 10.1046/j.1474-9728.2003.00046.x. [DOI] [PubMed] [Google Scholar]
- Jin K, Galvan V, Xie L, Mao XO, Gorostiza OF, Bredesen DE, Greenberg DA. Enhanced neurogenesis in Alzheimer's disease transgenic (PDGF-APPSw, Ind) mice. Proc Natl Acad Sci U S A. 2004a;101:13363–13367. doi: 10.1073/pnas.0403678101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, Peel AL, Mao XO, Xie L, Cottrell BA, Henshall DC, Greenberg DA. Increased hippocampal neurogenesis in Alzheimer's disease. Proc Natl Acad Sci U S A. 2004b;101:343–347. doi: 10.1073/pnas.2634794100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lleo A, Berezovska O, Growdon JH, Hyman BT. Clinical, pathological, and biochemical spectrum of Alzheimer disease associated with PS-1 mutations. Am J Geriatr Psychiatry. 2004;12:146–156. doi: 10.1097/00019442-200403000-00006. [DOI] [PubMed] [Google Scholar]
- Maiorano D, Lutzmann M, Mechali M. MCM proteins and DNA replication. Curr Opin Cell Biol. 2006;18:130–136. doi: 10.1016/j.ceb.2006.02.006. [DOI] [PubMed] [Google Scholar]
- Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760–1765. doi: 10.1126/science.1088417. [DOI] [PubMed] [Google Scholar]
- Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24:1726–1733. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacher J, Crespo C, McEwen BS. Doublecortin expression in the adult rat telencephalon. Eur J Neurosci. 2001;14:629–644. doi: 10.1046/j.0953-816x.2001.01683.x. [DOI] [PubMed] [Google Scholar]
- Overstreet-Wadiche LS, Westbrook GL. Functional maturation of adult-generated granule cells. Hippocampus. 2006;16:208–215. doi: 10.1002/hipo.20152. [DOI] [PubMed] [Google Scholar]
- Reaume AG, Howland DS, Trusko SP, Savage MJ, Lang DM, Greenberg BD, Siman R, Scott RW. Enhanced amyloidogenic processing of the beta-amyloid precursor protein in gene-targeted mice bearing the Swedish familial Alzheimer's disease mutations and a "humanized" Abeta sequence. J Biol Chem. 1996;271:23380–23388. doi: 10.1074/jbc.271.38.23380. [DOI] [PubMed] [Google Scholar]
- Rogers J, Webster S, Lue LF, Brachova L, Civin WH, Emmerling M, Shivers B, Walker D, McGeer P. Inflammation and Alzheimer's disease pathogenesis. Neurobiol Aging. 1996;17:681–686. doi: 10.1016/0197-4580(96)00115-7. [DOI] [PubMed] [Google Scholar]
- Rola R, Raber J, Rizk A, Otsuka S, VandenBerg SR, Morhardt DR, Fike JR. Radiation-induced impairment of hippocampal neurogenesis is associated with cognitive deficits in young mice. Exp Neurol. 2004;188:316–330. doi: 10.1016/j.expneurol.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Savage MJ, Kawooya JK, Pinsker LR, Emmons TL, Mistretta S, Siman R, Greenberg BD. Elevated Aβ levels in Alzheimer's disease brain are associated with selective accumulation of Aβ42 in parenchymal amyloid plaques and both Aβ40 and Aβ42 in cerebrovascular deposits. Amyloid Int J Exp Clin Invest. 1995;2:234–240. [Google Scholar]
- Schaffer DV, Gage FH. Neurogenesis and neuroadaptation. Neuromolecular Med. 2004;5:1–9. doi: 10.1385/NMM:5:1:001. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Siman R, Reaume AG, Savage MJ, Trusko S, Lin YG, Scott RW, Flood DG. Presenilin-1 P264L knock-in mutation: differential effects on abeta production, amyloid deposition, and neuronal vulnerability. J Neurosci. 2000;20:8717–8726. doi: 10.1523/JNEUROSCI.20-23-08717.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siman R, Salidas S. Gamma-secretase subunit composition and distribution in the presenilin wild-type and mutant mouse brain. Neuroscience. 2004;129:615–628. doi: 10.1016/j.neuroscience.2004.08.028. [DOI] [PubMed] [Google Scholar]
- St George-Hyslop PH. Genetic factors in the genesis of Alzheimer's disease. Ann N Y Acad Sci. 2000;924:1–7. doi: 10.1111/j.1749-6632.2000.tb05552.x. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. Twenty years of the Alzheimer's disease amyloid hypothesis: a genetic perspective. Cell. 2005;120:545–555. doi: 10.1016/j.cell.2005.02.008. [DOI] [PubMed] [Google Scholar]
- Wang R, Dineley KT, Sweatt JD, Zheng H. Presenilin 1 familial Alzheimer's disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience. 2004;126:305–312. doi: 10.1016/j.neuroscience.2004.03.048. [DOI] [PubMed] [Google Scholar]
- Wen PH, Friedrich VL, Jr, Shioi J, Robakis NK, Elder GA. Presenilin-1 is expressed in neural progenitor cells in the hippocampus of adult mice. Neurosci Lett. 2002;318:53–56. doi: 10.1016/s0304-3940(01)02485-5. [DOI] [PubMed] [Google Scholar]
- Wen PH, Hof PR, Chen X, Gluck K, Austin G, Younkin SG, Younkin LH, DeGasperi R, Gama Sosa MA, Robakis NK, Haroutunian V, Elder GA. The presenilin-1 familial Alzheimer disease mutant P117L impairs neurogenesis in the hippocampus of adult mice. Exp Neurol. 2004;188:224–237. doi: 10.1016/j.expneurol.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang Z, Haroutunian V, Ho L, Purohit D, Pasinetti GM. Microglia activation in the brain as inflammatory biomarker of Alzheimer's disease neuropathology and clinical dementia. Dis Markers. 2006;22:95–102. doi: 10.1155/2006/276239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Mak K, Vinters HV, Frautschy SA, Cole GM. Monoclonal antibody to the C-terminus of beta-amyloid. Neuroreport. 1994;5:2117–2120. doi: 10.1097/00001756-199410270-00032. [DOI] [PubMed] [Google Scholar]